

Abstract
Introduction
We wished to determine whether cholestasis induced by total parenteral nutrition (TPN) in preterm newborn infants is associated with increased oxidative stress secondary to increased reactive oxygen intermediates. We hypothesized that elevated urinary thiobarbituric-acid-reacting substances (TBARS), a marker of oxidative stress, would be associated with hepatocellular injury as measured by serum alanine transaminase (ALT) and aspartate transaminase (AST) levels.
Materials and methods
Preterm infants (<35 weeks' gestation) admitted to the neonatal intensive care unit were enrolled (with their parents' informed consent) in either the 'cholestasis' group (if their direct bilirubin was >2 mg/dl [34.2 μmol/l] and duration of TPN was ≥ 10 days [n = 27]) or in the control group. Urine samples for measurement of TBARS (proportionate to lipid peroxidation) and blood specimens for analysis of serum bilirubin, ALT, AST, and alkaline phosphatase were obtained within 24 hours of enrollment.
Results
The cholestasis and control groups were comparable with respect to gestational age, birth weight, Apgar score, maximum FiO2, and duration of supplemental oxygen administration. Median serum direct bilirubin concentrations in the cholestasis and control groups were, respectively, 3.3 mg/dl (56.4 μmol/l) and 1.7 mg/dl (29.1 μmol/l) (P < 0.001). Serum ALT and AST levels were also elevated in the cholestasis group, but alkaline phosphatase levels did not differ significantly between the groups. Urinary levels of TBARS in all the infants were correlated with ALT and AST but did not differ significantly between cholestatic and control infants.
Discussion
Our findings suggest that oxidant stress is associated with hepatocellular injury in preterm infants. This effect is not correlated with the degree of cholestasis.
Keywords: cholestasis, newborn, oxidant, peroxidation, premature
Introduction
The incidence of cholestasis related to total parenteral nutrition (TPN) among preterm infants has been estimated to be between 7% and 85%, depending on the population examined and the definition of cholestasis used [1]. In infants with necrotizing enterocolitis or short bowel syndrome, the prevalence of TPN-related cholestasis is 60–90% [2]. Although cholestasis is reversible in most patients after the successful advancement of enteral feeding, progressive liver fibrosis and cirrhosis occur in some patients even after complete enteral nutrition has been established [3]. Some studies have suggested that excessive amino acids, hepatotoxic bile acids, bacterial overgrowth, sepsis, micronutrient deficiency, and TPN contaminants all contribute to cholestatic liver injury [4,5,6]. Diminished volume of enteral feeds may also independently contribute to the development of cholestasis. However, the mechanisms of liver injury in cholestatic diseases in infants remain unclear.
Recent studies have supported the hypothesis that generation of reactive oxygen intermediates (ROIs) and elevated lipid hydroperoxides in the liver during cholestasis cause tissue injury. Exposure of isolated hepatocytes to hydrophobic bile acids leads to intracellular production of oxygen free radicals and lipid peroxides [7]. Animal studies using models of surgically induced extrahepatic biliary obstruction have also shown that lipid peroxidation products – specifically, malondialdehyde (MDA) – are increased in the cholestatic liver [8,9]. This increase is associated with decreased tissue antioxidant activity, increased leukocyte infiltration, and early evidence of collagen deposition; these effects are ameliorated by the administration of exogenous antioxidants [8,9,10]. Consistent with these findings, several forms of liver disease in humans have been shown to be associated with oxidative tissue injury. Specifically, products of lipid peroxidation are elevated in patients with hepatic hypoxia/reperfusion, obstructive liver disease, alcoholic liver disease, Wilson's disease, Alagille syndrome, sepsis, and inflammatory liver diseases [11,12,13,14,15]. In vitro, procollagen gene expression increases in human liver cells after exposure to lipid peroxide breakdown products [16]. Furthermore, deposition of lipofuscin, a by-product of lipid peroxidation, characterizes TPN-induced cholestasis in preterm infants [17].
The purpose of these studies was to determine whether TPN-induced cholestasis in preterm newborn infants is associated with increased oxidative stress secondary to increased ROIs. We hypothesized that elevation in markers of oxidative stress would be associated with increased liver injury, as measured by serum alanine transaminase (ALT) and aspartate transaminase (AST) levels. In order to quantify ROIs in infants, we measured urinary thiobarbituric-acid-reacting substances (TBARS). The most abundant of these substances is MDA, an aldehydic lipid peroxidation product formed by the action of ROIs on lipid membranes. The identification of ROIs as potential markers of liver injury in cholestatic preterm infants may aid in the diagnosis and management of those at highest risk for ongoing liver impairment.
