

Abstract
Background
To infer the tree of life requires knowledge of the common characteristics of each species descended from a common ancestor as the measuring criteria and a method to calculate the distance between the resulting values of each measure. Conventional phylogenetic analysis based on genomic sequences provides information about the genetic relationships between different organisms. In contrast, comparative analysis of metabolic pathways in different organisms can yield insights into their functional relationships under different physiological conditions. However, evaluating the similarities or differences between metabolic networks is a computationally challenging problem, and systematic methods of doing this are desirable. Here we introduce a graph-kernel method for computing the similarity between metabolic networks in polynomial time, and use it to profile metabolic pathways and to construct phylogenetic trees.
Results
To compare the structures of metabolic networks in organisms, we adopted the exponential graph kernel, which is a kernel-based approach with a labeled graph that includes a label matrix and an adjacency matrix. To construct the phylogenetic trees, we used an unweighted pair-group method with arithmetic mean, i.e., a hierarchical clustering algorithm. We applied the kernel-based network profiling method in a comparative analysis of nine carbohydrate metabolic networks from 81 biological species encompassing Archaea, Eukaryota, and Eubacteria. The resulting phylogenetic hierarchies generally support the tripartite scheme of three domains rather than the two domains of prokaryotes and eukaryotes.
Conclusion
By combining the kernel machines with metabolic information, the method infers the context of biosphere development that covers physiological events required for adaptation by genetic reconstruction. The results show that one may obtain a global view of the tree of life by comparing the metabolic pathway structures using meta-level information rather than sequence information. This method may yield further information about biological evolution, such as the history of horizontal transfer of each gene, by studying the detailed structure of the phylogenetic tree constructed by the kernel-based method.
Background
The availability of pathway databases such as Kyoto Encyclopedia of Genes and Genomes (KEGG), What is there? (WIT3), PathDB, and MetaCyc opens up various new possibilities for comparative analysis. In particular, information about metabolic pathways in different organisms yields important information about their evolution and offers a complementary approach to phylogenetic analysis. Here we present a comparative metabolomic approach to constructing phylogenetic trees that uses physiological functions of the organisms by computing the structural similarity of metabolic networks. The consideration of metabolic components complements the conventional approaches to phylogeny based on genome sequences. Recognizing the similarities and differences in metabolic functions between species may provide insights into other applications in biotechnology, ecology, and evolutionary studies. Several researchers have attempted to rebuild evolutionary history by comparing ribosomal RNA sequences [1], by phylogenomics [2], or by comparing whole genomes to overcome the limitations of the gene-sequence analyses [3-5].
Several recent studies have extended conventional phylogenetic analysis to incorporate metabolic pathway information. Forst and Schulten [6,7] presented one of the earliest approaches to extend the conventional sequence comparison and phylogenetic analysis of individual enzymes to metabolic networks. They also presented a method to calculate distances between metabolic networks based on sequence information of the biomolecules involved and information about the corresponding reaction networks. Dandekar et al. (1999) combined strategies in a systematic comparison of the enzymes and corresponding sequence information of the glycolytic pathway [8]. Other approaches involving the reconstructed phylogenies from gene-order data have been based on simulating genome evolution [9], and studying the genome evolution resulting from the metabolic adaptation of the organism to the surrounding environment. Liao et al. (2002) presented a method to group organisms by comparing the profiles of metabolic pathways, where the profiling was based simply on binary attributes (e.g., by denoting the presence or absence of pathways in the organisms) [10].
Whereas the previous approaches incorporated information about the additional metabolic pathways, systematic methods to calculate the similarities between metabolic networks are lacking or contain gaps in some of the biological assumptions. In this paper, we introduce the concept of graph kernels to calculate the similarities between two different network structures. The graph kernel-based approach can compute more efficiently the similarity of two graph structures by the kernel function that can extract important features from the graph. Our approach contrasts with that of Forst and Schulten [6,7] in that the graph kernel calculates the distance based on the network level instead of on its sequence information on the biomolecules involved.
In their comparative analysis of metabolic pathways, Heymans and Singh [11] showed that phylogenetic trees could be made from the graph similarities of metabolic networks. They applied a distance measure between metabolic graphs of the glycolytic pathway and the citric acid cycle from 16 organisms. However, some of their data on phylogenetic inference did not correspond entirely with the conventional taxonomy and did not provide a global view of the specialization of species according to the scale of analyzed species and metabolic pathways.
Several more recent attempts have reconstructed genome trees using different formalisms such as gene ordering [12,13], measuring gene contents [3,14], comparing sequence similarities [15,16], comparing proteome strings [17], and phylogenomics [18,19]. All are based on the principle of genome sequences, but none has applied the concepts of effectible physiology to the phylogenetic analyses. We report on our comparative results and discuss our findings.
