

Abstract
The activity of Hsp70 proteins is regulated by accessory proteins, among which the most studied are the members of the DnaJ-like protein family. BiP/GRP78 chaperones the translocation and maturation of secreted and membrane proteins in the endoplasmic reticulum. No DnaJ-like partner has been described so far to regulate the function of mammalian BiP/GRP78. We show here that murine BiP/GRP78 interacts with the lumenal J domain of the murine transmembrane protein MTJ1 (J-MTJ1). J-MTJ1 stimulates the ATPase activity of BiP/GRP78 at stoichiometric concentrations. The C-terminal tail of BiP/GRP78 is not required for the interaction with J-MTJ1, leaving the function of this portion of the molecule still unclear. Physical interactions between J-MTJ1 and BiP/GRP78 are stable and can be abolished by a single histidine → glutamine substitution in the highly conserved HPD motif shared by all DnaJ-like proteins. The J-MTJ1 fragment, but not the mutant J-MTJ1:H89Q fragment, stimulates the ATPase activity of Escherichia coli DnaK, although at a higher concentration than its genuine partner DnaJ. Full-length DnaJ does not stimulate BiP over the range of concentrations investigated. These results indicate that the J domain of MTJ1 is sufficient for its interaction with BiP/GRP78 and cannot be substituted by E. coli DnaJ.
The molecular chaperone BiP/GRP78, a member of the HSP70 family, is involved in many cellular processes, including regulation of calcium homeostasis (1, 2), translocation of newly synthesized polypeptides across the endoplasmic reticulum membrane (3–5), and their subsequent folding, maturation, transport or retrotranslocation (6, 7). BiP and other Hsp70 proteins consist of two domains: the 44-kDa N-terminal domain, which carries a weak ATPase activity (8, 9), and a C-terminal domain, which consists of a 20-kDa peptide-binding subdomain followed by a highly helical and variable 10-kDa C-terminal tail (10, 11). Although a C-terminal regulatory motif has been identified as important for the activity of the cytosolic Hsp70 (12), the function of the C-terminal tail of other Hsp70 proteins is still unknown.
Hsp70 proteins are regulated through the coupling of the ATPase activity of the N-terminal domain with the binding and release of unfolded polypeptides by the C-terminal domain (13). ATP binding onto the N-terminal domain leads to a low affinity state for unfolded peptides, characterized by fast exchange rates for the substrate (14–17). ATP hydrolysis induces change in the Hsp70 conformation that leads to the high affinity state, characterized by a slow off-rate of the peptidic substrate (14). It is believed that this cycle prevents the irreversible aggregation of polypeptides and promotes an overall more efficient folding process (18).
BiP/GRP78, and other Hsp70 proteins, self-associate into multiple oligomeric species (19–23). The C-terminal domain of BiP/GRP78 has been shown to be solely responsible for the oligomeric properties of the protein (22, 24). Binding of unfolded peptidic substrate onto the C-terminal domain, or binding of ATP onto the N-terminal domain, promotes depolymerization and stabilization of BiP monomers (21, 22, 24). The level of BiP basal ATPase activity varies with the degree of BiP oligomerization: the more oligomeric BiP is, the less active it is (21). Synthetic peptides that exhibit high affinity for BiP/GRP78 are also the most efficient in stimulating its catalytic activity and in stabilizing monomeric species (24).
The weak basal ATPase activity of Hsp70 proteins, with a turnover rate of about 0.02–0.5 min−1, is too slow to account by itself for the rate of assisted protein folding (13, 14). In Escherichia coli, two accessory proteins, DnaJ and GrpE, are essential for the chaperone functions of DnaK (25, 26). DnaJ specifically activates the ATP hydrolysis step of the reaction (27), and GrpE is a nucleotide-exchange factor that increases the rate of release of ADP (28). It has been demonstrated that, acting together, these two proteins can stimulate the steady-state ATPase activity of DnaK by 20- to 200-fold (17, 25, 29, 30). In yeast, the DnaJ-like transmembrane protein Sec63p is essential for the translocation process during which it interacts with Kar2p, the yeast homologue of BiP/GRP78 (31–33). The interaction between Sec63p and BiP/Kar2p is mediated by the conserved J domain, located near the N terminus of Sec63p (32, 34, 35). Similarly, the mammalian cytosolic DnaJ-like protein auxilin, which plays an important role in the dissociation of clathrin vesicles (36), stably interacts with the uncoating ATPase Hsc70 through its C-terminal J domain (37, 38).
An interaction of mammalian BiP with an endoplasmic reticulum-resident DnaJ homologue has not been characterized yet. The murine protein MTJ1 has been identified as a putative membrane DnaJ-like protein enriched in the microsomal and nuclear fractions of murine cells (39). We investigated whether MTJ1 constitutes an accessory protein able to regulate murine BiP/GRP78 catalytic activity. We present here biochemical evidence that the J domain of MTJ1 (J-MTJ1)1 interacts with murine BiP/GRP78 and stimulates its ATPase activity, and that the interaction between BiP/GRP78 and MTJ1 is mediated by an invariant motif found in all DnaJ-like proteins. J-MTJ1 can also activate the activity of the E. coli DnaK, although at higher concentrations. However, the E. coli DnaJ does not interact with murine BiP/GRP78.
EXPERIMENTAL PROCEDURES
Plasmids
pHis6-BiP was engineered from pT7B115 (21). The sequence coding for mature mouse BiP was amplified by polymerase chain reaction (PCR) with Pfu polymerase (Stratagene, La Jolla, CA) using the forward primer 5′-CCGCTCGAGGAGGAGGACAAGAAGGAG-3′ and the reverse primer 5′CCCAAGCTTTAATGCGGTAGTTTATC-3′. The PCR product was then digested with XhoI and HindIII enzymes (sites are underlined in the primers) and ligated into the pET15b vector (Novagen, Madison, WI) with T4 DNA ligase (MBI Fermentas, Amherst, NY). This vector allows expression of a six-histidine tag followed by a thrombin cleavage site (LVP(R/G)S) at the N-terminal extremity of the protein. The plasmid pHis6-BiP.ent was obtained from pFLAG-BiP.ent (22), by following the same strategy. The plasmid His10-BiP-ent-C19 encodes for a truncated form of BiP (residues 1–550) and will be described elsewhere.2 The plasmid pHis6-J-MTJ1 encodes the lumenal domain of MTJ1, from the start of the J domain following the first transmembrane segment, to the beginning of the second potential transmembrane segment (residues 46–148). The sequence coding for MTJ1 was amplified from a cDNA clone kindly provided by Dr. B. Zetter (Harvard Medical School, Boston, MA). After PCR amplification with Pfu polymerase using the forward primer Nde-MTJ1, 5′-GGGAATTCCATATGAGCGGAGACCTGGAGTTGTTC-3′, and the reverse primer Bam-MTJ1, 5′-GCGGATCCCTAAGCATTGCTCATTTTTCTCACTC-3′, the PCR product was digested with NdeI and BamHI enzymes (sites are underlined in the primers) and subcloned into pET15b. The plasmid pHis6-Jo-MTJ1 codes for the J domain only of MTJ1 (residues 46–129). The coding sequence of the J domain was amplified using the forward primer Nde-MTJ1 described above and the reverse primer, 5′-CGGGATCCCTAAAGTCCATTGATCAGAAC-3′. The PCR product was then digested with NdeI and BamHI enzymes and ligated into pET15b. The coding sequence of DnaJ was amplified from the plasmid pRLM232 (40), a kind gift of Dr. R. McMacken (Johns Hopkins University, Baltimore, MD), and subcloned into pET15b, in-frame with the hexahistidine N-terminal tag and the thrombin site.