Materials and methods
Patients
All preterm infants (<35 weeks' gestation) born at St Peter's University Hospital (New Brunswick, NJ, USA) and admitted to the hospital's neonatal intensive care unit between March 1997 and December 1998 were serially screened during the course of hospitalization for entry into the 'cholestasis' study group (Fig. 1). Infants with major congenital anomalies (including all gastrointestinal and liver anomalies) or with congenital or acquired infection were excluded. The criteria for entry into the cholestasis group were direct bilirubin >2 mg/dl (34.2 μmol/l) and duration of TPN ≥ 10 days. If the infant met these criteria, the parents' informed consent for entry into the study was requested. During this period, 36 infants qualified for the cholestasis group, and informed consent was obtained from the parents of 27. When each eligible infant with cholestasis was enrolled, a preterm infant without cholestasis was matched for gestational age, birth weight, and severity of respiratory illness, and the parents' informed consent was requested to enroll, the infant as a control subject. For the 27 infants enrolled in the cholestasis group, matched controls were identified for 24. Parental consent could not be obtained for 8, so 16 infants constituted the control group. Study personnel obtained demographic and medical information from infants medical records.
Figure 1.
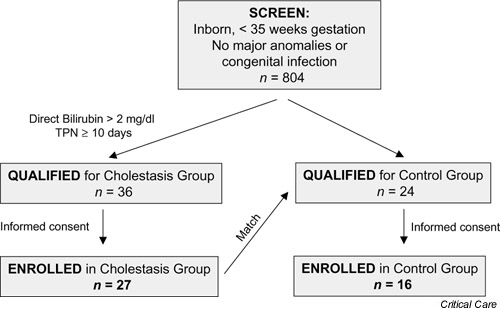
Patient recruitment. Preterm infants born at St Peter's University Hospital (New Brunswick, NJ, USA) who were admitted to the hospital's neonatal intensive care unit between March 1997 and December 1998 were serially screened during hospitalization for entry into the 'cholestasis' study group. Controls were matched for gestational age, birth weight, and severity of respiratory illness. TPN, total parenteral nutrition.
Determination of urinary thiobarbituric-acid-reacting substances
Urine samples were obtained under sterile conditions from all enrolled infants at the time of their entry into the study. Informed consent was obtained from parents for the acquisition of samples, and these studies were approved by the Committee for the Protection of Human Subjects in Research of St Peter's University Hospital. TBARS were measured as previously described [18,19]. Briefly, 200 μl of urine was combined with 10 μl of 5% butylated hydroxytoluene (in glacial acetic acid) and 300 l of a 0.5% aqueous thiobarbituric acid (TBA) solution. The samples were vortexed and were incubated at 100°C for 30 minutes, and the absorbance at 532 nm was measured using a PerkinElmer Lamba 3B spectrophotometer (PerkinElmer, Wellesley, MA, USA). The quantity of TBARS is proportionate to the amount of MDA, a lipid peroxidation product generated by the oxidation of membrane lipids by reactive oxygen species. MDA reacts with TBA to form a 1:2 MDA–TBA adduct that absorbs at 532 nm. In the present studies, MDA was confirmed to be the predominant TBA-reacting adduct by high-performance liquid chromatography analysis of representative samples. To control for urine concentration, data were normalized to urine creatinine concentrations, as previously described [20].
Determination of serum bilirubin, ALT, AST, alkaline phosphatase
Blood specimens were obtained from subjects within 24 hours of the urine specimen. Quantitative determinations of serum bilirubin, ALT, AST, and alkaline phosphatase levels were performed by the clinical laboratory at St Peter's University Hospital.
Statistical analysis
Direct bilirubin, ALT, AST, alkaline phosphatase, and urinary TBARS values were not normally distributed. The data are presented as median (25th, 75th percentile). Differences between the groups were analyzed for significance by one-way ANOVA using natural log transformations of the data, which are normally distributed. Differences were regarded as statistically significant at P values ≤ 0.05. Correlations of bilirubin, ALT, and AST with TBARS were calculated by regression using the log-transformed values to ensure normal distribution of all variables in those analyses.