Results and discussion
We chose nine pathways of carbohydrate metabolism and 81 species to perform a comparative analysis of metabolic networks that satisfied the most abundant dataset from KEGG (Table 1, 2 and 3). Our sample comprised 13 species of Archaea, eight species of Eukaryota, and 60 species of Eubacteria. The central pathways of metabolism include the glycolytic and pentose phosphate pathways, and the citric acid cycle, which generate biological energy and form the metabolic precursors essential for almost all living cells. To validate the data, we investigated the distribution of each enzyme in the nine pathways. Several enzymes appear at a high frequency in all species, and this frequency decreases rather exponentially as the value of x-axis increases (Figure 1), a phenomenon we observed in all pathways studied. Because the characteristics of the enzyme distribution did not differ, all pathways can be used in the phylogenetic analysis. We observed no obvious tendency of shift or deviation of the distributions from the overall pattern of pathways.
Table 1.
Statistics for the dataset according to the number of enzymes and their relationships
| enzyme | relation | |
| # of total occurrences | 35,134 | 17,567 |
| # of unique elements | 218 | 1,275 |
| max # per organism | 544 | 123 |
| min # per organism | 46 | 26 |
| avg # per organism | 68 | 217 |
| stdev across organisms | 26 | 133 |
Table 2.
The nine reference pathways used in the analysis
| MAP No. (KEGG) | pathway name |
| 00010 | glycolysis/gluconeogenesis |
| 00020 | citrate cycle (TCA cycle) |
| 00030 | pentose phosphate pathway |
| 00051 | fructose and mannose metabolism |
| 00052 | galactose metabolism |
| 00620 | pyruvate metabolism |
| 00630 | glyoxylate and dicarboxylate metabolism |
| 00640 | propanoate metabolism |
| 00650 | butanoate metabolism |
Table 3.
The 81 organisms included in the phylogenetic analysis. Full scientific names were abbreviated into three character notation (Abbr.) and their domain informations in phylogeny were also represented in single character that are Eubacteria (B), Archaea (A) and Eukaryota (E), respectively.
| Abbr. | Domain | Organism | Abbr. | Domain | Organism |
| Aae | B | Aquifex aeolicus | Mth | A | Methanobacterium thermoautotrophicum |
| Ana | B | Anabaena sp. | Mtu | B | Mycobacterium tuberculosis H37Rv |
| Atc | B | Agrobacterium tumefaciens C58 Cereon | Nma | B | Neisseria meningitidis serogroup A |
| Ath | E | Arabidopsis thaliana | Nme | B | Neisseria meningitidis serogroup B |
| Atu | B | Agrobacterium tumefaciens C58 UWash | Oih | B | Oceanobacillus iheyensis |
| Bha | B | Bacillus halodurans | Pab | A | Pyrococcus abyssi |
| Bme | B | Brucella melitensis | Pae | B | Pseudomonas aeruginosa |
| Bsu | B | Bacillus subtilis | Pai | A | Pyrobaculum aerophilum |
| Cac | B | Clostridium acetobutylicum | Pfu | A | Pyrococcus furiosus |
| Ccr | B | Caulobacter crescentus | Pho | A | Pyrococcus horikoshii |
| Cel | E | Caenorhabditis elegans | Pmu | B | Pasteurella multocida |
| Cje | B | Campylobacter jejuni | Rno | E | Rattus norvegicus |
| Cmu | B | Chlamydia muridarum | Rso | B | Ralstonia solanacearum |
| Cpa | B | Chlamydophila pneumoniae AR39 | Sam | B | Staphylococcus aureus MW2 |
| Cpe | B | Clostridium perfringens | Sau | B | Staphylococcus aureus N315 |
| Cpj | B | Chlamydophila pneumoniae J138 | Sav | B | Staphylococcus aureus Mu50 |
| Cpn | B | Chlamydophila pneumoniae CWL029 | Sce | E | Saccharomyces cerevisiae |
| Cte | B | Chlorobium tepidum | Sco | B | Streptomyces coelicolor |
| Ctr | B | Chlamydia trachomatis | Sme | B | Sinorhizobium meliloti |
| Dme | E | Drosophila melanogaster | Spg | B | Streptococcus pyogenes M3 |
| Dra | B | Deinococcus radiodurans | Spm | B | Streptococcus pyogenes M18 |
| Ece | B | Escherichia coli O157 EDL933 | Spo | E | Schizosaccharomyces pombe |
| Ecj | B | Escherichia coli K-12 W3110 | Spy | B | Streptococcus pyogenes |
| Eco | B | Escherichia coli K-12 MG1655 | Sso | A | Sulfolobus solfataricus |
| Ecs | B | Escherichia coli O157 Sakai | Stm | B | Salmonella typhimurium |
| Fnu | B | Fusobacterium nucleatum | Sto | A | Sulfolobus tokodaii |
| Hal | A | Halobacterium sp. | Sty | B | Salmonella typhi |
| Hin | B | Haemophilus influenzae | Syn | B | Synechocystis sp. |
| Hpj | B | Helicobacter pylori J99 | Tac | A | Thermoplasma acidophilum |
| Hpy | B | Helicobacter pylori 26695 | Tel | B | Thermosynechococcus elongatus |
| Hsa | E | Homo sapiens | Tma | B | Thermotoga maritima |
| Lin | B | Listeria innocua | Tpa | B | Treponema pallidum |
| Lla | B | Lactococcus lactis | Tte | B | Thermoanaerobacter tengcongensis |
| Lmo | B | Listeria monocytogenes | Tvo | A | Thermoplasma volcanium |
| Mac | A | Methanosarcina acetivorans | Vch | B | Vibrio cholerae |
| Mja | A | Methanococcus jannaschii | Xax | B | Xanthomonas axonopodis |
| Mle | B | Mycobacterium leprae | Xca | B | Xanthomonas campestris |
| Mlo | B | Mesorhizobium loti | Xfa | B | Xylella fastidiosa |
| Mma | A | Methanosarcina mazei | Ype | B | Yersinia pestis |
| Mmu | E | Mus musculus | Ypk | B | Yersinia pestis KIM |
| Mtc | B | Mycobacterium tuberculosis CDC1551 |
Figure 1.
