

Abstract
We have recently reported that cyclooxygenase (COX)-2-deficiency affects brain upstream and downstream enzymes in the arachidonic acid (AA) metabolic pathway to prostaglandin E2 (PGE2), as well as enzyme activity, protein and mRNA levels of the reciprocal isozyme, COX-1. To gain a better insight into the specific roles of COX isoforms and characterize the interactions between upstream and downstream enzymes in brain AA cascade, we examined the expression and activity of COX-2 and phospholipase A2 enzymes (cPLA2 and sPLA2 ), as well as the expression of terminal prostaglandin E synthases (cPGES, mPGES-1, and -2) in wild type and COX-1−/− mice. We found that brain PGE2 concentration was significantly increased, whereas thromboxane B2 (TXB2) concentration was decreased in COX-1−/− mice. There was a compensatory up-regulation of COX-2, accompanied by the activation of the NF-κB pathway, and also an increase in the upstream cPLA2 and sPLA2 enzymes. The mechanism of NF-κB activation in the COX-1−/− mice involved the up-regulation of protein expression of the p50 and p65 subunits of NF-κB, as well as the increased protein levels of phosphorylated IκBα and of phosphorylated IKKα/β. Overall, our data suggest that COX-1 and COX-2 play a distinct role in brain PG biosynthesis, with basal PGE2 production being metabolically coupled with COX-2 and TXB2 production being preferentially linked to COX-1. Additionally, COX-1 deficiency can affect the expression of reciprocal and coupled enzymes, COX-2, Ca2+-dependent PLA2, and terminal mPGES-2, to overcome defects in brain AA cascade.
Keywords: cyclooxygenase, prostaglandin E synthase, phospholipase A2, PGE2, NF-κB
Introduction
Prostaglandins (PGs) are important end-products of the arachidonic acid (AA) cascade, the metabolic pathway in which AA is first released from the sn-2 position of membrane phospholipids by a phospholipase A2 (PLA2) (Kudo and Murakami 2002) and then metabolized to bioactive eicosanoids, lipid mediators that have potent and diverse bioactivities in the central nervous system (CNS). PGs can modulate synaptic transmission and remodeling, neurotransmitter release, hypothalamic-pituitary-adrenal axis, sleep/wake cycle, and appetite (Adams et al. 1996; Kaufmann et al. 1997; Narumiya et al. 1999). In addition to these physiological roles, data indicate that PGs also participate in pathological processes in the CNS, including neurodegenerative disorders such as Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS) (Griffin et al. 1994; Montine et al. 1999; Almer et al. 2002; Ilzecka 2003), and psychiatric disorders such as schizophrenia and mood disorders (Nishino et al. 1989; Rapoport and Bosetti 2002; Sublette et al. 2004).
The PLA2 family of enzymes can be divided into two main classes, a Ca2+-independent iPLA2 and a Ca2+-dependent PLA2, which includes a cytosolic PLA2 (cPLA2) and a secretory PLA2 (sPLA2). cPLA2 is regulated post-transcriptionally by a low concentration of calcium and is activated by phosphorylation by protein kinase C and by mitogen-activated protein kinase (Leslie 1997). Cyclooxygenase (COX), which exists in two major isoforms, COX-1 and -2, catalyzes the second step in the conversion of AA to PGH2 (Smith et al. 2000). In most tissues, COX-1 is thought to be responsible for the production of PGs associated with homeostatic functions, whereas COX-2 is usually induced in several cell types in response to inflammatory stimuli (Kaufmann et al. 1997). However, COX-2 is expressed at relatively high basal levels in the brain, and is thought to contribute to synaptic plasticity and memory consolidation (Yamagata et al. 1993; Minghetti 2004).
The PGH2 intermediate can be subsequently metabolized by terminal PG synthases to biologically active PGs such as PGE2 and thromboxane A2 (TXA2) (Murakami et al. 2002; Wang and Kulmacz 2002). Recently, three distinct PGE2 synthase (PGES) enzymes have been identified, a cytosolic form (cPGES) (Tanioka et al. 2000) and two membrane-associated PGES isoforms (mPGES-1 and -2) (Jakobsson et al. 1999; Tanikawa et al. 2002). Since PGE2 exerts actions on the plasma membrane and intracellular compartments via it receptors (Tsuboi et al. 2002), and because its precursor PGH2 is extremely short lived, a coordinated interaction between COX and PGES is essential to produce PGE2 for actions on its vicinal receptors. A rather simplified model proposed that mPGES-1 is preferentially coupled with COX-2 (Murakami et al. 2000), whereas COX-1 is coordinately associated with the specific cPGES for PGE2 biosynthesis (Tanioka et al. 2000). In several cell lines mPGES-2 promotes PGE2 production via both COX-1 and COX-2 in the immediate and delayed responses (Murakami et al. 2003). However, the coordination and intracellular localization of the COXs and PGES are still unclear (Ivanov and Romanovsky 2004; Vazquez-Tello et al. 2004). In this regard, we have recently shown that in brain from COX-2−/− mice basal protein levels of mPGES-2 are selectively downregulated, whereas there is no change in the protein expression of either mPGES-1 and cPGES, suggesting that COX-2 is metabolically coupled with mPGES-2 under basal conditions (Bosetti et al. 2004).
Some of the enzymes controlling the various steps of the AA cascade are regulated at the transcriptional level (Lindstrom and Bennett 2004). In particular, NF-κB transcription factor seems to be involved in the transcriptional regulation of cPLA2, sPLA2 and COX-2 (Tay et al. 1994; Andreani et al. 2000; Tanabe and Tohnai 2002). In resting cells, NF-κB is sequestered in the cytoplasm in complexes with its endogenous inhibitor IκB. In response to various stimuli, IκB undergoes phosphorylation by IκB kinases (IKK), ubiquitination, and subsequent proteasome-dependent degradation. Then, free NF-κB dimers rapidly translocate to the nucleus to initiate transcription by binding to regulatory κB motifs on target genes (Chen 2005).
Little is known about the individual role of each COX isoform in the brain. An increasing number of observations indicate that physiological and pathophysiological functions of two COX enzymes are complex and interrelated (Parente and Perretti 2003; Schwab and Schluesener 2003). Recent data suggest that COX-1 activity may significantly contribute to PGs synthesis and oxidative damage in the brain (Pepicelli et al. 2005). The genetically modified COX-deficient (−/−) mice provide an alternative approach to nonsteroidal anti-inflammatory drug treatment for understanding the roles of COX-1 and COX-2 in both physiological and pathological conditions. We recently demonstrated that brain PGE2 and mPGES-2 expression are decreased in the COX-2−/− mice despite compensatory increases in COX-1 and Ca2+-dependent PLA2, suggesting that COX-2 plays a key role for physiological production of PGE2 (Bosetti et al. 2004). In order to elucidate the individual roles of COX isoforms in brain and their coupling to upstream and downstream enzymes in the AA cascade, we examined the expression and activity of COX-2 and terminal synthases (cPGES, mPGES-1, and -2), as well as the expression and activity of the PLA2 enzymes (cPLA2 and sPLA2) in wild type and COX-1−/− mice.