Results
Twenty-seven infants were enrolled in the cholestasis group and 16 infants served as controls. The cholestasis and control groups were not significantly different with respect to gestational age (29.3 ± 4.7 vs 27.1 ± 3.2 weeks, respectively) and birth weights (1276 ± 751 vs 1016 ± 392 g), as well as Apgar scores, maximum FiO2, and length of time for which supplemental oxygen was given (Table 1). Urine samples were collected at 48.3 ± 38.2 days of age in the cholestasis group and 38.4 ± 22.1 days in the control group (P = 0.34). At that time, infants in the cholestasis group had been advancing on enteral feedings in addition to parenteral nutrition for 11.3 ± 5.5 days (range 0–23 days) and were receiving 21.4 ± 12.3 ml/kg per day enterally. Control infants were on full enteral feedings at the time of study.
Table 1.
Demographic variables of infants studied
| Infants | |||
| Variable | With cholestatis (n = 27) | Controls (n = 16) | P |
| Birthweight (g) | 1276 ± 751 | 1016 ± 392 | 0.62 |
| Gestational age (weeks) | 29.3 ± 4.7 | 27.1 ± 3.2 | 0.59 |
| Apgar score (5 minutes) | 7.0 ± 2.0 | 8.1 ± 1.3 | 0.07 |
| Gender (males/females) | 18/8 | 13/4 | |
| Maximum FiO2 | 0.63 ± 0.31 | 0.58 ± 0.31 | 0.59 |
| Supplemental O2 (days) | 55.7 ± 54.1 | 44.4 ± 36.9 | 0.46 |
| TPN (days) | 59.6 ± 65.6* | 26.5 ± 17.0 | 0.04 |
Values are expressed as means ± standard deviation. TPN = total parenteral nutrition. * Significantly different from control group.
Median serum direct bilirubin concentrations were 3.3 mg/dl (56.4 μmol/l) in the cholestasis group and 1.7 mg/dl (29.1 μmol/l) in the control group (P < 0.001). Median serum ALT and AST levels were also elevated in the cholestasis group (32 vs 9 and 71 vs 33 U/l, respectively; P < 0.01). Values for alkaline phosphatase and mean urinary TBARS did not differ significantly between the groups (Table 2). Urinary TBARS were not significantly correlated with gestational age, gender, days on TPN, indirect bilirubin, or alkaline phosphatase (not shown). Likewise, urinary TBARS were not correlated with direct bilirubin (Fig. 2, top). In contrast, urinary TBARS levels among all infants were independently correlated with serum ALT and AST (Fig. 2, lower).
Table 2.
Indicators of hepatocellular injury and urinary thiobarbituric-acid-reacting substances (TBARS) in preterm infants studied
| Infants | |||
| Variable | With cholestatis (n = 27) | Controls (n = 16) | P |
| Direct bilirubin (mg/dl) | 3.3 (2.4,7.2)* | 1.7 (1.0,1.9) | <0.001 |
| ALT (U/l) | 32 (8,127)* | 9 (7,16) | 0.01 |
| AST (U/l) | 71 (40,189)* | 33 (22,39) | <0.001 |
| Alkaline phosphatase (U/l) | 383 (221,579) | 269 (199,450) | 0.57 |
| Urinary TBARS (ng/mg creatinine) | 2591 (1022,6445) | 3368 (1622,4625) | 0.93 |
Values are expressed as medians (25th,75th percentiles). *Significantly different from control group (P < 0.05 using one-way analysis of variance of natural log-transformed variables).
Figure 2.
