

Abstract
Myocardial dysfunction frequently accompanies severe sepsis and septic shock. Whereas myocardial depression was previously considered a preterminal event, it is now clear that cardiac dysfunction as evidenced by biventricular dilatation and reduced ejection fraction is present in most patients with severe sepsis and septic shock. Myocardial depression exists despite a fluid resuscitation-dependent hyperdynamic state that typically persists in septic shock patients until death or recovery. Cardiac function usually recovers within 7–10 days in survivors. Myocardial dysfunction does not appear to be due to myocardial hypoperfusion but due to circulating depressant factors, including the cytokines tumor necrosis factor alpha and IL-1β. At a cellular level, reduced myocardial contractility seems to be induced by both nitric oxide-dependent and nitric oxide-independent mechanisms. The present paper reviews both the clinical manifestations and the molecular/cellular mechanisms of sepsis-induced myocardial depression.
Keywords: contractility, cytokine, heart, myocardial depression, nitric oxide
Introduction
Sepsis and septic shock have been recognized as an increasingly serious clinical problem, accounting for substantial morbidity and mortality. The past four decades have seen the age-adjusted mortality of sepsis increase from 0.5 to 7 per 100,000 episodes despite major advances in the understanding of its pathophysiology [1]. The incidence of severe sepsis in the United States today is estimated at 750,000 cases per year, resulting in 215,000 deaths annually [2]. The majority of these sepsis patients die of refractory hypotension and of cardiovascular collapse.
Sepsis has been defined as the systemic inflammatory response to infection [3]. An infectious stimulus (e.g. endo-toxin or another microbiologic element) induces the release of local and systemic inflammatory mediators, especially tumor necrosis factor alpha (TNF-α) and IL-1β, from monocytes/macrophages and other cells [4]. These cytokines stimulate polymorphonuclear leukocytes, macrophages and endothelial cells to release a number of downstream inflammatory mediators, including platelet activating factor and nitric oxide (NO), further amplifying the inflammatory response. Several anti-inflammatory mediators are also released as part of this amplification cascade; namely, IL-10, transforming growth factor beta and IL-1 receptor antagonist. The relative contribution of these cytokines will determine the severity of the septic episode. If the inflammatory reaction is particularly intense, homeostasis of the cardiovascular system will be disrupted, leading to septic shock. One of the manifestations of cardiovascular dysfunction in septic shock is myocardial depression.
The present article reviews the clinical manifestations of cardiac dysfunction in sepsis, from the point of view of both the right and left ventricle, as well as cardiovascular prognostic factors in sepsis and septic shock. We will also review the potential pathophysiologic processes responsible for myocardial depression in sepsis, from the perspective of organ physiology and molecular biology.
Clinical manifestations of cardiovascular dysfunction
Historical perspectives
Our understanding of the cardiovascular manifestations of septic shock has evolved over the years, as new techniques to assess cardiovascular performance have become available. Before the introduction of the pulmonary artery catheter (PAC), two distinct cardiovascular clinical presentations of septic shock were described: a high cardiac output (CO) state, associated with warm, dry skin and a bounding pulse despite hypotension (warm shock); and a low CO state, associated with hypotension, cold, clammy skin and a thready pulse (cold shock) [5]. Clowes et al. [6], in a 1966 study, described these two clinical pictures as different stages of septic shock: patients were believed to initially experience a hyperdynamic phase (warm shock), and then to either recover or progress to hypodynamic shock (cold shock) and death. This view was reinforced by other clinical studies [5,7] that correlated survival with high cardiac index (CI). Only a few studies hinted at the relationship between volume status, the CI and outcome [8,9]. All these studies that supported the concept of terminal cold shock suffered from the fact that they used central venous pressure (CVP) as the best available estimate of left ventricular end-diastolic volume and adequacy of resuscitation. Evidence accumulated over the past 40 years shows that CVP, as a reflection of right ventricular preload, is a poor estimate of left ventricular preload in critically ill patients, and particularly in sepsis [10].