Materials and methods
Animals
Three-month-old male COX-1 wild-type (COX-1+/+) and homozygous (COX-1−/−) mice on a C57BL/6-129/Ola genetic background were used in this study (Langenbach et al. 1995). The mice were housed at 25°C in our animal facility with a 12 hr light/dark cycle with free access to food and water. The study was approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with NIH guidelines on the care and use of laboratory animals.
Determination of brain PGE2 and TXB2 concentrations
Mice used to measure PGE2 and TXB2 levels (eight for each group) were killed with an overdose of sodium pentobarbital (100 mg/kg, i.p.), then subjected to high-energy head-focused microwave irradiation (4.8 kW, 1.1 sec, Cober Electronics, Stanford, CT, USA) to stop metabolism. Microwaved brains were weighed and extracted with 18 volumes of hexane: 2-propanol (3:2, by volume) using a glass Tenbroeck homogenizer. The prostanoids were purified from the lipid extract as described by Powell (Powell 1985) and the concentrations of PGE2 and TXB2 were determined using specific enzyme-linked immunosorbent assay (ELISA) kits (Oxford Biomedical, Oxford, MI, USA).
Homogenization and Western blot analysis
Mice used for protein and mRNA analysis were killed with an overdose of sodium pentobarbital (100 mg/kg, i.p.) and immediately decapitated. The brains were removed, immediately frozen on dry ice, and stored at −80°C until used. Tissues were subsequently homogenized in a lysis buffer containing 25 mM Tris-HCl, pH 7.8, 150 mM NaCl, 1 mM EDTA with Complete Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA). The homogenates were incubated at 4°C for 30 min and then centrifuged at 14,000 × g for 20 min. Protein concentrations of the homogenates were measured by the bicinchoninic acid (BCA) method (Bio-Rad, Hercules, CA, USA).
For Western blotting, 50 μg of brain homogenates were separated on Criterion gels (Bio-Rad), and blotted onto a polyvinylidene difluoride membrane (Bio-Rad). The membranes were then incubated overnight at 4°C with specific primary antibodies (1:1,000 unless otherwise noted) for cPLA2 (Cell Signaling, Beverly, MA, USA), phospho-cPLA2 (Cell signaling), sPLA2 (Cayman, Ann Arbor, MI, USA), COX-2 (Cayman), cPGES (Cayman), mPGES-1 (1:200; Cayman), mPGES-2 (Cayman), p65 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), p50 (1:500; Santa Cruz), IκBα (Cell Signaling), phospho-IκBα (Cell Signaling), phospho-IKKα/β (Cell Signaling). An antibody against β-actin (1:4,000; Sigma-Aldrich, St. Louis, MO, USA) was used to confirm equal loading among the samples. Then the membranes were immunoblotted with a secondary anti-rabbit horseradish peroxidase-conjugated IgG antibody (Bio-Rad). The blots were visualized by using enhanced chemiluminescence plus western blotting detection system (Amersham Biosciences, Piscataway, NJ, USA). The protein level was quantified by measuring the integrated optical density of the bands and normalized to β-actin.
Measurement of PLA2 and COX-2 activity
The enzymatic activities of PLA2 and COX-2 were measured as reported (Bosetti et al. 2004) using specific cPLA2, sPLA2, or COX Assay kits (Cayman) as directed by the manufacturer. Briefly, each brain was homogenized in a lysis buffer containing 10 mM Tris-HCl, pH 7.8, 1% Nonidet P-40, 0.15 M NaCl, and 1 mM EDTA, then chilled on ice for 30 min and centrifuged at 10,000 × g for 15 min at 4°C. cPLA2 activity was determined in the supernatant in the presence of the specific inhibitors for iPLA2(10 μM bromoenol lactone, BEL) and sPLA2 (50 μM thioetheramide-PC, TE-PC), which were incubated with the samples for 15 min at 25°C prior to the assay. sPLA2 activity was measured using the 1,2-dithio analog of diheptanoyl phosphatidylcholine, which serves as a substrate for most PLA2 enzymes, with the exception of cPLA2. Bee venom PLA2 was run as a positive control in the same assay to show a linear increase in the absorbance over the time range chosen. Absorbance was measured every minute after adding the substrate, for a total of 11 min. cPLA2 and sPLA2 enzymatic activity was calculated by measuring the absorbance at 414 nm, using the DTNB extinction coefficient of 10.66 per mM per cm, and reported as nmol/min/g of cytosolic protein. The peroxidase activity of COX was assayed colorimetrically by monitoring the appearance of oxidized N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) at 590 nm.
RNA isolation and cDNA synthesis
Brain total RNA was extracted using RNeasy Lipid Tissue Midi Kit (Qiagen, Valencia, CA, USA) as directed by the manufacturer. Five micrograms of total RNA were reverse transcribed using a High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). Five micrograms of each RNA sample was incubated similarly in the absence of reverse transcriptase to ensure that PCR products resulted from amplification from the specific mRNA rather than from genomic DNA contamination.
Real-Time Polymerase Chain Reaction
The expression of COX-2, cPLA2, sPLA2, mPGES-1, and mPGES-2 was measured by real-time quantitative RT-PCR, using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Specific primers and probes were purchased from the available Assays-on-Demands (Applied Biosystems). To normalize the amount of cDNA present in each reaction, we used phosphoglyceratekinase 1 (pgk1) as an endogenous control. A reaction mix was prepared containing Taqman Universal PCR Master Mix, Assay-On-Demand primers and probe and Rnase-free water. Template cDNA (1 μl) was added, in triplicate, to each well of the MicroAMp optical 96-well reaction plate (Applied Biosystems), such that the final volume in each well was 20 μl. Appropriate no-template controls were run on each plate in triplicate. The thermal cycling conditions were set at 50°C for 2 min and 95°C for 10 min, followed by 15 sec at 95°C (melting step) and 1 min at 60°C (annealing/extending step) for 40 cycles. Data were analyzed using sequence detection systems software (Applied Biosystems). A threshold value was placed in the exponential phase of the amplification plot, where the levels of fluorescence were increasing linearly. Data were analyzed using the relative quantification technique. Results were expressed as relative levels of target gene in the COX-1−/−, compared with wild-type samples (the calibrator). The amount of target gene normalized to the endogenous control (pgk1) and relative to the wild-type was calculated using the ΔΔCT method (Livak and Schmittgen 2001). Briefly, the target amount = 2−ΔΔCT, where ΔΔCT = {[CT (target KO) − CT (pgk1 KO)] − [CT (target WT) − CT (pgk1 WT)]}. For the ΔΔCT method to be valid, the amplification efficacies of the target and endogenous control genes must be approximately equal (± 0.1), where the efficacies are calculated from the slopes of the standard curves of the target and the endogenous control using the formula efficacy = 10−1/slope.