Site-directed Mutagenesis
Site-directed mutagenesis was performed using two rounds of PCR in a procedure adapted from Morrison and Desrosiers (41). In the first set of reactions, the partial 5′ and 3′ fragments of J-MTJ1:H89Q were amplified from the pJ-MTJ1 plasmid with the Nde-MTJ1 and the Bam-MTJ1 outside primers, and the 5′-GCTTTCACTAACCTTACAACCAGACAAGAATAAAG-3′ and 5′-CTTTATTCTTGTCTGGTTGTAAGGTTAGTGAAAGC-3′ overlapping inside primers, which encode the H89Q mutation (underlined in the primers). The PCR products were gel-purified using low melting point agarose as described previously (42), mixed in equimolar concentrations, and used as templates for the second round of PCR amplification, together with the Nde-MTJ1 and the Bam-MTJ1 outside primers described above. The final product was digested with NdeI and BamHI enzymes and ligated into pET15b. The presence of the mutation was confirmed by DNA sequencing by using the Fentomol kit (Promega, Madison, WI) or the Sequenase kit (U. S. Biochemical Corp., Cleveland, OH).
Protein Expression and Purification
The recombinant proteins were expressed in E. coli BL21(DE3) cells. The cells were grown at 37 °C until A600 ~ 0.6, and synthesis of recombinant proteins was induced by addition of isopropyl-1-thio-β-d-galactopyranoside (Fischer Scientific, Hanover Park, IL) at 0.4 mm final concentration. Cells were harvested after 1-h induction for the different BiP proteins and after 2-h induction for J-MTJ1 or Jo-MTJ1 expressing cultures. Histidine-tagged proteins were purified on cobalt-chelating resin (Talon resin, CLONTECH, Palo Alto, CA). After binding to the resin in 20 mm Tris base, pH 8.0, 100 mm NaCl (buffer A), and a wash of nonspecific protein with 5 mm imidazole in buffer A, the bound proteins were eluted with 50 and 100 mm imidazole in buffer A. Eluted fractions were dialyzed overnight at 4 °C in 20 mm Tris base, pH 8.0, 50 mm KCl, 5 mm MgCl2 and loaded onto a MonoQ 5/5 HR anion exchange column connected to a fast protein liquid chromatography system (Amersham Pharmacia Biotech). Bound proteins were then eluted with a linear gradient of KCl (50–200 mm). Fractions were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) as described (43). Nucleotide-free BiP proteins were prepared as described (44). Briefly, purified BiP was mixed with activated charcoal (1 mg/ml) in 10 mm Na2EDTA, pH 8.0. After incubation for 4 h at 4 °C, the sample was centrifuged and the supernatant dialyzed exhaustively against 20 mm Hepes, pH 7.0, 75 mm KCl, 5 mm MgCl2 (assay buffer). Contamination by nucleotides was evaluated spectrophotometrically. An A280/A260 ratio of >1.5 indicated that the sample were free of nucleotides (45). For removal of the hexahistidine N-terminal tag, His6-J-MTJ1 or His6-DnaJ (30 μg) was incubated with 0.1 unit of thrombin (Sigma) at room temperature for 4 h. The cleaved proteins were then collected in the flow-through fraction of a Talon column. When necessary, the MTJ1-derived proteins were further purified by gel filtration on Superdex 75 (Pharmacia Amersham Biotech), equilibrated in assay buffer. All proteins were transferred into the assay buffer prior to analysis. Protein concentrations were estimated by the method of Bradford (46) or spectrophotometrically measured by using the extinction coefficients at 280 nm calculated by the method of Gill and von Hippel (47), i.e. 26,860 m−1 cm−1 and 13,730 m−1 cm−1 for BiP and J-MTJ1, respectively. E. coli DnaK was expressed from the plasmid pRLM163, a kind gift of Dr. R. McMacken and purified as described (48).
Far-UV Circular Dichroism Spectra
Circular dichroism (CD) spectra were obtained on a spectrometer (model 710, Jasco, Easton, MD). Spectra of native BiP (10 μm) were obtained in assay buffer. Spectra of native J-MTJ1 (11 μm) and mutant J-MTJ1:H89Q (8 μm) were obtained in 10 mm sodium phosphate, pH 7.0. Far-UV CD experiments were carried out at room temperature in the wavelength region of 250–190 nm with a 0.1-cm path length cell. Data were recorded at 0.5-nm intervals with a time constant of 1.0 s and a 1.0-nm constant spectral bandwidth. Data were averaged over five repetitive scans and fitted for protein concentration and optical path length. The mean residue ellipticity (θ) was calculated by using a mean residue weight of 110.9 and expressed in deg.cm2/dmol per residue. Data were analyzed by the spectral deconvolution method JFIT provided by Dr. B. Rupp (Lawrence Livermore National Laboratory, Livermore, CA).
ATPase Assays
Concentration-dependent assays were carried out as described (49). Time-dependent assays were done as follows: 70 μl of total assay buffer supplemented with 100 μm ATP, 1 μCi of [γ-32P]ATP (Amersham Pharmacia Biotech, 3000 mmol/Ci), and 0.2–0.3 μm BiP or DnaK was incubated at 37 °C with 0.4–0.6 μm J-MTJ1 protein. At various time points, 10 μl of the reaction mixtures was retrieved and quenched in SDS at 1% final concentration. The samples were then processed by thin layer chromatography or direct quantification of a phosphomolybdate complex as described (49). For assays in single turnover conditions, BiP (1 to 2 μm) was incubated with 10–30 nm ATP and 0.05 μCi of [α32P]ATP, in 20 mm Hepes, pH 7.0, 5 mm MgCl2, and 75 mM KCl or 50 mm NaCl. After incubation at 37 °C, 10-μl aliquots were quenched with 2 μl of 1 N HCl and put on ice. Conversion of ATP into ADP was determined by thin layer chromatography as described (49).
BiP-MTJ1 Binding Assays
His6-BiP (10 μg) in 20 mm Hepes, pH 7.0, 75 mm KCl, 5 mm MgCl2, 0.1% Tween 20, 2% glycerol, 1 mM phenylmethylsulfonyl fluoride (buffer B) was mixed with 20 μg of cleaved wild-type or mutant form of J-MTJ1, in the absence or presence of 1 mm ATP or ADP. After incubation at 37 °C for 30 min, 50 μl of the metal-chelating resin (50% suspension) was added and the samples were further incubated at 4 °C for 30 min, with gentle shaking. The resin was then washed five times with ice-cold buffer B, and the bound complexes were eluted by boiling after addition of SDS-PAGE sample buffer. Following denaturation, the samples were loaded onto a 15% SDS gel. After electrophoretic migration, the gels were stained with Coomassie blue and the quantity of proteins was estimated by densitometry scanning of the gel (1D analysis, Eastman Kodak, Rochester, NY). The data were then normalized to an equivalent background value.