The introduction of the PAC (which could measure pulmonary artery wedge pressure as a more accurate estimate of left ventricular preload) has allowed for better definition of the cardiovascular dysfunction in septic shock and has improved volume resuscitation. Several studies have shown that adequately resuscitated septic shock patients consistently manifest a hyperdynamic circulatory state with high CO and low systemic vascular resistance (SVR) [11,12]. In contrast to previous belief, this hyperdynamic state usually persists until death in nonsurvivors (Fig. 1) [13,14]. Despite the strong evidence characterizing sepsis as a hyperdynamic state, studies that examined myocardial performance still showed left ventricular dysfunction (illustrated by decreased left ventricular stroke work index) in properly resuscitated septic patients [15]. The depression in the Frank–Starling curve demonstrated in these studies, however, could be explained by either a change in myocardial contractility or compliance.
Figure 1.
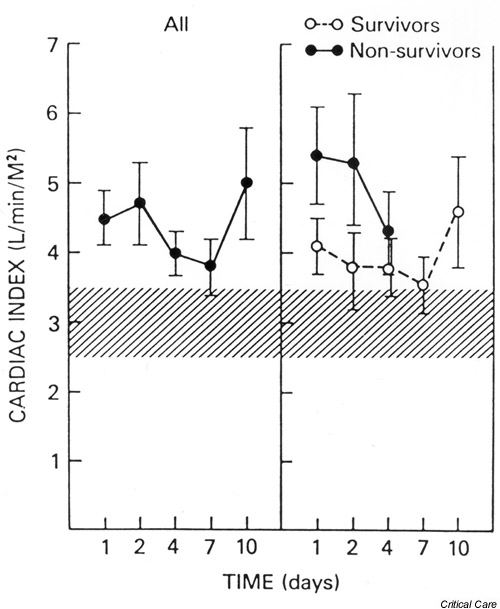
The mean (± SEM) cardiac index plotted against time for all patients, survivors, and nonsurvivors. The hatched areas show the normal range. All groups maintained an elevated cardiac index throughout the study period. The difference between the survivors and nonsurvivors was not statistically significant. Reproduced with permission from [16].
The development of portable radionuclide cineangiography and its application to critically ill patients has further improved our understanding of cardiovascular dysfunction in septic shock, by allowing differentiation between impaired contractility and impaired compliance.
Left ventricular function
Parker et al. [16] showed, using radionuclide cineangiography, that survivors of septic shock demonstrated a decreased left ventricular ejection fraction (LVEF) and an acutely dilated left ventricle, as evidenced by an increased left ventricular end-diastolic volume index (LVEDVI) (Fig. 2). These parameters returned to normal over 7–10 days in survivors. Nonsurvivors maintained normal LVEF and LVEDVI throughout the course of their illness until death. All patients in this study [16] had normal or elevated CI and low SVR, as measured by the PAC.
Figure 2.
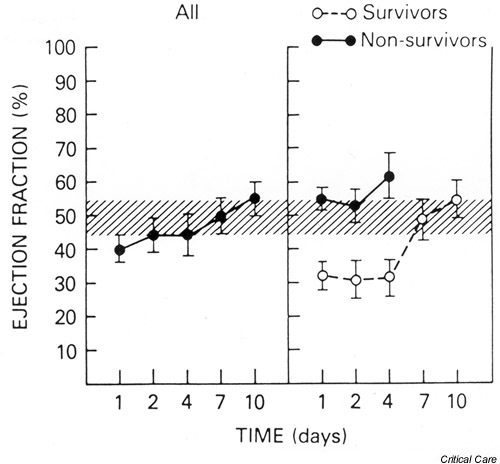
The mean (± SEM) left ventricular ejection fraction (LVEF) plotted versus time for all patients, survivors, and nonsurvivors. Overall, septic shock patients showed a decreased LVEF at the time of initial assessment. This effect was due to marked early depression of LVEF among survivors that persisted for up to 4 days and returned to normal within 7–10 days. Nonsurvivors maintained LVEF in the normal range. The hatched area represents the normal range. Reproduced with permission from [16].