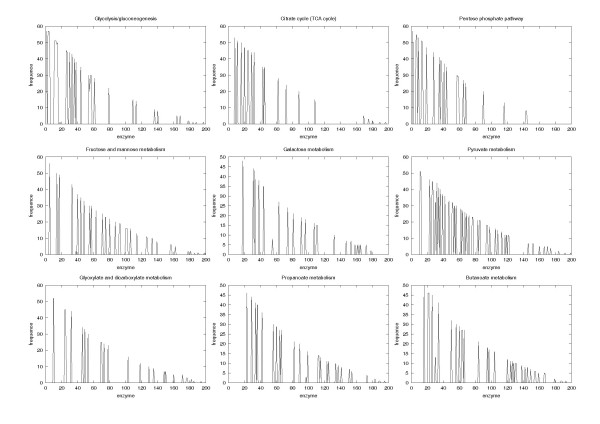
The distribution of enzymes in nine reference pathways. The x axis is the index of the maximum value of the order sorted by the frequency of enzyme over all pathways. All plots showed similar distributions.
The phylogeny took two directions: conventional taxonomy that focused on the morphological and physiological features to classify species, and a numerical taxonomy that stressed the historical changes in biological sequences. Phylogenies based on the ribosomal RNA molecules led to the proposal of a new tripartite scheme of three domains: Bacteria, Archaea, and Eukarya [20]. Although each approach is feasible on its own, it cannot provide a holistic view of the organism. Current phylogenetic studies indicate that horizontal gene transfer may have played a vital role in the evolution of major lineages [21]. Lake and Moore [22] also noted the pitfalls of comparative genomics based on molecular sequences. Our kernel-based method provides an alternative to the inference of an evolutionary scenario and allows for a higher-level comparison of the phylogenetic trees by measuring the distances between pathways using metabolic network data to infer an evolutionary scenario.
Consistency with conventional taxonomy
Figure 2 compares the phylogenetic tree constructed by the kernel method using (a) metabolic networks, and (b) by the sequence-based analysis of phosphoglycerate kinase and phosphopyruvate hydratase. The overall tree structure appears to be consistent with the current classification of the three domains, Eubacteria, Archaea, and Eukaryota (Figure 3). In the domain of Archaea, methanogens such as Methanosarcina, Methanococcus, and Methanobacterium are clustered in the early branch of Pyrococcus. Five thermophiles are also clustered in the same branch with a halophile, Halobacterium. The structure of collective carbohydrate pathways from Arabidopsis thaliana has the smallest distance from yeasts (Figure 2). As shown in Figure 2(a), the kernel-based method branches at the levels of family (Enterobacteriaceae), order (Bacillales), and class (Gammaproteobacterium) according to similar metabolic network structures. Figures 2(a) and 3 show that the domain of Archaea corresponded largely to that of conventional taxonomy http://www.ncbi.nih.gov/Taxonomy/taxonomyhome.html/; NCBI Taxonomy Browser). In Eukaryota, only the mouse deviated from the taxonomy consensus. Typically, the domain of Archaea always formed a single cluster in every case in our experiments, suggesting that the domain of Archaea has peculiar characteristics, at least in the carbohydrate metabolic networks. We conclude that the metabolic structure in Archaea is distinct from that of the other two domains.
Figure 2.
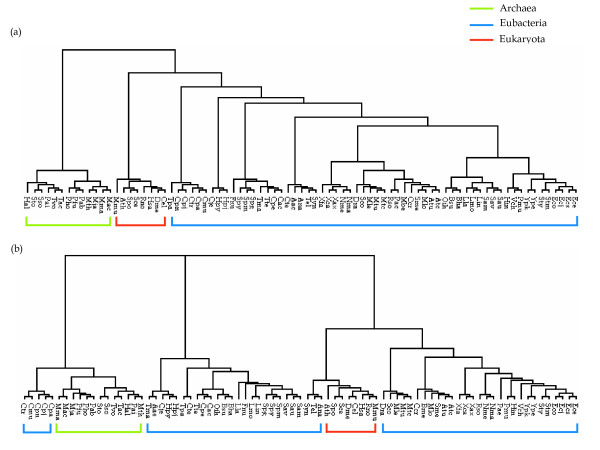
Comparison of phylogenetic trees constructed using (a) the kernel-based method and (b) the alignments of the following two enzyme sequences, together: phosphoglycerate kinase (EC 2.7.2.3) and phosphopyruvate hydratase (EC 4.2.1.11). The amino acid sequences of corresponding enzymes were retrieved from the GenBank database and analyzed by CLUSTAL with default parameters. The resultant trees were viewed with the TREEVIEW program [43].