RESULTS
Sequence Analysis of MTJ1
The in situ topology and cellular localization of MTJ1 has not been investigated yet. Using the most recent software available, we re-analyzed the primary sequence of MTJ1 and re-examined its putative topology (Figs. 1 and 2). An N-terminal signal sequence containing a putative cleavage site between amino acids 43 and 44, and a KKXX-like retrieval signal present near the C terminus, could account for MTJ1 retention in the endoplasmic reticulum (50). The N-terminal portion of MTJ1 carries a conserved J domain of about 70 amino acids (residues 60–117) that displays 42% identity with the J domain of yeast Sec63p. The J domain would be followed by one (model one, preferred model) or two (model two) putative transmembrane domains, exposing the C-terminal domain to the cytosolic or the luminal side, respectively (Fig. 2). In both models, the J domain of MTJ1 would be exposed in the lumen of the endoplasmic reticulum, similar to that of Sec63p (51), as proposed originally by Zetter and coworkers (39). Interestingly, another stretch of amino acids (residues 408–463) exhibits homology with Sec63p with 28% identity throughout this small region. This Sec63-homologous region is surrounded by two stretches of amino acids (residues 324–375 and residues 491–543) reminiscent of tryptophan-mediated repeats, also called the SANT domains, found in other DnaJ-like proteins such as MIDA-1, a murine protein involved in cell growth (52, 53), the Zuotin-related factors (54–56), the proto-oncogene c-myb (57–59), and the M-phase phosphoprotein 11 (60).
Fig. 1. Analysis of MTJ1 amino acid sequence (Swiss Protein Data Base accession number Q61712).

The transmembrane domains (long dashed underline) were predicted by using the TMPRED program. The signal peptide (dotted line) and cleavage site (arrow) were predicted with the program SignalP v1.1 (85). The DnaJ-like domain (triple underline) and the SANT domains (bold plain underline) were predicted by using the SMART program (86). The second region that presents homology with the yeast Sec63p (small dashed underline) has been identified using the alignment program SIM. The C-terminal endoplasmic reticulum retrieval signal (thick dotted line) was identified with the program PSORT II.
Fig. 2. Potential topology of murine MTJ1 and yeast Sec63p.
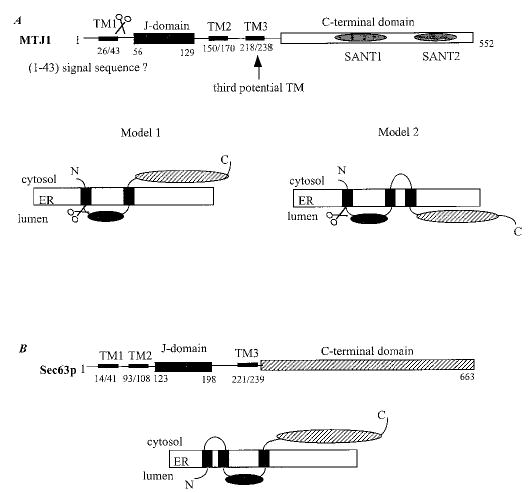
A, two models can be proposed for MTJ1 topology (see “Results”). B, the topology proposed by Feldheim and coworkers for Sec63p (51) is consistent with recent data obtained for a human homologue (80).
Expression of J-MTJ1
The lumenal domain of MTJ1 (residues 46–148) that encompasses the J domain was expressed in E. coli as a hexahistidine-tagged protein and found to be soluble and monomeric. After purification on a metal-chelating affinity column, the N-terminal histidine tag was removed by digestion with thrombin. The cleavage was fast and efficient, with a yield superior to 95% after 4 h of proteolytic digestion (Fig. 3, top panel). Cleaved J-MTJ1 was then submitted to far-UV CD spectroscopy (Fig. 3, bottom left panel). The spectra of wild-type J-MTJ1 presents the characteristic features of helical proteins: a high maximum at 195 nm followed by two minima at 208 and 222 nm. Deconvolution of the spectrum indicates that J-MTJ1 contains about 75% helical structures. These results demonstrate that wild-type MTJ1 is folded into a mostly helical domain, in agreement with the solution structure of the J domains of the E. coli DnaJ (61–63) and of the human HDJ1 (64).
Fig. 3. Purification and characterization of the J domain of MTJ1.
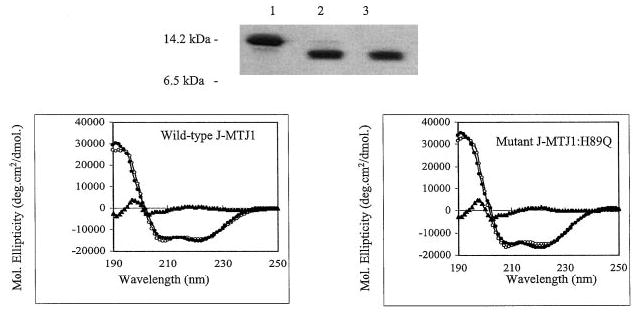
Top panel, 15% SDS-PAGE analysis of His6-J-MTJ1 before and after cleavage by thrombin. Lane 1, uncleaved His6-J-MTJ1; lane 2, cleaved J-MTJ1; lane 3, purified cleaved J-MTJ1 not retained by the metal-chelating resin. Bottom panel, far-UV CD spectra of J-MTJ1 wild-type (left panel) and J-MTJ1:H89Q mutant (right panel). The protein concentrations were 11 and 8 μm for the wild-type and the mutant, respectively, in 10 mm sodium phosphate, pH 7.0. The open circles, the closed circles, and the closed triangles represent the experimental data, the fitted data (as calculated by the JFIT method), and the difference between experimental and fitted data, respectively.
Steady-state ATPase Stimulation of BiP by J-MTJ1
His6-BiP was purified by affinity chromatography followed by ion exchange chromatography on MonoQ HR 5/5 (Amersham Pharmacia Biotech). His6-BiP is fully active in ATPase assays, and the kinetic parameters obtained in steady-state conditions are of the same order of magnitude as values determined for BiP purified from other constructs (Table I). His6-BiP purified in these conditions was found to be mostly monomeric (data not shown), contrary to our previously described His10-BiP that has a high tendency to oligomerize (22), and exhibits a slightly lower basal ATPase activity (Table I). Large variations in the basal ATPase activity of His10-BiP preparations correlate with the extent of aggregation/polymerization: lower activities were observed for highly polymerized samples, and higher activities were obtained for mostly monomeric samples. Single turnover experiments conducted with His6-BiP indicate that the rate of ATP hydrolysis (k2 ~ 0.30 min−1, data not shown) is similar to the steady-state value (kcat ~ 0.34 min−1), indicating that the rate of ATP hydrolysis is the rate-limiting step in BiP activity. J-MTJ1 causes a 2-fold stimulation of the steady-state ATPase activity of His6-BiP when used at a concentration as low as two molar excess over BiP (Table I). Because His10-BiP has a lower basal activity than His6-BiP (Table I), we investigated whether J-MTJ1 can stimulate His10-BiP ATPase activity more efficiently. Concentration dependence experiments show that the steady-state ATPase activity of His10-BiP is indeed stimulated by a 4- to 5-fold factor at a saturating concentration of J-MTJ1 (Fig. 4A). From the double-reciprocal plot (Fig. 4B), we calculated that the apparent affinity constant of J-MTJ1 for 0.3 μm BiP is equal to a Km value for MTJ1 of 0.31 μm. Thus, the J domain of MTJ1 stimulates efficiently the ATPase activity of BiP/GRP78 at stoichiometric concentrations.
Table I. Catalytic constants determined in steady-state conditions for BiP and DnaK in the presence of a synthetic peptide or J-MTJ1.