NF-κB activation assay
Nuclear proteins were prepared by using a compartmental protein extraction kit (Chemicon, Temecula, CA, USA) according to the manufacturer's protocol. NF-κB activation was assayed using Active Motif (Carlsbad, CA, USA) ELISA-based transactivation TransAM kit containing a 96-well plate with immobilized oligonucleotides encoding an NF-κB consensus site (5′-GGGACTTTCC-3′). The active form of NF-κB contained in the nuclear extract specifically binds to this oligonucleotide. The primary antibody used to detect NF-κB recognizes an epitope on p65 that is accessible only when NF-κB is activated and bound to its target DNA. A horseradish peroxidase (HRP)-conjugated secondary antibody provides a sensitive colorimetric readout that is quantified by a spectrophotometer at 450 nm with a reference wavelength of 655 nm. For competition experiments, NF-κB wild type and mutated consensus oligonucleotides were used at a concentration of 40 pmol per well.
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed using unpaired t-test. P values < 0.05 were considered statistically significant.
Results
Brain prostaglandin concentrations in wild type and COX-1−/− mice
To examine the effect of COX-1-deficiency on COX-derived products, the concentrations of PGE2 and TXB2 were determined in microwaved brains of wild type and COX-1−/− mice. Endogenous brain PGE2 levels showed a 36.6 % increase (p < 0.05) in COX-1−/− mice (Fig. 1A). In contrast, brain TXB2 levels were decreased by 64.8 % (p < 0.05) in COX-1−/− compared with wild type mice (Fig. 1B).
Fig. 1.
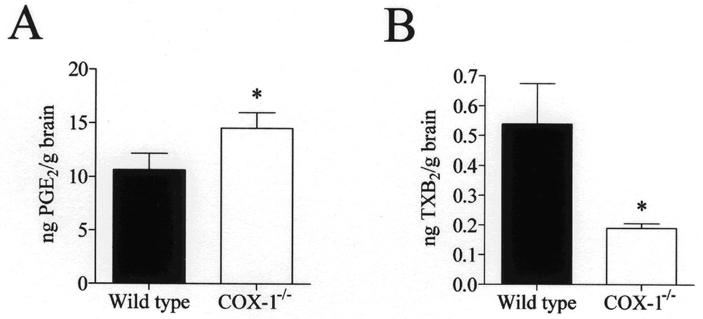
Brain endogenous PGE2 (A) and TXB2 (B) levels in wild type and COX-1-/- mice. Prostaglandins were analyzed using specific immunoassay kits. Results are expressed as mean ± SEM of eight independent samples, each assayed in duplicate. *p ≤ 0.05 versus wild type.
Compensatory increase in the protein expression of COX-2 in COX-1−/− mice
To see whether there was a compensatory activation of the reciprocal isoenzyme in COX-1−/−mice, we examined gene and protein expression of COX-2 in wild type and COX-1−/− mice using real-time PCR and Western blot analysis. COX-2 protein level was significantly increased in COX-1−/− mice (p < 0.05, Fig. 2B). However, the expression of COX-2 mRNA was not significantly changed (Fig. 2A), suggesting that the compensatory effects on COX-2 occurred at the post-transcriptional level. Supporting the observation of increased COX-2 protein level, COX-2 activity also was increased in COX-1−/− mice compared with wild type (p < 0.05, Fig. 2C).
Fig. 2.
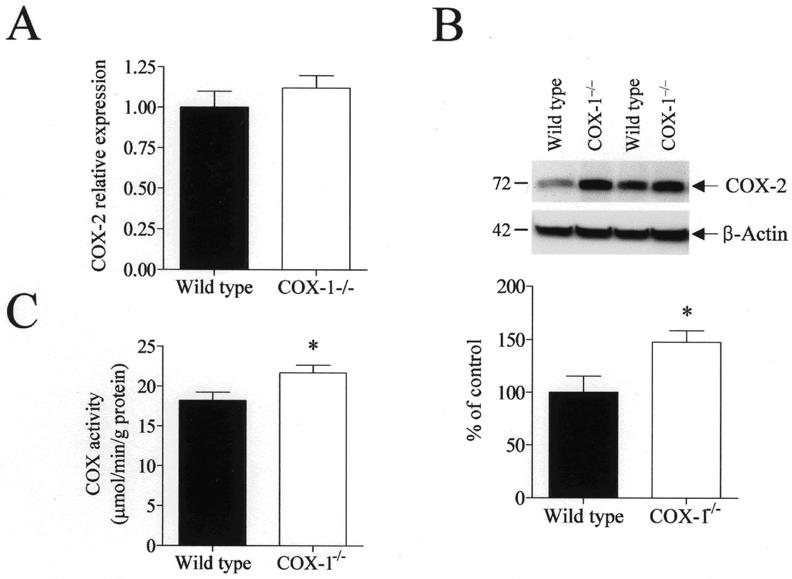
Brain COX-2 mRNA, protein and enzymatic activity levels in wild type and COX-1−/− mice. (A) Brain COX-2 mRNA expression was measured using real-time PCR. COX-2 mRNA levels were normalized to an endogenous reference (pgk1) in the COX-1−/− mice relative to the wild type, using the ΔΔCT method. The results are expressed as mean ± SEM of six independent samples, each assayed in triplicate. (B) Representative immunoblots illustrating COX-2 (72 kDa) and β-actin (42 kDa) protein levels. Relative integrated density values of COX-2 protein level normalized to β-actin protein level in brain from COX-1−/− mice is expressed as percentage of protein levels in wild type mice. Each sample derived from an individual mouse brain (n = 12). *p ≤0.05 versus wild type. (C) Brain COX-2 enzymatic activity in wild type and COX-1−/− mice was assessed colorimetrically using a commercial COX activity assay kit that measures the peroxidase activity of COX. The results are expressed as mean ± SEM of twelve independent samples, each assayed in duplicate.