Figure 3.
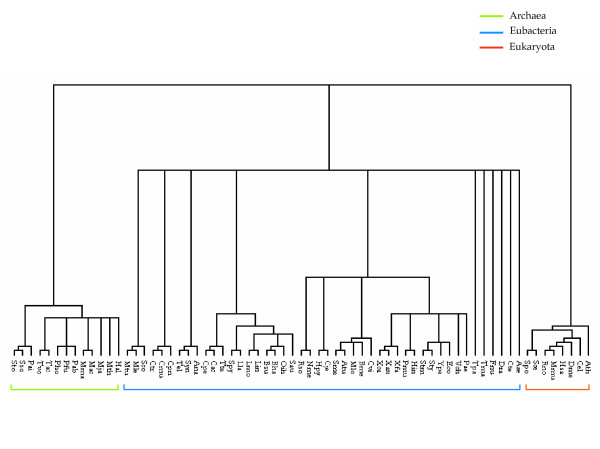
Current classification of biological taxonomy. The tree was reconstructed from part of the data in the NCBI (National Center for Biotechnology Information) Taxonomy [44] and viewed with the TREEVIEW program [43].
Figure 2(a) shows that archaeal metabolic networks are more closely related to the eukaryotic networks than with the eubacterial networks. This corresponds with a comparison of the information-transfer pathways and pathway-level organization between two domains [23], whereas eukaryotic metabolic enzymes are primarily of bacterial origin [24].
Inferring hidden order by network clustering
The conventional sequence-based analysis passes over or does not embrace the discordant evolution of each species or the horizontal gene transfer [25]. Our method can cope with this limitation by taking into account the structural features of individual metabolic networks. The disagreement between the molecular sequence data of operational genes and the rRNA tree suggests that different genes have different evolutionary histories [26,27]. To address this problem, Li studied the mitochondrial genomes in relation to the problem of whole-genome phylogeny, where evolutionary events, such as genetic rearrangements that include gene transfer from the exterior, make genome alignments difficult [28].
To compare the kernel-based comparative analysis of metabolic networks to the sequence-based phylogenetic analysis, we analyzed two enzyme sequences that participate in carbohydrate metabolism in all 81 species together using a multiple sequence alignment (Figure 2(b)). In the resulting phylogenetic tree, Archaea and Eukaryota are clustered at each terminal; however, short-distance neighboring node members belong to fairly distant taxonomic groups. The overall structure of the tree eventually becomes remote from not only that of the kernel-based method (Figure 2(a)), but also from that of current taxonomy (Figure 3). Although the phylogenetic tree constructed from the multiple alignment of two enzyme sequences shows a few unusual characteristics, our approach provides a good solution. The cluster mainly comprised archaeal species including the bacterial members Chlamydia and Chlamydophila, and had long branches at the root of the tree. Three eubacterial members (Ana, Tel, and Syn) are more closely related to Eukaryota than Eubacteria. Moreover, eubacterial groups are separated over the topology, and the Eukaryota are inserted between them (Figure 2(b)).
This result shows an example of the sequence-based phylogenetic approach when we intend to perform phylogenetic analyses for as many species as possible. The sequence-based phylogenetic analysis can fail to precisely represent evolutionary history without an analysis using a set of whole sequences. Unfortunately, analyses using whole sequences require massive computing power and are highly complex. A sequence-based phylogenetic analysis can still be limited to cover a number of species, although alternative approaches exist. However, our method can easily solve this problem by utilizing given resources. In order to measure the quality of our constructed trees, we compared the phylogenetic tree based on our graph kernel method with that of Heymans and Singh [11] in terms of their similarity to conventional taxonomy. We used a software tool, 'Cousins' [31], which compares two alternative phylogenetic trees based on common cousin pairs in the trees. The comparison by cousin pairs is said to more focus on local similarity between two trees because it evaluates the similarity based on a cousin pair within a certain degree. Table 4 shows the similarity score of our kernel-based method in comparison with [11] for the glycolysis pathways of 65 organisms. Our method shows a better result in terms of the similarity score with the conventional NCBI taxonomy.
Table 4.
Comparison of similarity scores with respect to NCBI taxonomy for 65 organisms with the glycolysis pathway (β = 0.8).
| Method | Similarity score |
| Our method | 0.196 |
| [11] | 0.154 |
In this paper, we intended to present a meta-level analysis of biological systems to construct a unitary phylogenetic tree that could be used to interpret the context of biological evolution. Our results suggest that the phylogenetic analysis with submetabolic network information might also allow us to infer horizontal or lateral gene transfer. Our results also support the tripartite scheme of the three domains, Bacteria, Archaea and Eukaryota [20].
Comparing the pathogenic bacterial genomes by focusing on the pathways of bacterial and eukaryotic aminoacyl-tRNA synthesis showed that this pathway is uniquely prokaryotic/archaeal and that it is found widely among the pathogenic bacteria. This suggests that members of this pathway can be used as targets for novel antimicrobial drugs [32]. Metabolic analysis of pathogenic organisms may play a critical role in the selective treatment or prevention of diseases caused by these organisms by using this innovative concept to develop new drugs.