BiP and DnaK ATPase assays were performed as described under Experimental Procedures, in the absence (basal kcat) or presence of various concentrations of Pep2 (0–2 mm) or J-MTJ1 (0–10 μm).
| Proteins | Basal kcat | kcat Pep2 | Stimulation | kcat J-MTJ1 | Stimulation |
|---|---|---|---|---|---|
| min−1 | min−1 | -fold | min−1 | -fold | |
| pHis10-BiP | 0.20 ± 0.10 | 0.57 ± 0.14 | 2.85 | 0.95 ± 0.14 | 4.75 |
| pHis10-BiP.ent | 0.40 ± 0.09 | 0.47 ± 0.2 | 1.20 | 0.82 ± 0.02 | 2.05 |
| pHis10-BiP-ent-C19 | 0.32 ± 0.1 | 0.62 ± 0.11 | 1.95 | 0.67 ± 0.016 | 2.10 |
| pHis6-BiP | 0.34 ± 0.04 | 0.52 ± 0.06 | 1.55 | 0.74 ± 0.09 | 2.20 |
| pHis6-BiP.ent | 0.45 ± 0.06 | 0.51 ± 0.04 | 1.15 | 0.74 ± 0.15 | 1.65 |
| DnaK | 0.05 ± 0.007 | NDa | NDa | 0.40 ± 0.03 | 8.0 |
ND, not determined.
Fig. 4. Concentration-dependent stimulation of BiP ATPase activity by J-MTJ1.
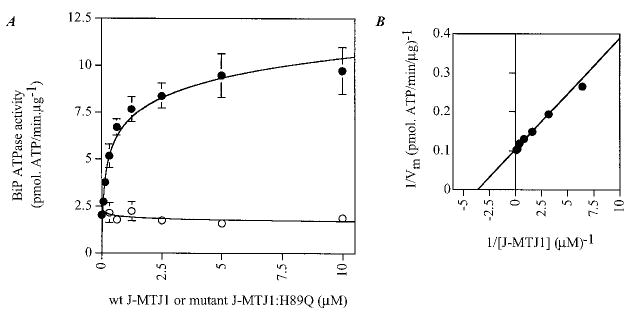
A, stimulation of His10-BiP steady-state ATPase activity by increasing concentrations of wild-type J-MTJ1 and mutant J-MTJ1:H89Q. His10-BiP (0.2 μm) was incubated with increasing concentrations (0.03–10 μm) of wild-type J-MTJ1 (closed circles) or mutant J-MTJ1:H89Q (open circles) and assayed for ATPase activity as described under “Experimental Procedures.” B, double reciprocal plot of BiP ATPase activity stimulated by J-MTJ1.
The J Domain Is Only Sufficient to Stimulate BiP/GRP78 Activity
The fragment J-MTJ1 corresponds to the putative luminal domain, most of which corresponds to the Sec63-like J domain (Fig. 1). However, the C-terminal region of J-MTJ1 contains one tryptophan (Trp-132), one phenylalanine (Phe-137), and two tyrosines (Tyr-138 and Tyr-139), residues that are highly enriched in peptides that bind to BiP with high affinity (24, 65, 66). To ensure that such an interaction was not taking place here, we expressed and assayed the J domain only (Jo-MTJ1, residues 46–129) for its ability to stimulate BiP ATPase activity. The fragment Jo-MTJ1 (0.4 μm) stimulates 2-fold the activity of His6-BiP (0.2 μm), with a kcat value of 0.47 min−1 versus kcat = 0.27 min−1 for the unstimulated BiP (data not shown).
Effect of J-MTJ1 on BiP-ent and BiP-ent-C19
We have described earlier the engineering of a FLAG-BiP-ent protein, in which the N- and C-terminal domains are linked by an enterokinase site and showed that FLAG-BiP-ent displays a slightly higher basal ATPase activity than His10-BiP and was slightly stimulated by a specific unfolded peptide, i.e. Pep2 (LSVKFLT) (22). We proposed that the insertion of one extra residue in the hinge between the two domains somehow disengaged the two domains from each other, therefore alleviating the inhibitory effect of the C terminus on the N-terminal catalytic domain (22). However, FLAG-BiP-ent had a high tendency to oligomerize and aggregate (22). We replaced the N-terminal FLAG epitope by a hexahistidine or decahistidine tag and assayed whether His6-BiP-ent and His10-BiP-ent could be efficiently stimulated by millimolar concentrations of a synthetic peptide and by submicromolar concentrations of J-MTJ1. As observed previously, the insertion of the enterokinase site slightly increased the basal activity of BiP (Table I). Interestingly, the peptide Pep2 does not efficiently stimulate BiP-ent (Table I), most likely because the purified BiP-ent proteins are enriched in monomeric species and the peptide binding does not cause further monomerization and apparent stimulation of BiP ATPase activity. When His6-BiP-ent (0.2 μm) was incubated in the presence of a 2-fold molar excess of J-MTJ1, its ATPase activity was stimulated by a factor of 1.65 (Table I). In similar conditions, His10-BiP-ent was stimulated by J-MTJ1 by a factor of 2 (Table I). The stoichiometry of BiP:J-MTJ1 interaction, and the action of J-MTJ1 on BiP-ent, a mostly monomeric protein that is not efficiently stimulated by millimolar concentrations of peptide, indicates that the J-MTJ1 stimulation of BiP ATPase activity is distinct from the peptide-induced stimulation caused by monomerization of inactive oligomers (22, 24).
The C-terminal domain of Hsp70 proteins is actually composed of two subdomains: A β-sandwich of 18–19 kDa that is responsible for the binding of unfolded peptidic substrates and a 10- to 11-kDa C-terminal helical tail of still unknown function (10, 67). Some observations indicate that the C-terminal tail of Hsp70 is involved in the regulation of Hsp70 ATPase activity as well as its chaperone function (12). To determine whether that region was important for BiP:MTJ1 interactions, we engineered a 60-kDa BiP-ent protein (residues 1–550, BiP-ent-C19) in which part of the C-terminal tail was deleted. Within experimental errors, His10-BiP-ent and His10-BiP-C19 have comparable basal and Pep2-stimulated activities (Table I). Both proteins are stimulated as efficiently by submicromolar concentrations of J-MTJ1 (Table I). This indicates that the C-terminal tail of BiP is not necessary for ATPase stimulation by the J domain, in agreement with other studies on Hsc70 and RDJ1 (68).
J-MTJ1 Physically Interacts with BiP
Next, we studied the physical interaction between BiP and the J domain of MTJ1. We used a binding assay in which His6-BiP and wild-type J-MTJ1 were incubated in the absence, or presence, of adenosine nucleotides and then immobilized onto a metal-chelating nickel resin. The unbound material was washed away, and the bound proteins were analyzed by SDS-PAGE. Our data indicate that His6-BiP and J-MTJ1 can form stable complexes in all conditions (Fig. 5, left panels).
Fig. 5. Complex formation between BiP and J-MTJ1.