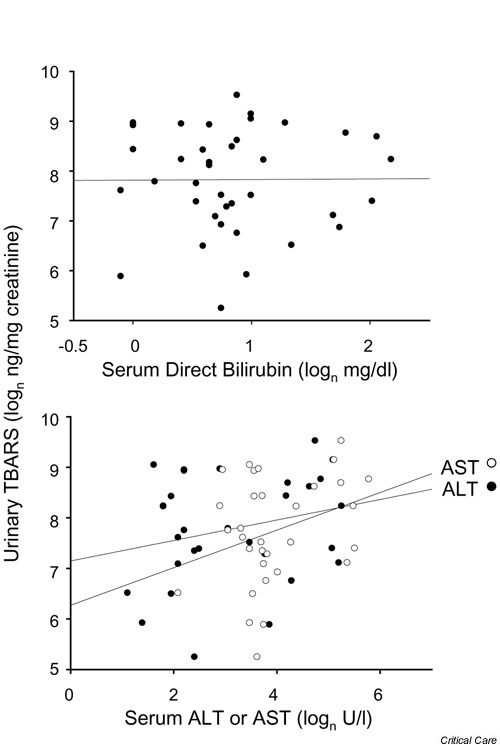
Correlation of serum direct bilirubin and transaminase levels with urinary TBARS. Urine and serum specimens were obtained from cholestatic (n = 27) and control (n = 16) infants, as described in Materials and methods. Urinary TBARS levels of infants (n = 43) were plotted against (top) serum direct bilirubin (NS; analysis done after exclusion of three outlying points) or (bottom) ALT (r = 0.43, P = 0.04) or AST (r = 0.44, P = 0.02). Natural logarithm transformations of variables are used, and regression lines are displayed. ALT, alanine transaminase; AST, aspartate transaminase; TBARS, thiobarbituric-acid-reacting substances.
Discussion
We found that elevated liver transaminases are associated with increased oxidative stress. These findings suggest that oxidant stress (as indicated by elevated TBARS) is associated with hepatocellular injury in preterm infants. Although there is ample evidence that oxidant stress follows cholestasis, our findings suggest that oxidative injury in the liver may be induced by mechanisms that are independent of cholestasis [7,8,9,10,11]. For example, the production of ROIs in the liver may be linked to inflammation, which has emerged as a primary mechanism of liver injury after pathophysiological insults. Activated Kupffer cells and neutrophils release ROIs and proteases in response to inflammatory cytokines in the liver [21]. ROIs in excess inactivate proteins, disrupt DNA, and oxidize lipids [22]. Preterm newborn infants may be particularly susceptible to such injury because they exhibit an imbalance between antioxidant- and oxidant-generating systems. For example, such infants exhibit decreased levels in the liver of superoxide dismutase, vitamin E, and β-carotene [23,24,25]. Antioxidant capacity in preterm infants is also compromised by relative deficiencies of selenium and taurine, as well as reduced ability to synthesize sufficient glutathione [26].
Despite longer TPN courses and elevated serum transaminases, infants with cholestasis or elevated serum direct bilirubin did not display elevated urinary TBARS. Our findings are consistent with previous reports indicating an inconsistent relation between bilirubin levels and the degree of histologic liver injury [27,28]. This inconsistency suggests that cholestasis and hepatic impairment in preterm infants receiving TPN are induced by mechanisms that are not dependent on oxidant-mediated hepatocellular injury. For example, lack of enteral feeding plays an important role in the development of cholestasis in the neonatal period, possibly mediated by direct hepatotoxic activity of bile salts [5]. Bile salts are thought to exert adverse effects on signal transduction and gene transcription in hepatocytes and cholangiocytes and to activate Fas-induced apoptosis [21]. TPN-induced cholestasis in infants also appears to be related directly to developmental immaturity of bile flow and production and to possible infection [29].
TBARS measurements provide a measure of membrane lipid peroxidation and, as such, may provide a direct assessment of the progression of liver injury at the cellular level. Although there was a statistically significant relationship between urinary TBARS and liver transaminases, a large degree of overlap existed between the groups. Furthermore, the association between TBARS levels and liver injury does not necessarily indicate causality. Despite physiologic evidence that ROIs play a central role in tissue injury during inflammation, it is possible that elevated TBARS occur secondary to other mechanisms of hepatocellular injury [30,31]. Larger longitudinal and/or interventional (e.g. antioxidant administration) studies will be necessary to determine whether there is a causal relationship between lipid peroxidation and TPN-induced liver disease in preterm infants.
Key messages
• Serum transaminases are elevated in preterm infants (<35 weeks' gestational age) with cholestasis and prolonged administration of total parenteral nutrition (TPN), indicating that cholestasis is associated with hepatocellular injury
• Urinary levels of thiobarbituric-acid-reacting substances (TBARS), which are proportional to lipid peroxidation and oxidant stress, are correlated with serum transaminase levels. Our findings suggest that oxidant stress is associated with hepatocellular injury in preterm infants
• Urinary levels of TBARS do not differ significantly between infants with cholestasis and control infants
• Our findings suggest that cholestasis in preterm infants receiving TPN is induced, in large part, by mechanisms that are not dependent on oxidant-mediated hepatocellular injury
Competing interests
None declared.