PG synthases expression in wild type and COX-1−/− mice
Previous in vitro studies have indicated a functional coupling between COX enzymes and different terminal PG synthases (Murakami et al. 2000; Tanioka et al. 2000). Additionally, we recently demonstrated a selective downregulation of mPGES-2, but not of mPGES-1 or cPGES, in brain from COX-2−/− mice under basal conditions (Bosetti et al. 2004). To see the effect of COX-1 deficiency on downstream enzymes, we examined the expression of different PGES and thromboxane synthase (TXS) in wild type and COX-1−/− mice. The protein expression of mPGES-1 or cPGES was not significantly different in COX-1−/− mice compared with wild type (Fig. 3A and B). In contrast, we observed a significant reduction of brain mPGES-2 protein in COX-1−/− mice (p < 0.05, Fig. 3A). The mRNA level of mPGES-2, normalized to the level of endogenous control pgk1 and compared with the wild-type, was also decreased in the COX-1−/− mice (p < 0.05, Fig. 3B).
Fig. 3.
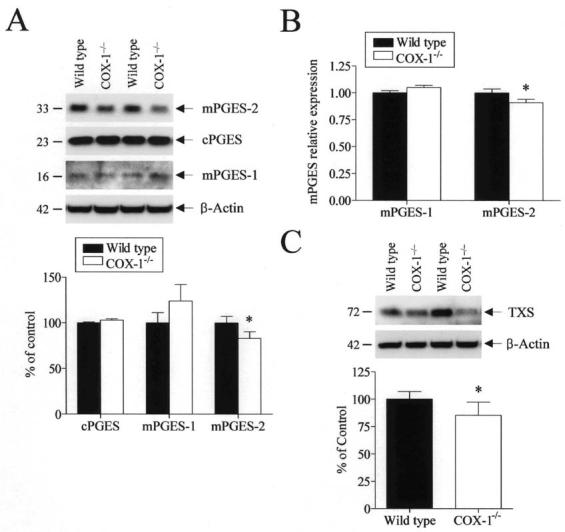
Brain PGES and TXS expression in wild type and COX-1−/− mice. (A) Representative immunoblots of the different PGES enzymes. Relative integrated density values of brain PGES enzymes, normalized to β-actin protein levels, in COX-1−/− mice were expressed as percentage of wild type. Each sample derived from an individual mouse brain (n = 12). *p < 0.05 versus wild-type. (B) Brain mPGES-1 and -2 mRNA levels measured by real-time PCR and normalized to an endogenous reference (pgk1) in the COX-1−/− mice relative to the wild type, using the ΔΔCT method. Results are mean ± SEM of six independent samples, each assayed in duplicate. (C) Representative immunoblot of TXS protein. Relative integrated density values of TXS protein levels normalized to β-actin protein level in brain from COX-1−/− mice are expressed as percentage of values in wild type mice. Each sample derived from an individual mouse brain (n = 12).
In line with our observation that brain TXB2 concentration was decreased in COX-1−/− mice, levels of TXS protein were significantly decreased in COX-1−/− mice compared with wild type (p < 0.05, Fig. 3C), suggesting a selective coupling of COX-1 with the TXB2 biosynthetic pathway.
Brain Ca2+-dependent PLA2 activity and expression in wild type and COX-1−/− mice
Since PLA2 plays a key role in initiating and regulating PG biosynthesis by supplying AA to COX-1 and COX-2, we next investigated the expression and enzymatic activity of PLA2 in wild type and COX-1−/− mice. The enzymatic activity of both cPLA2 and sPLA2 was increased in the COX-1−/− mice compared with wild-type (p < 0.05, Fig. 4A).
Fig. 4.
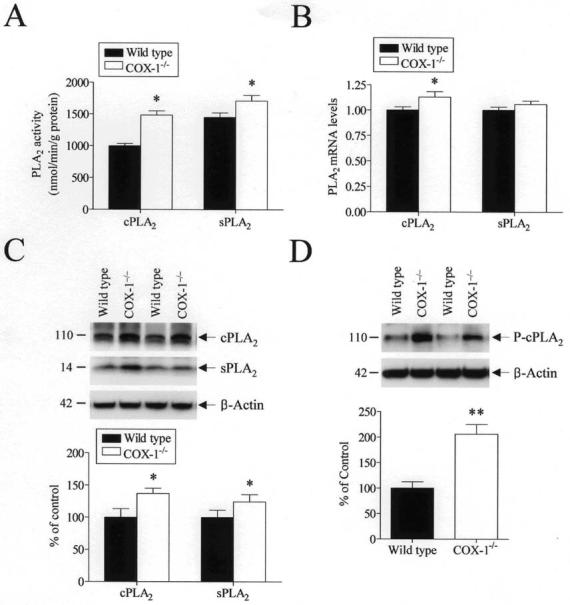
Brain Ca2+-dependent PLA2 enzymatic activity, protein and mRNA expression in wild type and COX-1−/− mice. (A) Brain Ca2+-dependent cPLA2 and sPLA2 activity was assessed using cPLA2 and sPLA2 activity assay kits. The results are expressed as mean ± SEM of twelve independent samples, each assayed in duplicate. (B) Brain cPLA2 and sPLA2 mRNA expression measured by real-time PCR and normalized to an endogenous reference (pgk1) in the COX-1−/− mice relative to the wild type, using the ΔΔCT method. The results are means ± SEM of six independent samples, each assayed in duplicate. (C) Representative immunoblots illustrating cPLA2 (110 kDa), sPLA2 (14 kDa), and β-actin (42 kDa) proteins. Relative integrated density values of cPLA2 and sPLA2 in brain from COX-1−/− mice are reported as percentage of values in wild type mice. Each sample derived from an individual mouse brain (n = 12). (D) Representative immunoblots illustrating phospho-cPLA2 (110 kDa) and β-actin (42 kDa) protein. Relative integrated density values of phospho-cPLA2 in brain from COX-1−/− mice are expressed as percentage of wild type. Each sample derived from an individual mouse brain (n = 12). *p ≤ 0.05, **p ≤ 0.01 versus wild type.
To account for the increased cPLA2 and sPLA2 activities, we measured the mRNA and protein levels of each enzyme by Western blotting and real-time PCR. Brain cPLA2 mRNA and protein levels were significantly increased in the COX-1−/− mice compared with wild-type (p < 0.05, Fig. 4B and C). Western blot analysis and real-time PCR showed that also sPLA2 protein levels, but not mRNA levels, were increased in the COX-1−/− compared with wild-type (p < 0.05, Fig. 4B and C).