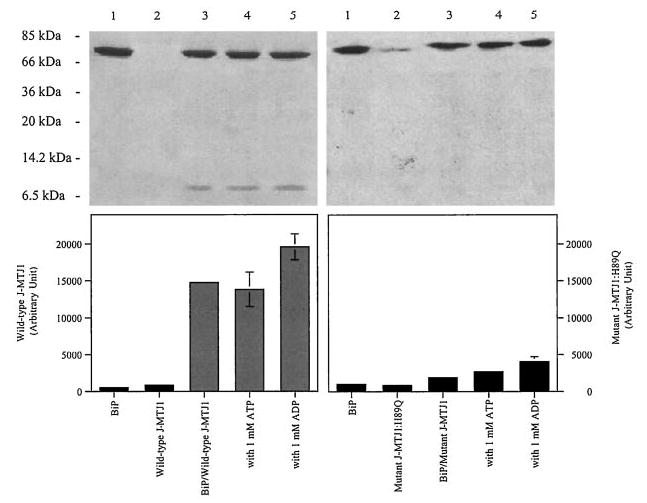
Interaction of His6-BiP with J-MTJ1 (left panels) or J-MTJ1: H89Q (right panels) was assayed as described under “Experimental Procedures.” Top panels, Coomassie-stained 15% SDS-PAGE. Lane 1, His6-BiP; lane 2; cleaved J-MTJ1 (or, right panel, mutant J-MTJ1:H89Q); lane 3, His6-BiP with wild-type J-MTJ1 (or, right panel, mutant J-MTJ1:H89Q); lane 4, His6-BiP with wild-type (or, right panel, mutant J-MTJ1:H89Q) in the presence of 1 mm ATP; lane 5, His6-BiP with wild-type (or, right panel, mutant J-MTJ1:H89Q) in the presence of 1 mm ADP. Bottom panel, densitometry scanning of the SDS-PAGE. The y axis represents the average of three independent experiments.
Mutation in the Conserved HPD Sequence Abolishes BiP: MTJ1 Interaction
From bacteria to high eukaryotes, all DnaJ-like proteins share a conserved HPD motif, whose integrity is essential for interaction with their Hsp70 partner, as demonstrated in E. coli (69), in yeast (32, 70) and human (71). We have expressed and purified a mutant form of J-MTJ1 in which the conserved histidine residue (in bold in Fig. 1) was substituted by a glutamine (J-MTJ1:H89Q). Circular dichroism analysis in the far-UV region shows that the mutant protein folds into a structure similar to that of wild-type J-MTJ1 (Fig. 3, bottom right panel). Deconvolution of the spectrum indicates that the mutant J-MTJ1:H89Q contains about 80% of helical structures, a value close to the 75% obtained for the wild-type protein. The mutant J-MTJ1:H89Q does not stimulate His10-BiP ATPase activity (Fig. 4, open circles) and does not interact with His6-BiP (Fig. 5, right panels). This indicates that the single substitution in the conserved HPD motif of J-MTJ1 totally abolishes interactions with murine BiP.
J-MTJ1 Stimulates Efficiently the E. coli DnaK, But the E. coli DnaJ Does Not Stimulate BiP Activity
Hsp70 proteins are not interchangeable in yeast (72, 73), and the efficiency of stimulation of ATPase activity in yeast Hsp70 proteins is most efficient in the presence of the genuine DnaJ-like partner (34, 74). These observations suggest that J-MTJ1 would not efficiently stimulate the ATPase activity of a foreign Hsp70. To investigate this hypothesis, we tested whether the E. coli Hsp70, i.e. DnaK, could be stimulated by J-MTJ1. Surprisingly, J-MTJ1, but not the mutant J-MTJ1:H89Q, stimulates the basal ATPase activity of DnaK by a factor of 8 (Fig. 6A and Table I). The double reciprocal plot (Fig. 6B) allows graphic determination of an apparent affinity equal to 1.17 μm for 0.3 μm DnaK: thus J-MTJ1 stimulates DnaK activity but with an apparent affinity that is at least four times lower than the one obtained for BiP. It should be noted that the basal activity of unstimulated DnaK (kcat = 0.05 min−1) is much lower than the kcat obtained for any of the BiP used in the present and previous studies (kcat = 0.2–0.5 min−1, Table I and Refs. 21, 24, and 49). J-MTJ1 stimulates DnaK activity by a factor of 8, so even stimulated DnaK functions at a rate that is at least 2-fold slower than that of BiP.
Fig. 6. Effect of J-MTJ1 and J-MTJ1:H89Q on DnaK ATPase activity.
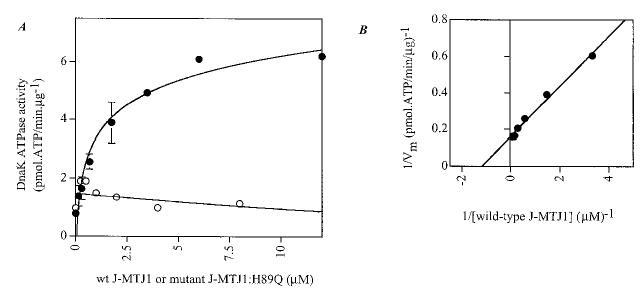
A, steady-state stimulation of DnaK activity by J-MTJ1 or J-MTJ1:H89Q. DnaK (0.3 μm) was incubated with increasing concentrations (0.03–10 μm) of wild-type J-MTJ1 (closed circles) or J-MTJ1:H89Q (open circles) and assayed for ATPase activity. B, double reciprocal plot of J-MTJ1 stimulated ATPase activity of DnaK.
We next assayed whether the full-length E. coli DnaJ could stimulate BiP as well as its genuine partner DnaK. Fig. 7 clearly shows that DnaJ stimulates DnaK at substoichiometric concentrations (0.12 μm DnaJ for 0.3 μm DnaK) but has no effect on BiP activity (Fig. 7, open circles).
Fig. 7. Effect of DnaJ on DnaK and BiP ATPase activity.
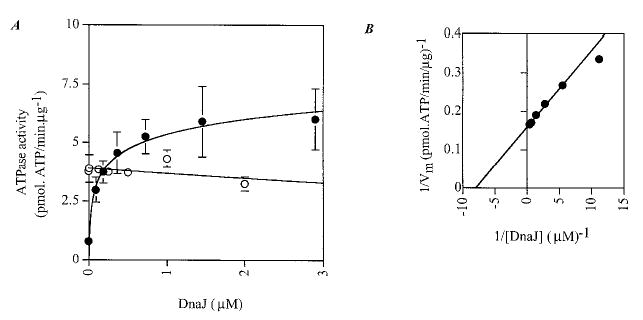
A, steady-state stimulation of DnaK and BiP activity by DnaJ. 0.3 μm DnaK (closed circles) or His10-BiP (open circles) was incubated with increasing concentrations of DnaJ and assayed for ATPase activity. B, double reciprocal plot of DnaJ-stimulated ATPase activity of DnaK.
DISCUSSION
We report here the characterization of the interaction of murine BiP with the luminal domain of the murine DnaJ-like protein MTJ1. Several years ago, MTJ1 was identified as a putative transmembrane protein enriched in the heavy microsomal and nuclear fractions (39). MTJ1 is widely expressed in all tissues, but its function has not been investigated. Re-examination of the MTJ1 amino acid sequence allowed us to propose two models for its topology. Both models place the J domain of MTJ1 in the lumen of the endoplasmic reticulum, consistent with the lumenal localization of the J domain of the yeast Sec63p (51). This makes MTJ1 a potential partner for the molecular chaperone BiP/GRP78. The predicted topology of the C-terminal domain is less obvious, because the MTJ1 sequence may contain one or two additional putative transmembrane domains. The C-terminal domain does not have any homology with other known proteins, except for two DNA binding domains homologous to SANT domains (or Trp-repeat domain) found in two other DnaJ-like proteins, MIDA1 and the zuotin-related factor ZRF1, but also in the proto-oncogene c-myb and the M-phase phosphoprotein 11 and related genes. These domains are thought to be involved in substrate binding (52–59) and may represent the site of interaction between MTJ1 and potential partners. Interestingly, the C-terminal domain of the E. coli DnaJ and of the yeast Ydj1p also contain a zinc finger-like domain (75–77). It has been proposed that the zinc finger domain of DnaJ is able to form a hydrophobic pocket in which unfolded peptidic substrates bind before being further delivered to the peptide binding pocket of the molecular chaperone DnaK (75). We are currently testing if the C-terminal domain of MTJ1 can bind unfolded polypeptides.