Abbreviations
ALT = alanine transaminase; AST = aspartate transaminase; MDA = malondialdehyde; ROIs = reactive oxygen intermediates; TBA = thiobarbituric acid; TBARS = thiobarbituric-acid-reacting substances; TPN = total parenteral nutrition.
References
- Bell RL, Ferry GD, Smith EO, Shulman RJ, Christensen BL, Labarthe DR, Wills CA. Total parenteral nutrition-related cholestasis in infants. J Parenter Enteral Nutr. 1986;10:356–359. doi: 10.1177/0148607186010004356. [DOI] [PubMed] [Google Scholar]
- Caniano DA, Starr J, Ginn-Pease ME. Extensive short-bowel syndrome in neonates: outcome in the 1980's. Surgery. 1989;105:119–124. [PubMed] [Google Scholar]
- Dahms BB, Halpin TC. Serial liver biopsies in parenteral nutrition-associated cholestasis in early infancy. Gastroenterology. 1981;81:136–144. [PubMed] [Google Scholar]
- Vileises RA, Inwood RJ, Hunt CE. Prospective controlled study of parenteral nutrition-associated cholestatic jaundice: effect of protein intake. J Pediatr. 1980;96:893. doi: 10.1016/s0022-3476(80)80573-7. [DOI] [PubMed] [Google Scholar]
- Brown MR, Thunberg BJ, Golub L, Maniscalco WM, Cox C, Shapiro DL. Decreased cholestasis with enteral instead of intravenous protein in the very low-birth weight infant. J Ped Gastroenerol Nutr. 1989;9:21–27. [PubMed] [Google Scholar]
- Manginello FP, Javitt NB. Parenteral nutrition and neonatal cholestasis. J Pediatr. 1979;94:296–298. doi: 10.1016/s0022-3476(79)80849-5. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Winklhofer-Roob BM, Devereaux MW, McKim JM., Jr Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gas-troenterology. 1995;109:1249–1256. doi: 10.1016/0016-5085(95)90585-5. [DOI] [PubMed] [Google Scholar]
- Parola M, Leonarduzzi G, Robino G, Albano E, Poli G, Dianzani MU. On the role of lipid peroxidation in the pathogenesis of liver damage induced by long-standing cholestasis. Free Rad Biol Med. 1996;20:351–359. doi: 10.1016/0891-5849(96)02055-2. [DOI] [PubMed] [Google Scholar]
- Orellana M, Rodrigo R, Thielemann L, Guajardo V. Bile duct ligation and oxidative stress in the rat: effects in liver and kidney. Comp Biochem Physiol Part C. 2000;126:105–111. doi: 10.1016/S0742-8413(00)00102-X. [DOI] [PubMed] [Google Scholar]
- Krahenbuhl S, Talos C, Lauterburg BH, Reichen J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology. 1995;22:607–612. doi: 10.1002/hep.1840220234. [DOI] [PubMed] [Google Scholar]
- Tsai LY, Lee KT, Tai SM, Lee SC, Yu HS. Changes of lipid peroxide levels in blood and liver tissue of patients with obstructive jaundice. Clin Chim Acta. 1993;215:41–50. doi: 10.1016/0009-8981(93)90247-2. [DOI] [PubMed] [Google Scholar]
- Bjorneboe A, Bjorneboe GE. Antioxidant status and alcohol-related diseases. Alcohol Alcohol. 1993;28:111–116. [PubMed] [Google Scholar]
- Sokol RJ, Twedt D, McKim JM, Jr, Devereaux MW, Karrer FM, Kam I, von Steigman G, Narkewicz MR, Bacon BR, Britton RS. Oxidant injury to hepatic mitochondria in patients with Wilson's disease and Bedlington terriers with copper toxicosis. Gastroenterology. 1994;107:1788–1798. doi: 10.1016/0016-5085(94)90822-2. [DOI] [PubMed] [Google Scholar]
- Davit-Spraul A, Cosson C, Couturier M, Hadchouel M, Legrand A, Lemonnier F, Therond P. Standard treatment of α-tocopherol in Alagille patients with severe cholestasis is insufficient. Pediatr Res. 2001;49:232–236. doi: 10.1203/00006450-200102000-00017. [DOI] [PubMed] [Google Scholar]