Conclusion
Biological classification, taxonomy, and systematics are the profound themes in biology. Using phylogeny in evolutionary classification implies functional and morphological innovation, adaptive range, parallelism, and convergence. We have used a method based on the graph kernel to compare information on each metabolic network including cardinality, distance, and topology relating to metabolic networks as a type of undirected graph. Our results showed that our approach has potential in the macroscopic analysis of phylogenetic relationships among organisms in relation to horizontal gene transfer. To obtain information about each causal mechanism in the context of a similar phenotype, one should first analyze the phenomena at the level of a protein network. The analysis of a metabolic network is an example of this type of analysis. Biological entities that interact with the environment and eventually influence adaptation are a function of the activity of proteins and other bioactive molecules rather than gene order or genetic history.
The overall structure of the phylogenetic tree constructed from our experiments supports the tripartite scheme of the three domains Archaea, Eubacteria, and Eukaryota as described in an early report of Woese et al. [20]. The structures of metabolic pathway deduced from Archaea are more similar to those from Eukaryota than to those from Eubacteria. This agrees with the rooted universal tree of life [33,34] and the tree of life [35,36]. The metabolic network structures of organisms reflect their functional relationship with the environment, and the similarity might provide a measure of the organism's physiological functions. The trajectory of an organism's adaptation can be explained using the structure of its metabolic contents. Our approach can be extended to more organisms and applied to other types of biomolecular interactions, such as physical protein interactions in regulatory networks, to provide a basis for understanding the functional relationships between biological networks in different organisms.
Methods
We attempt to cluster organisms by comparing sets of metabolic pathways. Our basic assumption is that different species exhibit overlapping components of metabolic pathways. To construct phylogenetic trees, the features of the organism-specific pathways are automatically extracted by considering the reference pathway. Here, the features represent the connection information between two enzymes. Figure 4 summarizes the procedure for the phylogenetic clustering of organisms by metabolic pathways using four steps:
Figure 4.
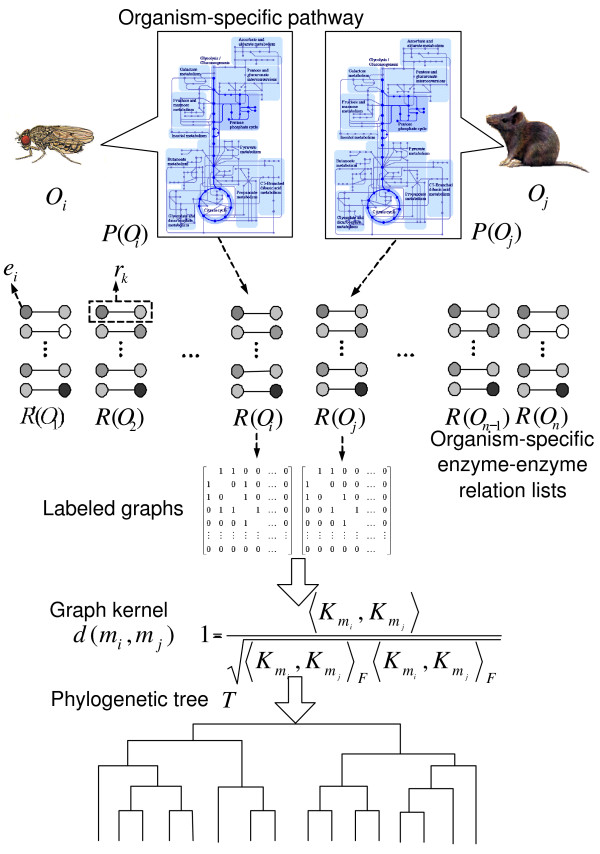
The procedure for data processing for phylogenetic tree construction from the metabolic networks.
- Step 1: Build the enzyme-enzyme relation lists.
- Step 2: Convert the lists to graph structures.
- Step 3: Compute similarity by graph kernels.
- Step 4: Build the phylogenetic trees.
We evaluated this method using known experimental data on a collection of nine metabolic pathways from 81 representative organisms. Figure 5 shows the simple concept used to compute the distance between two metabolic networks. The resulting phylogenetic trees were cross-compared for consistency with existing methods to analyze phylogenies. The next section describes the datasets collected and the preparation methods, followed by the definition of graph kernels and their use in comparing and clustering metabolic networks for phylogenetic analysis.
Figure 5.
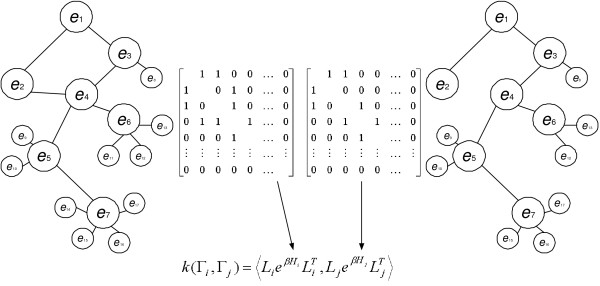
The simple concept for computing the similarity between two metabolic networks using the kernel method.
Data preparation
Dataset
We chose the KEGG database [37] as the resource for previous phylogenetic analysis. KEGG provides both an online map of pathways and the ability to focus on metabolic reactions in specific organisms. Each reaction may be uni- or bidirectional.