The J domain of MTJ1 (J-MTJ1) was successfully expressed in E. coli and found to fold in a mostly α-helical conformation, as observed for the J domains of the bacterial DnaJ (63) and of the human HDJ1 (64). Interaction with equimolar concentrations of J-MTJ1 led to the stimulation of the basal ATPase activity of BiP/GRP78 up to 5-fold. Single turnover experiments indicated that J-MTJ1 enhances the rate of ATP hydrolysis in the BiP catalytic cycle,3 in agreement with recent kinetic studies on DnaK:DnaJ interaction (27, 78).
Several pieces of evidence indicate that MTJ1 interacts with BiP/GRP78 in a native manner that is distinct from the interactions of the BiP C terminus with unfolded peptides (24, 65). First, CD spectroscopy gave us a good indication that J-MTJ1 was folded and, therefore, would not interact with BiP as an unfolded substrate. Second, BiP is stimulated up to almost 5-fold by stoichiometric concentrations of J-MTJ1 (J-MTJ1, Km = 0.31 μm for 0.3 μm BiP) but is stimulated 2- to 3-fold by much higher concentrations of short synthetic peptides used as models for denatured substrates (peptide, Km = 5–100 μm) (Table I and Refs. 22, 24). Third, a form of BiP that contains an engineered enterokinase site between the N-terminal catalytic domain and the peptide binding domain is stimulated about 2-fold by submicromolar concentrations of J-MTJ1 but is only slightly stimulated by millimolar concentrations of Pep2 (1.15-to 1.2-fold stimulation, Table I). Therefore, the insertion of one residue to create the enterokinase site at the junction between the two domains does not alter the ATPase stimulation by the J domain as much as it affects the coupling between the peptide binding and the catalytic domains. One explanation is that J-MTJ1 may bind to a conserved channel present in the catalytic domain, as described for DnaJ interaction with DnaK (79), and that the interaction is not being totally impaired by the insertion of one amino acid in the hinge region. Other studies, however, showed that insertion or four or more amino acids impairs the DnaJ:DnaK interaction (27) and that DnaJ binding enables coupling between the catalytic domain and the peptide binding domain of DnaK (79). Taken together, these findings suggest that DnaJ-like proteins interact strongly with the catalytic domain of Hsp70 proteins through their conserved J domain but may also make contact with the peptide binding domain.
Finally, the integrity of the highly conserved HPD motif is essential for BiP:J-MTJ1 physical interactions and for J-MTJ1-stimulated ATPase activity of BiP. BiP/GRP78 peptide binding domain recognizes specifically stretches of amino residues rich in large and/or aromatic hydrophobic amino acids (24, 66) and totally excludes charged residues such as histidine or aspartate found in the HPD motif. This strongly indicates that BiP/GRP78 binds to unfolded polypeptides and to J-MTJ1 through different type of interactions.
Our results also demonstrate that the C-terminal tail of BiP is not necessary for BiP:J-MTJ1 interaction. This is in agreement with studies on the rat RDJ1 J domain and the 60-kDa proteolytic fragment of Hsc70 (68) and with studies showing that a truncated form of DnaK is efficiently stimulated by DnaJ (79). Apart from the regulatory role of the EEVD motif present at the C terminus of Hsp70 (12), the C-terminal tail of BiP and of other Hsp70 proteins has not been shown to be involved in any cellular mechanism, and its function remains obscure.
We show that BiP and J-MTJ1 are able to interact physically. This is consistent with the results obtained by Brodsky and coworkers (34) on the J domain of Sec63p with yeast BiP/kar2p and of Ungewickell and co-workers (37, 38) that demonstrate that the J domain of auxilin interacts with Hsc70. However, studies on DnaJ indicate that the glycine/phenylalanine-rich region linking the J domain to the C-terminal domain is essential for interaction with DnaK (69). The E. coli DnaJ belongs to class I of DnaJ-like proteins, but auxilin, MTJ1, Sec63, and its newly identified human homologue (80) all belong to class III (81). It appears that class III DnaJ-like proteins may not require elements other than the J domain to regulate the activity of their Hsp70 partner. The J domain of MTJ1 and Sec63p is exposed in the lumen of the endoplasmic reticulum. It is reasonable to think that, for transmembrane DnaJ-like proteins like Sec63 and MTJ1, the J domain alone is sufficient to recruit their Hsp70 counterpart near the translocation channel where it can perform its function.
We do not observe that the physical interaction between BiP and J-MTJ1 requires a high concentration of ATP, contrary to what has been observed for Sec63p or auxilin (32, 34, 38). Because our studies were performed on nucleotide-free BiP (see “Experimental Procedures”), this may represent a specific feature of murine BiP and/or J-MTJ1. BiP self-associates into multiple oligomeric species (21), and ATP binding induces rapid monomerization (22). The His6-BiP used in the present study has a lower tendency to self-associate than the His10-BiP previously described and was isolated as a mostly monomeric species. The requirement for ATP may not be necessary, because His6-BiP is already in a fully monomeric conformation.
The J domain of MTJ1 stimulates very efficiently the E. coli DnaK (Table I), although a 3- to 4-fold molar excess is required to reach the Vmax level (J-MTJ1, Km = 1.17 μm for 0.3 μm DnaK), indicating that J-MTJ1 has a lower affinity for DnaK than for the murine BiP/GRP78 (Table I). Interestingly, full-length DnaJ does not stimulate BiP activity, even when present at a 7-fold molar excess (Fig. 7). It seems that the J domain of MTJ1 has been extremely conserved throughout evolution and can still stimulate DnaK, but the domains adjacent to the J domain in DnaJ prevent its interaction with the murine BiP/GRP78. We conclude that the J domain can be sufficient for interaction with an Hsp70 partner despite its origin and localization, but other structural determinants may play an important role in the specificity and stability of the complex. Our data are consistent with other studies that showed that J domains can be substituted by others and still lead to a functional DnaJ homologue (82, 83). Some slight modification in the sequence of the J domain may, however, be necessary to restore fully functional activity in vivo, as demonstrated in yeast (84).
The newly characterized MTJ1 may prove to be a genuine partner for BiP/GRP78. Further studies on the cellular localization and topology of MTJ1 and the role of its C-terminal domain are currently in progress. This should help improve the understanding of the function of murine BiP and MTJ1 in the endoplasmic reticulum of mammalian cells.
Acknowledgments
We are very grateful to Dr. Bruce Zetter (Harvard Medical School, Boston, MA) for sending us the MTJ1 clone and to Dr. Roger McMacken (Johns Hopkins University, Baltimore, MD) for supplying DnaK, DnaJ, and GrpE overexpressing strains as well as very detailed protocols and plasmid maps. We thank Mary Hall for purifying DnaK, Sunghyouk Park for his help in the deconvolution of circular dichroism spectra, and LaShaunda King and Michael Berg for critical reading of the manuscript.
Footnotes
This work was supported in part by National Institutes of Health Grant GM58107 (to S. Y. B.).