Since phosphorylation of serine residues, and in particular of serine 505 is important in the activation of cPLA2 (Clark et al. 1995), levels of phospho-cPLA2 were measured by Western blotting. Brain Ser505-phosphorylated cPLA2 was significantly increased in the COX-1−/− mice compared with wild-type (p < 0.01, Fig. 4D).
Brain NF-κB activation in the COX-1−/− mice
To gain insight into the potential mechanism by which COX-1 deficiency causes the up-regulation of reciprocal and upstream enzymes in the AA cascade, we investigated the DNA binding activity of NF-κB, which has been shown to be essential for the induction of the COX-2, cPLA2, and type IIA sPLA2 genes in many cell types [reviewed in:(Andreani et al. 2000; Lindstrom and Bennett 2004)]. Nuclear and cytosolic proteins were isolated from wild-type and COX-1−/− mice and analyzed using an ELISA-based TransAM NF-κB kit. As shown in Fig. 5A, activation of NF-κB was significantly increased in COX-1−/− compared with wild-type mice (p < 0.05). To confirm the specificity of the NF-κB assay, we preincubated nuclear proteins with oligonucleotides containing either the specific NF-κB-binding consensus sequences (wt competitor) or mutant sequences (mut competitor) prior to the NF-κB activity assay. Nuclear proteins preincubated with an excess of oligonucleotide having NF-κB-binding consensus sequences showed a marked decrease in NF-κB-binding activity in both wild type and COX-1−/− mice (Fig. 5A, p < 0.01), whereas mutant oligonucleotides were ineffective in competing with the binding of NF-κB to its target sequences (Fig. 5A).
Fig. 5.
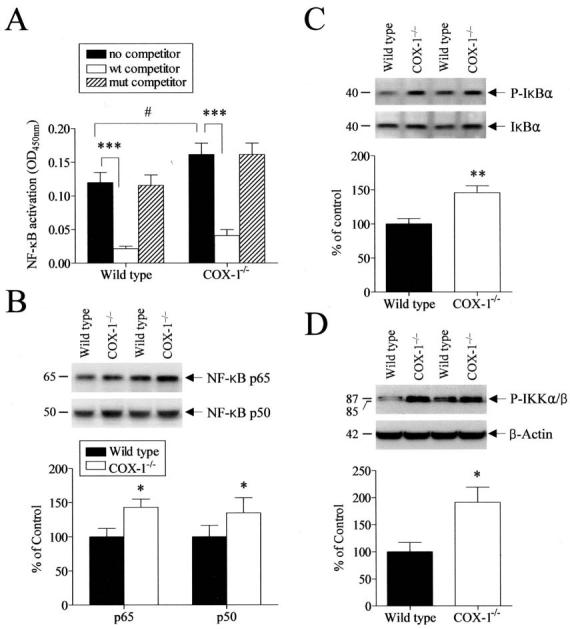
Brain NF-κB pathway in wild type and COX-1−/− mice. (A) NF-κB DNA binding activity was assayed using an ELISA-based TransAM NF-κB kit. The results are expressed as mean ± SEM of six independent samples, each assayed in duplicate. #p ≤ 0.05 versus wild type. ***p ≤ 0.001 versus no competitor. (B) Representative immunoblots illustrating p65 and p50 subunits of NF-κB. Relative integrated density values of p65 and p50 protein, normalized to β-actin protein levels in brain from COX-1−/− mice, are expressed as percentage of values in wild type mice. Each sample derived from an individual mouse brain (n = 12). (C) Representative immunoblots of Phospho-IκBα and IκBα proteins. Integrated density values of Phospho-IκBα protein relative to IκBα protein levels in brain from COX-1−/− mice are expressed as percentage of wild type. Each sample derived from an individual mouse brain (n = 12). (D) Representative immunoblots of Phospho-IKKα/β and β-actin proteins. Relative integrated density values of Phospho-IKKα/β protein in brain from COX-1−/− mice are expressed as percentage of wild type. Each sample derived from an individual mouse brain (n = 12). *p ≤ 0.05, **p≤ 0.01 versus wild type.
To examine whether the increase in NF-κB activation was accompanied by an increased expression of NF-κB subunits, we measured cytosolic protein levels of p65 and p50 protein. Western blotting showed a significant increase in both cytosolic p65 and p50 protein expression (p < 0.05, Fig. 5B), consistent with the observed increase in NF-κB activity.
Since NF-κB activation also depends on the phosphorylation and degradation of IκB, we also examined protein levels of phospho-IκBα and of the upstream kinases phospho-IKKα/β in wild type and COX-1−/−mice by Western blot. The protein level of phospho-IκBα was significantly increased in the COX-1−/− compared with wild-type mice (p < 0.05, Fig. 5C). However, there was no significant change in total cellular IκBα expression (Fig. 5C), resulting in a markedly increase in the ratio of phospho-IκBα to total IκBα. As shown in Fig. 5D, protein levels of phospho-IKKα/β were also increased in COX-1−/− mice compared with wild type (p < 0.05).
Discussion
In this study, we found that brain PGE2 concentration was significantly increased in COX-1−/− mice, likely as a result of a compensatory up-regulation of COX-2 and of the up-regulation of the expression and activity of the upstream cPLA2 and sPLA2 enzymes. These effects were accompanied by the activation of the NF-κB pathway. In contrast, COX-1 deficiency caused a significant decrease in brain TXB2 levels, suggesting a metabolic coupling of COX-1 with TX synthase. Our findings indicate that COX-1 deficiency can affect the expression of reciprocal and coupled enzymes: COX-2, PLA2, and terminal PGES to overcome defects in the brain prostaglandin synthesis, and suggest a possible direct modulation of the NF-κB pathway by the COX-2 derived PGE2.
The ability of COX-2 to compensate for the loss of COX-1 activity and PGs production in the COX-1−/− mouse is controversial and may reflect tissue-specific functional importance of the individual COX enzymes (Zhang et al. 2002). A previous report using qualitative immunohistochemistry showed no apparent compensatory increase of COX-2 immunoreactivity in the frontal cortex of COX-1−/− mice (Li et al. 1999). The compensatory up-regulation of COX-1 that we have previously reported in brain of COX-2−/− mice (Bosetti et al. 2004) and the up-regulation of COX-2 that we found in this study are in agreement with previous reports indicating an increase in COX-1 and COX-2 mRNA expression in brain of COX−/− mice (Zhang et al. 2002). The different effect of COX-1 and COX-2 deficiency on the brain levels of PGE2 indicates that the two COX isoforms play a unique and indispensable function in the brain. In this regard, data on brain PGE2 concentration in COX-1−/− mice are discrepant. A recent study reported that the brains of COX-1−/− mice had 70% lower levels of PGE2 than those of wild type animals (Ayoub et al. 2004). On the other hand, the present study as well as another report using stimulated and unstimulated immortalized lung fibroblasts (Kirtikara et al. 1998) showed that COX-2-dependent PGE2 concentration was higher in COX-1−/− mice than in wild type animals. It is possible that differences in the animal strains, experimental conditions (e.g., microwaved versus non-microwaved brain tissue), as well as tissue-specific functional parameters could account for such differences.