Representation of organisms
Let O = {O1,..., ON} be a set of N organisms and P = {P1,..., PM} be a set of M reference pathways. Here a reference pathway contains all known alternatives of reaction paths. The set of organism-specific pathways is defined as P' = {,...,}, which contains organism-specific reactions. If we define a set of enzyme-enzyme relations as R = {r1,...,rK}, then a subset of R constitutes Pj or (1 ≤ j ≤ M). Here, rk (1 ≤ k ≤ K) is a pair of enzymes {eu, ev}, which means that eu directly connects with ev. The specific organism Oi (1 ≤ i ≤ N) contains a set of pathways P, defined as P'(Oi) and including a subset of R for the specific organism, R'(Oi).
Enzyme-enzyme relation lists of organisms
The pathways provided in KEGG are visualized on manually drawn pathway maps or XML-based graphics. To construct enzyme-enzyme relation lists, we used information about chemical compounds and chemical reactions contained in the LIGAND database [38]. The LIGAND database provides detailed molecular information about one type of the generalized protein-protein interaction, namely, the enzyme-enzyme relation. LIGAND is a composite database of ENZYME and COMPOUND. The ENZYME section contains information about enzymatic reactions and enzyme molecules, and the COMPOUND section contains more than 6,000 chemical compounds. The enzyme-enzyme relationship can be extracted from information about enzymes contained in the COMPOUND entries. We automatically extracted information about enzymes of a specific organism from the ENZYME section.
The enzyme-enzyme relationships of a specific organism were extracted from the enzyme-enzyme relation list. If two enzymes of rk in an enzyme-enzyme relation list existed in the enzyme list of a specific organism, we inserted rk into R'(Oi). P'(Oi) can be constructed by R'(Oi).
Data analysis
Metabolic networks as labeled graphs
Our approach to estimate the distance between two metabolic networks is based on the graph comparison. Using the relation list of enzymes, the metabolic network of each organism is represented by a labeled graph Γ = (, , f), where is a vertex set and is an edge set. f is a vertex-labeling function f: → , where = {ℓl} is a set of possible labels for vertices.
For an organism Oi, each vertex v ∈ i corresponds to an enzyme of Oi, and the cardinality |i| is equal to the number of distinct enzymes in the enzyme-enzyme relation list R(Oi) of the organism. When an entry for two enzymes eu and ev is found in R(Oi), the corresponding vertices u and v are directly connected by an edge (u, v) ∈ i (denoted by u ~ v). The set contains the unique identifiers (i.e., EC numbers) of all enzymes found in of all selected organisms.
A matrix representation of a labeled graph Γi can be given by an adjacency matrix Hi and a label matrix Li and, where Hi is a |i| × |i| square matrix and Li is a || × |i| matrix. Each element Hi(a, b) is given by
where is the weight of the edge (va, vb). Whenever the vertices va and vb are joined by an edge, we set the weight such that w(va, vb) = , where deg(va) is the degree of va and is a constant for the graph Γi. Then, w(va, vb) can be thought to be proportional to a probability (1/deg(va)) to visit vb in one step in a random walk starting from va. We set , which makes Hi such that its total sum of elemets is still same to the number of edges in a bidirectional representation of Γi.
An element of the matrix Li defined as
with 1 ≤ l ≤ ||, 1 ≤ a ≤ |i|. This means that Li(l, a) is 1 only when the label of vertex va is ℓl. Since we represent a metabolic pathway in such a way that every vertex (enzyme) in it has a unique EC number, every column sum of Li is 1, that is, ∑lLi(l, a) = 1, (∀ a). And, in terms of rows of L, ∑aLi(l, a) = 1 if ℓl = f(v) (∃ v ∈ i); ∑aLi(l, a) = 0 otherwise. To compare the structures between metabolic networks of two organisms represented in graphs as described above, we adopted a kernel-based approach called the exponential graph kernel [39].
Comparison of metabolic networks: graph kernel
Given two graphs Γi = (Li, Hi) and Γj = (Lj, Hj), the first simple approach to the graph comparison is to count the common vertices with the same labels in both Γi and Γj. This similarity (or kernel) can be calculated by k(Γi, Γj) = <, >, where the inner product <Mi, Mj> between two matrices of the same dimension is defined as
Based on the definition of the label matrix in Equation (2), the matrix Mi = is a || × || diagonal matrix where Mi(l, l) = 1 only when f(v) = ℓl (∃v ∈ i), and Mi(l, l) = 0 otherwise. However, this approach considers only the presence or absence of vertices (enzymes) but does not consider the structure of the graph, such that the successive enzymes or reaction steps cannot be considered when comparing metabolic networks. To capture the structure of the graph, one must also consider vertices that can be reached from a vertex by a subsequent traverse.
In the exponential graph-kernel method, the similarity between two graphs Γi and Γj is defined as
where β (≥ 0) is a real-valued parameter and its value is chosen by performing many tries. When β = 0, it recovers the simple common vertex-counting measure since exp(0H) = I, the || × || identity matrix. Each element Hn(a, b) of the matrix Hn in Equation (5) represents the number of walks of length n (admitting cycles) from va to vb, and allows the representation of the global structure of a graph.