The abbreviations used are: J-MTJ1, J domain of protein MTJ1; PCR, polymerase chain reaction; PAGE, polyacrylamide gel electrophoresis; CD, circular dichroism.
L. King, M-G. Berg, C. Wang, A. Carey, M. Chevalier, E. C. Elguindi, and S. Y. Blond, manuscript in preparation.
M. Chevalier, unpublished data.
References
- 1.Lievremont JP, Rizzuto R, Hendershot L, Meldolesi J. J Biol Chem. 1997;272:30873–30879. doi: 10.1074/jbc.272.49.30873. [DOI] [PubMed] [Google Scholar]
- 2.Hamman BD, Hendershot LM, Johnson AE. Cell. 1998;92:747–758. doi: 10.1016/s0092-8674(00)81403-8. [DOI] [PubMed] [Google Scholar]
- 3.Brodsky JL, Goeckeler J, Schekman R. Proc Natl Acad Sci U S A. 1995;92:9643–9646. doi: 10.1073/pnas.92.21.9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corsi AK, Schekman R. J Biol Chem. 1996;271:30299–30302. doi: 10.1074/jbc.271.48.30299. [DOI] [PubMed] [Google Scholar]
- 5.Lyman SK, Schekman R. Cell. 1997;88:85–96. doi: 10.1016/s0092-8674(00)81861-9. [DOI] [PubMed] [Google Scholar]
- 6.Hammond C, Helenius A. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- 7.Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA. J Biol Chem. 1999;274:3453–3460. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- 8.Chappell TG, Konforti BB, Schmid SL, Rothman JE. J Biol Chem. 1987;262:746–751. [PubMed] [Google Scholar]
- 9.Flaherty KM, DeLuca-Flaherty C, McKay D. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Kurochkin AV, Pang Y, Hu W, Flynn GC, Zuiderweg ER. Biochemistry. 1998;37:7929–7940. doi: 10.1021/bi9800855. [DOI] [PubMed] [Google Scholar]
- 12.Freeman BC, Myers MP, Schumacher R, Morimoto RI. EMBO J. 1995;14:2281–2292. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukau B, Horwich AL. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 14.Schmid D, Baici A, Gehring H, Christen P. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 15.McCarty JS, Buchberger A, Reinstein J, Bukau B. J Mol Biol. 1995;1249:126–137. doi: 10.1006/jmbi.1995.0284. [DOI] [PubMed] [Google Scholar]
- 16.Theyssen H, Schuster HP, Packschies L, Bukau B, Reinstein J. J Mol Biol. 1996;263:657–670. doi: 10.1006/jmbi.1996.0606. [DOI] [PubMed] [Google Scholar]
- 17.Pierpaoli EV, Sandmeier E, Baici A, Schonfeld HJ, Gisler S, Christen P. J Mol Biol. 1997;269:757–768. doi: 10.1006/jmbi.1997.1072. [DOI] [PubMed] [Google Scholar]
- 18.Fedorov AN, Baldwin TO. J Biol Chem. 1997;272:32715–32718. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- 19.Freiden PJ, Gaut JR, Hendershot LM. EMBO J. 1992;11:63–70. doi: 10.1002/j.1460-2075.1992.tb05028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlino A, Toledo H, Skaleris D, DeLisio R, Weissbach H, Brot N. Proc Natl Acad Sci U S A. 1992;89:2081–2085. doi: 10.1073/pnas.89.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blond-Elguindi S, Fourie AM, Sambrook JF, Gething MJ. J Biol Chem. 1993;268:12730–12735. [PubMed] [Google Scholar]
- 22.Chevalier M, King L, Wang C, Gething MJ, Elguindi E, Blond SY. J Biol Chem. 1998;273:26827–26835. doi: 10.1074/jbc.273.41.26827. [DOI] [PubMed] [Google Scholar]
- 23.Fouchaq B, Benaroudj N, Ebel C, Ladjimi MM. Eur J Biochem. 1999;259:379–384. doi: 10.1046/j.1432-1327.1999.00053.x. [DOI] [PubMed] [Google Scholar]
- 24.King L, Chevalier M, Blond SY. Biochem Biophys Res Commun. 1999;263:181–186. doi: 10.1006/bbrc.1999.1321. [DOI] [PubMed] [Google Scholar]
- 25.Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Proc Natl Acad Sci U S A. 1991;88:2874–2878. doi: 10.1073/pnas.88.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wickner S, Skowyra D, Hoskins J, McKenney K. Proc Natl Acad Sci U S A. 1992;89:10345–10349. doi: 10.1073/pnas.89.21.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Proc Natl Acad Sci U S A. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Packschies L, Theyssen H, Buchberger A, Bukau B, Goody RS, Reinstein J. Biochemistry. 1997;36:3417–3422. doi: 10.1021/bi962835l. [DOI] [PubMed] [Google Scholar]
- 29.Gamer J, Multhaup G, Tomoyasu T, McCarty JS, Rudiger S, Schonfeld HJ, Schirra C, Bujard H, Bukau B. EMBO J. 1996;15:607–617. [PMC free article] [PubMed] [Google Scholar]
- 30.Banecki B, Zylicz M. J Biol Chem. 1996;271:6137–6143. doi: 10.1074/jbc.271.11.6137. [DOI] [PubMed] [Google Scholar]
- 31.Scidmore MA, Okamura HH, Rose MD. Mol Biol Cell. 1993;4:1145–1159. doi: 10.1091/mbc.4.11.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corsi AK, Schekman R. J Cell Biol. 1997;137:1483–1493. doi: 10.1083/jcb.137.7.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matlack KE, Misselwitz B, Plath K, Rapoport TA. Cell. 1999;97:553–564. doi: 10.1016/s0092-8674(00)80767-9. [DOI] [PubMed] [Google Scholar]
- 34.McClellan AJ, Endres JB, Vogel JP, Palazzi D, Rose MD, Brodsky JL. Mol Biol Cell. 1998;9:3533–3545. doi: 10.1091/mbc.9.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misselwitz B, Staeck O, Matlack KE, Rapoport TA. J Biol Chem. 1999;274:20110–20115. doi: 10.1074/jbc.274.29.20110. [DOI] [PubMed] [Google Scholar]
- 36.Ungewickell E, Ungewickell H, Holstein SEH, Lindner R, Prasad K, Barouch W, Martin B, Greene LE, Eisenberg E. Nature. 1995;378:632–635. doi: 10.1038/378632a0. [DOI] [PubMed] [Google Scholar]
- 37.Holstein SE, Ungewickell H, Ungewickell E. J Cell Biol. 1996;135:925–937. doi: 10.1083/jcb.135.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ungewickell E, Ungewickell H, Holstein SE. J Biol Chem. 1997;272:19594–19600. doi: 10.1074/jbc.272.31.19594. [DOI] [PubMed] [Google Scholar]
- 39.Brightman SE, Blatch GL, Zetter BR. Gene. 1995;153:249–254. doi: 10.1016/0378-1119(94)00741-a. [DOI] [PubMed] [Google Scholar]
- 40.Karzai AW, McMacken R. J Biol Chem. 1996;271:11236–11246. doi: 10.1074/jbc.271.19.11236. [DOI] [PubMed] [Google Scholar]
- 41.Morrison HG, Desrosiers RC. BioTechniques. 1993;14:454–457. [PubMed] [Google Scholar]