In 1988, Ognibene et al. compared left ventricular performance curves (plotting left ventricular stroke work index versus LVEDVI) of septic and nonseptic critically ill patients (Fig. 3). They showed a flattening of the curve in septic shock patients, with significantly smaller left ventricular stroke work index increments in response to similar LVEDVI increments when compared with nonseptic critically ill controls [17]. Subsequent studies have confirmed the presence of significant left ventricular systolic dysfunction in septic patients [18,19].
Figure 3.
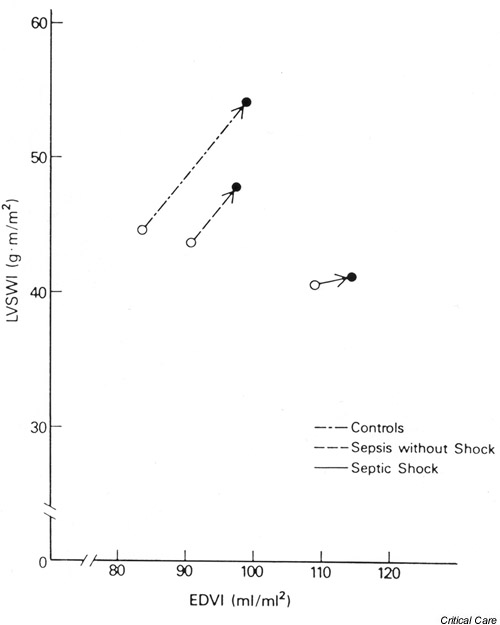
Frank–Starling ventricular performance relationship for each of the three patient groups. Data points plotted represent the mean prevolume and postvolume infusion values of end-diastolic volume index (EDVI) and left ventricular stroke work index (LVSWI) for each patient group. Control patients showed a normal increase of EDVI and LVSWI in response to volume infusion. The absolute increases of EDVI and LVSWI in patients with sepsis without shock were less than those of control subjects, but the slope of the curve is similar to control patients. Patients with septic shock had a greatly diminished response and showed a marked rightward and downward shift of the Frank–Starling relationship. Reproduced with permission from [17].
Left ventricular diastolic function in septic shock is not as clearly defined. The dilatation of the left ventricle [16] and the lack of discordance between pulmonary artery wedge pressure (PAWP) and left ventricular end-diastolic volume [17] both argue against significant diastolic dysfunction in sepsis. More recent studies using echocardiography, however, have demonstrated slower left ventricular filling [20] and aberrant left ventricular relaxation [21,22] in septic patients, suggesting that impaired compliance may significantly contribute to myocardial depression in sepsis.
Right ventricular function
Low peripheral vascular resistance in sepsis leads to decreased left ventricular afterload. However, the right ventricular afterload is frequently elevated due to increased pulmonary vascular resistance from acute lung injury [23]. These different physiologic conditions mean that the right ventricle cannot be expected to behave like the left ventricle in septic patients. Multiple studies have therefore specifically examined right ventricular function in sepsis.
A number of studies have documented right ventricular systolic dysfunction in volume-resuscitated septic patients, as evidenced by decreased right ventricular ejection fraction (RVEF) and right ventricular dilation [24,25,26,27]. Kimchi et al. [24] and Parker et al. [26] also showed that right ventricular dysfunction occurred independently of pulmonary vascular resistance and pulmonary artery pressure, suggesting that increased right ventricular afterload could not be the dominant cause of right ventricular depression in septic shock. Parker et al. [26] further demonstrated a close temporal parallel between right ventricular and left ventricular dysfunction in sepsis. In their study, survivors experienced significant right ventricular dilation and decreased RVEF and right ventricular stroke work index, all of which returned to normal within 7–14 days (Fig. 4). Nonsurvivors had moderate right ventricular dilation and a marginally decreased RVEF, neither of which improved throughout their illness.
Figure 4.