- Fabris C, Pirisi M, Panozzo MP, Soardo G, Toniutto P, Hocza V, Bartoli E. Intensity of inflammatory damage and serum lipid peroxide concentrations in liver disease. J Clin Path. 1993;46:364–367. doi: 10.1136/jcp.46.4.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola M, Pinzani M, Casini A, Albano E, Poli G, Gentilini A, Gen-tilini P, Dianzani MU. Stimulation of lipid peroxidation or 4-hydroxynonenal treatment increases procollagen α1(I) gene expression in human liver fat-storing cells. Biochem Biophys Res Commun. 1993;194:1044–1050. doi: 10.1006/bbrc.1993.1927. [DOI] [PubMed] [Google Scholar]
- Berger HM, Den Ouden AL, Calame JJ. Pathogenesis of liver damage during parenteral nutrition: is lipofuscin a clue? Arch Dis Child. 1985;60:774–776. doi: 10.1136/adc.60.8.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- Valenzuela A. The biological significance of malondialdehyde determination in the assessment of tissue oxidative stress. Life Sci. 1991;48:301–309. doi: 10.1016/0024-3205(91)90550-u. [DOI] [PubMed] [Google Scholar]
- Coulthard MG, Hey EN, Ruddock V. Creatinine and urea clearances compared to inulin clearance in preterm and mature babies. Early Hum Dev. 1985;11:11–19. doi: 10.1016/0378-3782(85)90114-8. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- Davis KJA. Oxidative damage and repair: introduction and overview. In: Davis KJA, editor. In Oxidative Damage and Repair: Chemical, Biological, and Medical Aspects. Elmsford, NY: Perga-mon Press; 1991. pp. xvii–xxvii. [Google Scholar]
- McElroy MC, Postle AD, Kelly KG. Catalase, superoxide dismu-tase and glutathione peroxidase activities of lung and liver during human development. Biochim Biophys Acta. 1992;1117:153–158. doi: 10.1016/0304-4165(92)90073-4. [DOI] [PubMed] [Google Scholar]
- Lindeman JH, van Zoeren-Grobben D, Schrijver J, Speek AJ, Poorthuis BJ, Berger HM. The total free radical trapping ability of cord blood plasma in preterm and term babies. Pediatr Res. 1989;26:20–24. doi: 10.1203/00006450-198907000-00008. [DOI] [PubMed] [Google Scholar]
- Sullivan JL, Newton RB. Serum antioxidant activity in neonates. Arch Dis Child. 1988;l63:748–757. doi: 10.1136/adc.63.7_spec_no.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubman TR, Halliday HL, McMaster D. Glutathione peroxidase and selenium levels in the preterm infant. Biol Neonate. 1990;58:305–310. doi: 10.1159/000243284. [DOI] [PubMed] [Google Scholar]
- Moss RL, Das JB, Raffensberger JG. Total parenteral nutrition-associated cholestasis: clinical and histopathologic correlation. J Pediatr Surg. 1993;28:1270–1274. doi: 10.1016/s0022-3468(05)80311-2. [DOI] [PubMed] [Google Scholar]
- Beath SV, Davies P, Papadopoulou A, Khan AR, Buick RG, Corkery JJ, Gornall P, Booth IW. Parenteral nutrition-related cholestasis in postsurgical neonates: multivariate analysis of risk factors. J Pediatr Surg. 1996;31:604–606. doi: 10.1016/s0022-3468(96)90507-2. [DOI] [PubMed] [Google Scholar]
- Shulman RJ. New developments in total parenteral nutrition for children. Curr Gastroenterol Rep. 2000;2:253–258. doi: 10.1007/s11894-000-0069-x. [DOI] [PubMed] [Google Scholar]
- Alric L, Orfila C, Carrere N, Beraud M, Carrera G, Lepert JC, Duffaut M, Pipy B, Vinel JP. Reactive oxygen intermediates and eicosanoid production by Kupffer cells and infiltrated macrophages in acute and chronic liver injury induced in rats by CCl4. Inflamm Res. 2000;49:700–707. doi: 10.1007/s000110050649. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Pendino KJ. Macrophages and inflammatory mediators in tissue injury. Annu Rev Pharmacol Toxicol. 1995;35:655–677. doi: 10.1146/annurev.pharmtox.35.1.655. [DOI] [PubMed] [Google Scholar]