Our observations that basal brain PGE2 concentration is decreased in the COX-2−/− mice, despite the compensatory increase in COX-1 expression, and that in contrast it is increased in COX-1−/− mice, where COX-2 is upregulated, indicates that COX-2 is the major contributor to the basal PGE2 production in brain. Since the brain PGE2 level in COX-1−/− mice was higher than that observed in wild-type mice, one possibility is that the increased availability of AA substrate due to increased PLA2 activity and expression, combined with the compensatory upregulation of the constitutive COX-2 protein and activity, leads to a synergistic increase in PGE2 formation, which not only compensates for the loss of COX-1 activity but also accounts for the higher PGE2 concentration in the brain of COX-1−/− mice. Since nitric oxide (NO), has been shown to stimulate COX activity (Di Rosa et al. 1996; Salvemini 1997), we also investigated a possible involvement of NO in the up-regulation of COX-2 activity by examining gene expression of neuronal nitric oxide synthase (nNOS) and inducible NOS (iNOS) in the brain of wild type and COX-1−/− mice using real-time PCR. However, the expression of nNOS and of iNOS was not significantly changed in the brain of COX-1−/− mice (data not shown). Factors other than increased PLA2- derived AA availability might also contribute to the increased COX-2 activity, for instance reactive oxygen species (Fujimoto et al. 2004).
Induction of COX-2 in COX-1−/− mice could lead to an increased PGH2 availability for PG biosynthesis. However, we found that brain TXB2 concentration was significantly reduced in COX-1−/− mice, and this decrease was accompanied by a down-regulation of TXS enzyme, suggesting that thromboxane production is preferentially coupled with the upstream COX-1 isoenzyme. This finding is consistent with our previous report indicating that valproate, when administered chronically to rats at therapeutically relevant concentrations, downregulates both COX-1 and COX-2 protein levels, as well as brain concentrations of both PGE2 and TXB2 (Bosetti and Weerasinghe 2003). Our data suggest that, providing an increase in AA availability through an up-regulation of the PLA2 enzymes, metabolic coupling between upstream and downstream enzymes and, possibly, compartmentalization of the substrate and the enzymes, drive brain PG biosynthesis towards specific end-products. Therefore, although further studies are required to determine whether compensatory changes of AA cascade reflect peripheral changes in COX-deficient mice, compensatory expression of the reciprocal COX isoenzyme appears to be organ-specific and may reflect tissue-specific functional importance of the individual COX enzyme. It should also be noted that, although we examined the whole brain tissue, regional disparities might occur as a consequence of the different regional distribution of COX-1 and COX-2 in specific brain regions.
Although levels of PGE2 were increased in brain of COX-1−/− mice, we did not find a significant change in mPGES-1 or cPGES protein expression. In contrast, we observed a significant reduction of mPGES-2 expression in COX-1−/− mice. The selective down-regulation of mPGES-2 expression compared with mPGES-1 and cPGES, in both COX-1−/− and COX-2−/− mice (Bosetti et al. 2004) suggests that mPGES-2 may be metabolically coupled with both COX-1 and COX-2 for basal PGE2 formation.
Although evidence supports a compensatory up-regulation of COX-2 in COX-1−/− mice, the mechanism that controls this compensation has not been fully elucidated. Kanekura and colleagues (Kanekura et al. 2002) have reported that constitutive COX-2 activity was transcriptionally enhanced in fibroblasts derived from COX-1−/− mice through the activation of NF-κB and that COX-1−/− cells contained higher levels of activated NF-κB than either wild type or COX-2−/− cells. In the present study we show that the increase in brain NF-κB activation in the COX-1−/− mice involves the up-regulation of the NF-κB subunits as well as both IκBα and upstream IKKα/β phosphorylation. These results are consistent with our recent study showing that brain NF-κB DNA binding activity, as well as the phosphorylation state of both IκBα and p65 proteins, were decreased in the COX-2−/− mice (Rao et al. 2005).
Since we have previously reported that brain PGE2 level is decreased in the COX-2−/− mice (Bosetti et al. 2004), our combined results from COX-1−/− and COX-2−/− mice suggest that in addition to the well-known capability of NF-κB to regulate COX-2 expression, PGE2 levels, in turn, also might modulate NFκB activity. Although further studies are needed to understand the mechanism of the direct interaction between PGE2 and NF-κB, it is possible that the increase in brain basal PGE2 level can lead to increased p65 expression and NF-κB activity in COX-1−/− mice, whereas a decrease in brain basal PGE2 level in COX-2−/− mice can lead to decreased activation of p65 and NF-κB activity (Rao et al. 2005). Consistent with this observation, a previous study reported that COX-2 and PG metabolites may affect NF-κB activation through a positive or negative feedback control mechanism dependent on different PG metabolites (Poligone and Baldwin 2001). For instance, PGE2 may activate the transcriptional activation domain of p65 in order to regulate NF-κB activity and PGJ2 can inhibit NF-κB activation via the inhibition of the IKK enzyme. Furthermore, a recent report indicates a positive feedback regulation of COX-2 and cPLA2 expression by PGs (Vichai et al. 2005), suggesting that PG levels play an important role in mediating the up-regulation of COX-2 protein expression and activity that we observe in COX-1−/− mice. Although we cannot exclude that NF-κB activation is the primary cause of the compensatory up-regulation of COX-2 and PGE2, the absence of a change in the gene expression of other target genes of NF-κB such as IL-1β, iNOS and nNOS, in the COX-1−/− mice (data not shown) suggests that COX-1 deficiency does not cause a general NF-κB dependent gene-induction, but it appears to be a more specific, PG-dependent, process. Since the mRNAs of these target genes are highly unstable, it is possible that compensatory post-transcriptional mechanisms are activated to keep their mRNA levels normal.