- 42.Sambrook, J. F., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 43.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 44.Ha J, McKay DB. Biochemistry. 1994;33:14625–14635. doi: 10.1021/bi00252a031. [DOI] [PubMed] [Google Scholar]
- 45.Farr CD, Slepenkov SV, Witt SN. J Biol Chem. 1998;273:9744–9748. doi: 10.1074/jbc.273.16.9744. [DOI] [PubMed] [Google Scholar]
- 46.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 47.Gill SC, Hippel PHv. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 48.Russell R, Jordan R, McMacken R. Biochemistry. 1998;37:596–607. doi: 10.1021/bi972025p. [DOI] [PubMed] [Google Scholar]
- 49.Chevalier M, King L, Blond S. Methods Enzymol. 1998;290:384–409. doi: 10.1016/s0076-6879(98)90033-7. [DOI] [PubMed] [Google Scholar]
- 50.Teasdale RD, Jackson MR. Annu Rev Cell Dev Biol. 1996;12:27–54. doi: 10.1146/annurev.cellbio.12.1.27. [DOI] [PubMed] [Google Scholar]
- 51.Feldheim D, Yoshimura K, Admon A, Schekman R. Mol Biol Cell. 1993;4:931–939. doi: 10.1091/mbc.4.9.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shoji W, Inoue T, Yamamoto T, Obinata M. J Biol Chem. 1995;270:24818–24825. doi: 10.1074/jbc.270.42.24818. [DOI] [PubMed] [Google Scholar]
- 53.Inoue T, Shoji W, Obinata M. Biochem Biophys Res Commun. 1999;266:147–151. doi: 10.1006/bbrc.1999.1779. [DOI] [PubMed] [Google Scholar]
- 54.Kanei-Ishii C, Sarai A, Sawazaki T, Nakagoshi H, He DN, Ogata K, Nishimura Y, Ishii S. J Biol Chem. 1990;265:19990–19995. [PubMed] [Google Scholar]
- 55.Howe KM, Reakes CF, Watson RJ. EMBO J. 1990;9:161–169. doi: 10.1002/j.1460-2075.1990.tb08092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gabrielsen OS, Sentenac A, Fromageot P. Science. 1991;253:1140–1143. doi: 10.1126/science.1887237. [DOI] [PubMed] [Google Scholar]
- 57.Zhang S, Lockshin C, Herbert A, Winter E, Rich A. EMBO J. 1992;11:3787–3796. doi: 10.1002/j.1460-2075.1992.tb05464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan W, Schilke B, Pfund C, Walter W, Kim S, Craig EA. EMBO J. 1998;17:4809–4817. doi: 10.1093/emboj/17.16.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hugues R, Chan FY, White RA, Zon LI. Genomics. 1995;29:546–550. doi: 10.1006/geno.1995.9969. [DOI] [PubMed] [Google Scholar]
- 60.Matsumoto-Taniura N, Pirollet F, Monroe R, Gerace L, Westendorf JM. Mol Biol Cell. 1996;7:1455–1469. doi: 10.1091/mbc.7.9.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hill RB, Flanagan JM, Prestegard JH. Biochemistry. 1995;34:5587–5597. doi: 10.1021/bi00016a033. [DOI] [PubMed] [Google Scholar]
- 62.Szyperski T, Pellecchia M, Wall D, Georgopuolos C, Wüthrich K. Proc Natl Acad Sci U S A. 1994;91:11343–11347. doi: 10.1073/pnas.91.24.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pellecchia M, Szyperski T, Wall D, Georgopoulos C, Wuthrich K. J Mol Biol. 1996;260:236–250. doi: 10.1006/jmbi.1996.0395. [DOI] [PubMed] [Google Scholar]
- 64.Qian YQ, Patel D, Hartl FU, McColl DJ. J Mol Biol. 1996;260:224–235. doi: 10.1006/jmbi.1996.0394. [DOI] [PubMed] [Google Scholar]
- 65.Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething MJ. Cell. 1993;75:717–728. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- 66.Fourie AM, Sambrook JF, Gething MJH. J Biol Chem. 1994;269:30470–30478. [PubMed] [Google Scholar]
- 67.Bertelsen EB, Zhou H, Lowry DF, Flynn GC, Dahlquist FW. Protein Sci. 1999;8:343–354. doi: 10.1110/ps.8.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leng CH, Brodsky JL, Wang C. Protein Sci. 1998;7:1186–1194. doi: 10.1002/pro.5560070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wall D, Zylicz M, Georgopoulos C. J Biol Chem. 1994;269:5446–5451. [PubMed] [Google Scholar]
- 70.Tsai J, Douglas MG. J Biol Chem. 1996;271:9347–9354. doi: 10.1074/jbc.271.16.9347. [DOI] [PubMed] [Google Scholar]
- 71.Liu JS, Kuo SR, Makhov AM, Cyr DM, Griffith JD, Broker TR, Chow LT. J Biol Chem. 1998;273:30704–30712. doi: 10.1074/jbc.273.46.30704. [DOI] [PubMed] [Google Scholar]
- 72.Brodsky JL, Hamamoto S, Feldheim D, Schekman R. J Cell Biol. 1993;120:95–102. doi: 10.1083/jcb.120.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.James P, Pfund C, Craig EA. Science. 1997;275:387–389. doi: 10.1126/science.275.5298.387. [DOI] [PubMed] [Google Scholar]
- 74.Cyr DM, Douglas MG. J Biol Chem. 1994;269:9798–9804. [PubMed] [Google Scholar]
- 75.Szabo A, Korzun R, Hartl FU, Flanagan J. EMBO J. 1996;15:408–417. [PMC free article] [PubMed] [Google Scholar]
- 76.Banecki B, Liberek K, Wall D, Warrzynow A, Georgopoulos C, Bertoli E, Tanfani F, Zylicz M. J Biol Chem. 1996;271:14840–14848. doi: 10.1074/jbc.271.25.14840. [DOI] [PubMed] [Google Scholar]
- 77.Lu Z, Cyr DM. J Biol Chem. 1998;273:5970–5978. doi: 10.1074/jbc.273.10.5970. [DOI] [PubMed] [Google Scholar]
- 78.Russell R, Wali Karzai A, Mehl AF, McMacken R. Biochemistry. 1999;38:4165–4176. doi: 10.1021/bi9824036. [DOI] [PubMed] [Google Scholar]
- 79.Gassler CS, Buchberger A, Laufen T, Mayer MP, Schroder H, Valencia A, Bukau B. Proc Natl Acad Sci U S A. 1998;95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Skowronek MH, Rotter M, Haas IG. Biol Chem. 1999;380:1133–1138. doi: 10.1515/BC.1999.142. [DOI] [PubMed] [Google Scholar]
- 81.Cheetham ME, Caplan AJ. Cell Stress Chaperones. 1998;3:28–36. doi: 10.1379/1466-1268(1998)003<0028:sfaeod>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, Pipas JM, Silver PA, Roberts TM, Schaffhausen BS, DeCaprio JA. Genes Dev. 1997;11:1098–1110. doi: 10.1101/gad.11.9.1098. [DOI] [PubMed] [Google Scholar]
- 83.Kelley WL, Georgopoulos C. Proc Natl Acad Sci U S A. 1997;94:3679–3684. doi: 10.1073/pnas.94.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schlenstedt G, Harris S, Risse B, Lill R, Silver P. J Cell Biol. 1995;129:979–988. doi: 10.1083/jcb.129.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 86.Schultz J, Milpetz F, Bork P, Ponting CP. Proc Natl Acad Sci U S A. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]