Substituting Equation (5) into Equation (4), we can decompose the kernel function k(Γi, Γj) into two meaningful parts, k(Γi, Γj) = k1(Γi, Γj) + k2(Γi, Γj), where
The kernel function k1 contributes by considering walks of the same length in both graphs, and k2 can take into account the insertion or deletion of vertices in the graph [39]. As the number of movements in a graph increases, the significance of walks of length n decreases by . Eventually, the exponential matrix eβH can be interpreted as the product of a continuous process H, from which the identity matrix expands gradually to the matrix of the global structure of Γ [40].
The exponential graph kernel requires the exponentiation of square matrices Hs. This can be performed by matrix diagonalization, with time complexity of about O(|i|3) for Hi thus Γi. [39]. The time complexity of the element-wise product of two matrices in k(Γi, Γj) is O(max (|i|2, |j|2)). With N graphs, finally, the total time complexity for constructing the kernel matrix K = {kij} (1 ≤ i, j ≤ N) is O(NV3 + N2V2) where V = maxi |i|.
From the kernel k(Γi, Γj), the dissimilarity metric is defined in the standard manner, that is,
If we use the normalized kernel,
then the distance metric is simplified as
To summarize, metabolic networks constructed from reference pathways of N organisms were first converted to labeled undirected graphs. Each graph Γi (1 ≤ i ≤ N) was then represented by two matrices: the vertex-label matrix Li and the adjacency matrix Hi. Using these two matrices, we can take into account only the local structure (the direct connectivities between enzymes in pathways) of networks. To compare networks in terms of their global structure, we adopted a kernel-based method, which we named the exponential graph kernel. Finally, the distance matrix acquired from the kernel function was fed into a hierarchical clustering algorithm to construct the phylogenetic trees.
Constructing phylogenetic trees
The distance between two organisms was calculated by comparing their metabolic networks using the measures mentioned earlier. To construct a phylogenetic tree, we used an unweighted pair-group method with arithmetic mean (UPGMA) [41,42], a hierarchical agglomerative clustering algorithm. Given N organisms, the algorithm starts by initializing N clusters, each of which contains exactly one distinct organism, and proceeds by iteratively merging the two nearest clusters until only one cluster (called the root of the tree) remains. The dendrogram derived from UPGMA is a binary tree, which we consider may represent a binary phylogenetic tree.
Authors' contributions
SJO proposed the idea, organized overall procedure, built the data set for computational experiments and carried out an analysis of experimental results. JGJ built the data set for computational experiments and carried out an analysis of experimental results. JHC carried out implementation of the computational method on graph kernels, computational experiments and analysis. BTZ developed the idea, provided intellectual guidance and mentorship. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This research was supported by the National Research Laboratory (NRL) Program (M1041200095-04J0000-03610) of Korean Ministry of Science and Technology and the Inje University research grant.
Contributor Information
S June Oh, Email: o@biophilos.org.
Je-Gun Joung, Email: jgjoung@bi.snu.ac.kr.
Jeong-Ho Chang, Email: jhchang@bi.snu.ac.kr.
Byoung-Tak Zhang, Email: btzhang@bi.snu.ac.kr.
References
- Whiting MF, Carpenter JC, Wheeler QD, Wheeler WC. The Strepsiptera problem: phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst Biol. 1997;46:1–68. doi: 10.1093/sysbio/46.1.1. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nature Rev Genet. 2005;6:361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- Fitz-Gibbon ST, House CH. Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 1999;27:4218–4222. doi: 10.1093/nar/27.21.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Gerstein M. Whole-genome trees based on the occurrence of folds and orthologs: Implications for comparing genomes on different levels. Genome Res. 2000;10:808–818. doi: 10.1101/gr.10.6.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otu HH, Sayood K. A new sequence distance measure for phylogenetic tree construction. Bioinformatics. 2003;19:2122–2130. doi: 10.1093/bioinformatics/btg295. [DOI] [PubMed] [Google Scholar]
- Forst CV, Schulten K. Evolution of metabolisms: A new method for the comparison of metabolic pathways using genomics information. J Comp Biol. 1999;6:343–360. doi: 10.1089/106652799318319. [DOI] [PubMed] [Google Scholar]
- Forst CV, Schulten K. Phylgenetic analysis of metabolic pathways. J Mol Evol. 2001;52:471–489. doi: 10.1007/s002390010178. [DOI] [PubMed] [Google Scholar]
- Schuster DandekarTT, Snel B, Huynen M, Bork P. Pathway alignment: application to the comparative analysis of glycolytic enzymes. Biochem J. 1999;343:115–124. doi: 10.1042/0264-6021:3430115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret BME, Wang LS, Warnow T, Wyman SK. New approaches for reconstructing phylogenies from gene order data. Bioinformatics. 2001;17:S165–S173. doi: 10.1093/bioinformatics/17.suppl_1.s165. [DOI] [PubMed] [Google Scholar]
- Liao L, Kim S, Tomb JF. Genome comparisons based on profiles of metabolic pathways. Proceedings of the Sixth International Conference on Knowledge-based Intelligent Information & Engineering Systems. 2002. pp. 469–476.