It is possible that the increase in PGE2 levels or other PG metabolites may trigger a positive feedback mechanism that can enhance COX-2 expression. Growing evidence indicates that post-transcriptional mechanisms are important in controlling COX-2 expression. An AU-rich element within the 3′-untranslated region (3′-UTR) of COX-2 mRNA can affect both mRNA stability and protein translation (Dixon et al. 2000; Cok and Morrison 2001). Additionally, the strength and duration of the induction of COX-2 mRNA, protein, and PGE2 release induced by IL-1β has been reported to be primarily the result of PGE2-dependent stabilization of COX-2 mRNA and stimulation of translation (Faour et al. 2001). Similarly, COX-2 protein level and enzyme activity are elevated by increasing its mRNA stability and its effect is controlled by various signaling pathways, including NF-κB (Tamura et al. 2002). Therefore, it is possible that posttranscriptional regulation affecting mRNA degradation, stabilization, or translation may be responsible for the changes in the COX-2 protein levels and enzymatic activity in the COX-1−/− mice, although it should be noted that results obtained under activating conditions may not necessarily reflect what occurs under basal conditions.
Taken together, our data suggest that brain basal PGE2 production is preferentially coupled with COX-2, whereas TXB2 production is preferentially coupled with COX-1, and that the compensatory overexpression of COX-2 in the COX-1−/− mice involves the activation of the NF-κB pathway with a mechanism involving increased expression of p50 and p65 subunits, increased phospho-IκBα and increased phospho-IKKα/β. Considering the increasingly recognized role of NF-κB in normal physiological conditions of the CNS, including behavior, neurotransmitter release and synaptic activity (Meffert and Baltimore 2005), the differential effect of COX-1 and COX-2 deficiency on this important pathway is likely to have several functional consequences on gene expression of other NF-κB brain target genes, synaptic activity, and behavior. Furthermore, the differential regulation of NF-κB pathway in COX-1−/− and COX-2−/− mice could also account for their different response to pathological conditions such as ischemia, excitotoxicity, and inflammation. The use of NF-κB inhibitors in COX-1−/− and COX-2−/− mice could help to clarify these issues. Further studies are required to elucidate the individual roles of the different COX enzymes, as well as of coupled upstream and downstream enzymes in brain signal transduction and in pathological conditions involving the activation of the arachidonic acid cascade, such as neuroinflammation and excitotoxicity.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. We thank to Dr. Stanley I. Rapoport and Dr. Myriam Gorospe for helpful discussion on this manuscript, Dr. Saba Aid, Christopher D. Toscano, and Richard P. Bazinet for useful experimental suggestions.
Abbreviations used:
- AA
arachidonic acid
- cPLA2
cytoslic phospholipase A2
- sPLA2
secretory phospholipase A2
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- PGE2
prostaglandin E2
- TXB2
thromboxane B2
- mPGES
microsomal prostaglandin E synthase
- cPGES
cytosolic prostaglandin E synthase
- TXS
thromboxane synthase
- NF-κB
nuclear factor kappa B
- IKK
IkappaB kinase
References
- Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- Almer G, Teismann P, Stevic Z, Halaschek-Wiener J, Deecke L, Kostic V, Przedborski S. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology. 2002;58:1277–1279. doi: 10.1212/wnl.58.8.1277. [DOI] [PubMed] [Google Scholar]
- Andreani M, Olivier JL, Berenbaum F, Raymondjean M, Bereziat G. Transcriptional regulation of inflammatory secreted phospholipases A(2) Biochim Biophys Acta. 2000;1488:149–158. doi: 10.1016/s1388-1981(00)00117-7. [DOI] [PubMed] [Google Scholar]
- Ayoub SS, Botting RM, Goorha S, Colville-Nash PR, Willoughby DA, Ballou LR. Acetaminophen-induced hypothermia in mice is mediated by a prostaglandin endoperoxide synthase 1 gene-derived protein. Proc Natl Acad Sci U S A. 2004;101:11165–11169. doi: 10.1073/pnas.0404185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosetti F, Weerasinghe GR. The expression of brain cyclooxygenase-2 is down-regulated in the cytosolic phospholipase A2 knockout mouse. J Neurochem. 2003;87:1471–1477. doi: 10.1046/j.1471-4159.2003.02118.x. [DOI] [PubMed] [Google Scholar]
- Bosetti F, Langenbach R, Weerasinghe GR. Prostaglandin E2 and microsomal prostaglandin E synthase-2 expression are decreased in the cyclooxygenase-2-deficient mouse brain despite compensatory induction of cyclooxygenase-1 and Ca2+-dependent phospholipase A2. J Neurochem. 2004;91:1389–1397. doi: 10.1111/j.1471-4159.2004.02829.x. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JD, Schievella AR, Nalefski EA, Lin LL. Cytosolic phospholipase A2. J Lipid Mediat Cell Signal. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- Cok SJ, Morrison AR. The 3′-untranslated region of murine cyclooxygenase-2 contains multiple regulatory elements that alter message stability and translational efficiency. J Biol Chem. 2001;276:23179–23185. doi: 10.1074/jbc.M008461200. [DOI] [PubMed] [Google Scholar]
- Di Rosa M, Ialenti A, Ianaro A, Sautebin L. Interaction between nitric oxide and cyclooxygenase pathways. Prostaglandins Leukot Essent Fatty Acids. 1996;54:229–238. doi: 10.1016/s0952-3278(96)90053-8. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- Faour WH, He Y, He QW, de Ladurantaye M, Quintero M, Mancini A, Di Battista JA. Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J Biol Chem. 2001;276:31720–31731. doi: 10.1074/jbc.M104036200. [DOI] [PubMed] [Google Scholar]
- Fujimoto Y, Uno E, Sakuma S. Effects of reactive oxygen and nitrogen species on cyclooxygenase-1 and -2 activities. Prostaglandins Leukot Essent Fatty Acids. 2004;71:335–340. doi: 10.1016/j.plefa.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Griffin DE, Wesselingh SL, McArthur JC. Elevated central nervous system prostaglandins in human immunodeficiency virus-associated dementia. Ann Neurol. 1994;35:592–597. doi: 10.1002/ana.410350513. [DOI] [PubMed] [Google Scholar]
- Ilzecka J. Prostaglandin E2 is increased in amyotrophic lateral sclerosis patients. Acta Neurol Scand. 2003;108:125–129. doi: 10.1034/j.1600-0404.2003.00102.x. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Romanovsky AA. Prostaglandin E2 as a mediator of fever: synthesis and catabolism. Front Biosci. 2004;9:1977–1993. doi: 10.2741/1383. [DOI] [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura T, Goorha S, Kirtikara K, Ballou LR. The involvement of NF-kappaB in the constitutive overexpression of cyclooxygenase-2 in cyclooxygenase-1 null cells. Biochim Biophys Acta. 2002;1542:14–22. doi: 10.1016/s0167-4889(01)00162-8. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- Kirtikara K, Morham SG, Raghow R, Laulederkind SJ, Kanekura T, Goorha S, Ballou LR. Compensatory prostaglandin E2 biosynthesis in cyclooxygenase 1 or 2 null cells. J Exp Med. 1998;187:517–523. doi: 10.1084/jem.187.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- Leslie CC. Properties and regulation of cytosolic phospholipase A2. J Biol Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- Li S, Wang Y, Matsumura K, Ballou LR, Morham SG, Blatteis CM. The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2(−/−), but not in cyclooxygenase-1(−/−) mice. Brain Res. 1999;825:86–94. doi: 10.1016/s0006-8993(99)01225-1. [DOI] [PubMed] [Google Scholar]
- Lindstrom T, Bennett P. Transcriptional regulation of genes for enzymes of the prostaglandin biosynthetic pathway. Prostaglandins Leukot Essent Fatty Acids. 2004;70:115–135. doi: 10.1016/j.plefa.2003.04.003. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63:901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ, 2nd, Morrow JD. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999;53:1495–1498. doi: 10.1212/wnl.53.7.1495. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68-69:383–399. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, Ohmiya Y, Watanabe K, Kudo I. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278:37937–37947. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ueno R, Ohishi K, Sakai T, Hayaishi O. Salivary prostaglandin concentrations: possible state indicators for major depression. Am J Psychiatry. 1989;146:365–368. doi: 10.1176/ajp.146.3.365. [DOI] [PubMed] [Google Scholar]
- Parente L, Perretti M. Advances in the pathophysiology of constitutive and inducible cyclooxygenases: two enzymes in the spotlight. Biochem Pharmacol. 2003;65:153–159. doi: 10.1016/s0006-2952(02)01422-3. [DOI] [PubMed] [Google Scholar]
- Pepicelli O, Fedele E, Berardi M, Raiteri M, Levi G, Greco A, Ajmone-Cat MA, Minghetti L. Cyclo-oxygenase-1 and -2 differently contribute to prostaglandin E2 synthesis and lipid peroxidation after in vivo activation of N-methyl-D-aspartate receptors in rat hippocampus. J Neurochem. 2005;93:1561–1567. doi: 10.1111/j.1471-4159.2005.03150.x. [DOI] [PubMed] [Google Scholar]
- Poligone B, Baldwin AS. Positive and negative regulation of NF-kappaB by COX-2: roles of different prostaglandins. J Biol Chem. 2001;276:38658–38664. doi: 10.1074/jbc.M106599200. [DOI] [PubMed] [Google Scholar]
- Powell WS. Reversed-phase high-pressure liquid chromatography of arachidonic acid metabolites formed by cyclooxygenase and lipoxygenases. Anal Biochem. 1985;148:59–69. doi: 10.1016/0003-2697(85)90628-1. [DOI] [PubMed] [Google Scholar]
- Rao JS, Langenbach R, Bosetti F. Down-regulation of brain nuclear factor-kappa B pathway in the cyclooxygenase-2 knockout mouse. Brain Res Mol Brain Res. 2005;139:217–224. doi: 10.1016/j.molbrainres.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Rapoport SI, Bosetti F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch Gen Psychiatry. 2002;59:592–596. doi: 10.1001/archpsyc.59.7.592. [DOI] [PubMed] [Google Scholar]
- Salvemini D. Regulation of cyclooxygenase enzymes by nitric oxide. Cell Mol Life Sci. 1997;53:576–582. doi: 10.1007/s000180050074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab JM, Schluesener HJ. Cyclooxygenases and central nervous system inflammation: conceptual neglect of cyclooxygenase 1. Arch Neurol. 2003;60:630–632. doi: 10.1001/archneur.60.4.630. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Sublette ME, Russ MJ, Smith GS. Evidence for a role of the arachidonic acid cascade in affective disorders: a review. Bipolar Disord. 2004;6:95–105. doi: 10.1046/j.1399-5618.2003.00094.x. [DOI] [PubMed] [Google Scholar]
- Tamura M, Sebastian S, Yang S, Gurates B, Fang Z, Bulun SE. Interleukin-1beta elevates cyclooxygenase-2 protein level and enzyme activity via increasing its mRNA stability in human endometrial stromal cells: an effect mediated by extracellularly regulated kinases 1 and 2. J Clin Endocrinol Metab. 2002;87:3263–3273. doi: 10.1210/jcem.87.7.8594. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002;68-69:95–114. doi: 10.1016/s0090-6980(02)00024-2. [DOI] [PubMed] [Google Scholar]
- Tanikawa N, Ohmiya Y, Ohkubo H, Hashimoto K, Kangawa K, Kojima M, Ito S, Watanabe K. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem Biophys Res Commun. 2002;291:884–889. doi: 10.1006/bbrc.2002.6531. [DOI] [PubMed] [Google Scholar]
- Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275:32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- Tay A, Maxwell P, Li Z, Goldberg H, Skorecki K. Isolation of promoter for cytosolic phospholipase A2 (cPLA2) Biochim Biophys Acta. 1994;1217:345–347. doi: 10.1016/0167-4781(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002;68-69:535–556. doi: 10.1016/s0090-6980(02)00054-0. [DOI] [PubMed] [Google Scholar]
- Vazquez-Tello A, Fan L, Hou X, Joyal JS, Mancini JA, Quiniou C, Clyman RI, Gobeil F, Jr., Varma DR, Chemtob S. Intracellular-specific colocalization of prostaglandin E2 synthases and cyclooxygenases in the brain. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1155–1163. doi: 10.1152/ajpregu.00077.2004. [DOI] [PubMed] [Google Scholar]
- Vichai V, Suyarnsesthakorn C, Pittayakhajonwut D, Sriklung K, Kirtikara K. Positive feedback regulation of COX-2 expression by prostaglandin metabolites. Inflamm Res. 2005;54:163–172. doi: 10.1007/s00011-004-1338-1. [DOI] [PubMed] [Google Scholar]
- Wang LH, Kulmacz RJ. Thromboxane synthase: structure and function of protein and gene. Prostaglandins Other Lipid Mediat. 2002;68-69:409–422. doi: 10.1016/s0090-6980(02)00045-x. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Zhang J, Goorha S, Raghow R, Ballou LR. The tissue-specific, compensatory expression of cyclooxygenase-1 and -2 in transgenic mice. Prostaglandins Other Lipid Mediat. 2002;67:121–135. doi: 10.1016/s0090-6980(01)00177-0. [DOI] [PubMed] [Google Scholar]