- Heymans M, Singh AK. Deriving phylogenetic trees from the similarity analysis of metabolic pathways. Bioinformatics. 2003;19:i138–i146. doi: 10.1093/bioinformatics/btg1018. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Tatusov RL, Koonin EV. Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol. 2001;1:8. doi: 10.1186/1471-2148-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Snel B, Huynen MA, Bork P. SHOT: a web server for the construction of genome phylogenies. Trends Genet. 2002;18:158–162. doi: 10.1016/S0168-9525(01)02597-5. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Lazcano A, Dujon B. The genomic tree as revealed from whole proteome comparison. Genome Res. 1999;9:550–557. [PMC free article] [PubMed] [Google Scholar]
- Grishin NV, Wolf YI, Koonin EV. From complete genomes to measures of substitution rate variability within and between proteins. Genome Res. 2000;10:991–1000. doi: 10.1101/gr.10.7.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC. Whole-genome prokaryotic phylogeny. Bioinformatics. 2005;21:2329–2335. doi: 10.1093/bioinformatics/bth324. [DOI] [PubMed] [Google Scholar]
- Qi J, Wang B, Hao BI. Whole proteome prokaryote phylogeny without sequence alignment: a K-string composition approach. J Mol Evol. 2004;58:1–11. doi: 10.1007/s00239-003-2493-7. [DOI] [PubMed] [Google Scholar]
- Daubin V, Gouy M, Perriere G. A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 2002;12:1080–1090. doi: 10.1101/gr.187002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King L, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Woese CR, Kandler O, L WM. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2128. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Lake JA, Moore JE. Phylogenetic analysis and comparative genomics. Trends Guide to Bioinformatics. 1998.
- Podani J, Oltvai ZN, Jeong H, Tombor B, Barabasi AL. Comparable system-level organization of Archaea and Eukaryotes. Nat Genet. 2001;29:54–56. doi: 10.1038/ng708. [DOI] [PubMed] [Google Scholar]
- Rivera MC, Jain R, Moore JE, Lake JA. Genomic evidence for two functionally distinct gene classes. Proc Natl Acad Sci USA. 1998;95:6239–6244. doi: 10.1073/pnas.95.11.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canback B, Andersson SGE, Kurland CG. The global phylogeny of glycolytic enzymes. Proc Natl Acad Sci USA. 2002;99:6097–6102. doi: 10.1073/pnas.082112499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. Lateral genomics. Trends Cell Biol. 1999;9:M5–M8. doi: 10.1016/S0962-8924(99)01664-5. [DOI] [PubMed] [Google Scholar]
- Li M, Badger JH, Chen X, Kwong S, Kearney P, Zhang H. An information-based sequence distance and its application to whole mitochondrial genome phylogeny. Bioinformatics. 2001;17:149–154. doi: 10.1093/bioinformatics/17.2.149. [DOI] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Lateral transfer at the gene and subgenic levels in the evolution of eukaryotic enolase. Proc Natl Acad Sci USA. 2001;98:10745–10750. doi: 10.1073/pnas.191337098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye TMW, Lio P, Gilks WR. A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics. 2006;22:117–119. doi: 10.1093/bioinformatics/bti720. [DOI] [PubMed] [Google Scholar]
- Zhang K, Wang JTL, Shasha D. On the editing distance between undirected acyclic graphs. Int J Foundations Comput Sci. 1996;7:43–57. doi: 10.1142/S0129054196000051. [DOI] [Google Scholar]
- Fritz B, Raczniak GA. Bacterial genomics: potential for antimicrobial drug discovery. Biodrugs. 2002;16:331–337. doi: 10.2165/00063030-200216050-00002. [DOI] [PubMed] [Google Scholar]
- Doolittle WF, Brown JR. Tempo, mode, the progenote, and the universal root. Proc Natl Acad Sci USA. 1994;91:6721–6728. doi: 10.1073/pnas.91.15.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Logsdon JM., Jr Archaeal genomics: Do archaea have a mixed heritage? Curr Biol. 1998;8:R209–R211. doi: 10.1016/S0960-9822(98)70127-7. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Koonin EV. Genome trees and the tree of life. Trends Genet. 2002;18:472–479. doi: 10.1016/S0168-9525(02)02744-0. [DOI] [PubMed] [Google Scholar]
- Tree of Life http://tolweb.org
- Ogata H, Goto SK, Fujibuchi H, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Okuno Y, Hattori M, Nishioka T, Kanehisa M. LIGAND: database of chemical compounds and reactions in biological pathways. Nucleic Acids Res. 2002;30:402–404. doi: 10.1093/nar/30.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner T. Exponential and Geometric Kernels for Graphs. NIPS 2002 Workshop on Unreal Data: Principles of Modeling Nonvectorial Data. 2002.
- Kondor RI, Lafferty J. Diffusion kernels on graphs and other discrete input spaces. Proceedings of 19th International Conference on Machine Learning. 2002. pp. 315–322.
- Jain AK, Dubes RC. Algorithms for Clustering Data. 2. address in USA: Prentice Hall; 1988. [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press; 1998. [Google Scholar]
- Page RDM. TREEVIEW: An application to display phylogenetic trees on personal computers. CABIOS. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- NCBI taxonomy http://www.ncbi.nlm.nih.gov/Taxonomy/