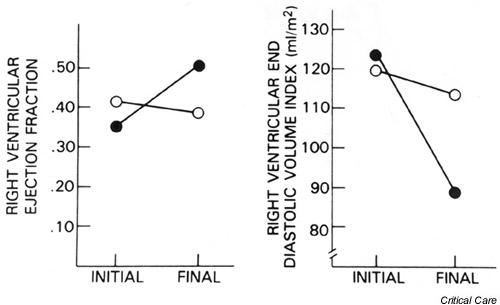
Serial changes in right ventricular ejection fraction and end-diastolic volume index during septic shock in humans. (a) Mean initial and final right ventricular ejection fractions for survivors (closed circles, P < 0.001) and nonsurvivors (open circles, P < 0.001). (b) Mean initial and final right ventricular end-diastolic volume index for survivors (closed circles, P < 0.05) and nonsurvivors (open circles, P = not significant). The right ventricle, similar to the left ventricle, undergoes dilation with a drop in ejection fraction with the acute onset of septic shock. In 7–10 days, right ventricular dilation and decreased ejection fraction revert to normal in survivors. Data from [26]; adapted with permission [69].
There is also evidence of right ventricular diastolic dysfunction in septic patients. Kimchi et al. [24] noticed a lack of correlation between right atrial pressure and right ventricular end-diastolic volume, suggesting altered right ventricular compliance. Schneider et al. [25] similarly identified a subgroup of patients who failed to exhibit an increased right ventricular end-diastolic volume index in response to volume loading, despite a rise in CVP. However, the relative contribution of systolic and diastolic dysfunction to right ventricular depression in sepsis remains largely unknown.
Cardiovascular prognostic factors in septic shock
Early studies of cardiovascular dysfunction in septic shock suggested that a low or decreasing CI invariably carried a poor prognosis [5,6,7,8]. As previously discussed, these studies relied on CVP measurements to assess volume status. It is now known that CVP is a poor reflection of left ventricular preload in critical illness and cannot accurately determine adequacy of resuscitation. Introduction of the PAC showed that adequately volume-resuscitated septic shock patients (as measured by PAWP) predictably exhibited a high CI and low SVR, including nonsurvivors [11,12]. CI is therefore not a reliable predictor of mortality in sepsis.
Recognition of the significant post-resuscitation peripheral vasodilation (low SVR) in septic shock led to the theory that peripheral vascular failure could be a major determinant of mortality in septic shock. Baumgartner et al. [28] noticed that patients with an extremely high CI (>7.0 l/min/m2) and low SVR had a uniformly poor outcome. Groeneveld et al. [29] retrospectively examined data from septic shock patients. They found that, for equivalent CI, nonsurvivors had a lower SVR than survivors. They concluded that peripheral vascular resistance was closely linked to outcome in septic shock.
Parker et al. [13] reviewed hemodynamic data from septic shock patients on presentation and at 24 hours to identify prognostic value. They found that, on presentation, only a heart rate <106 beats/min suggested a favorable outcome. At 24 hours, a heart rate <95 beats/min, a SVR index >1529 dynes s cm5/m2, a decrease in heart rate >18 beats/min and a decrease in CI >0.5 l/min/m2 all predicted survival. In a subsequent study [14], the same authors confirmed previous findings of decreased LVEF and increased LVEDVI in survivors of septic shock but not in non-survivors. However, although nonsurvivors as a whole did not exhibit left ventricular dilation, they could be divided into two groups: the first with decreasing LVEDVI and decreasing CI on serial measurements, and the other group with progressively increasing LVEDVI and maintained CI.
There are three hemodynamic patterns of death in septic shock [13]. Early deaths are due either to distributive shock (low SVR and refractory hypotension despite preserved CI) or to a cardiogenic form of septic shock (decreased CI). Late deaths are due to multisystem organ failure. Correlating this with Parker et al.'s [14] findings, it is possible that nonsurvivors who are unable to dilate their left ventricle (decreasing LVEDVI and CI) succumb to the cardiogenic form of septic shock. Those who have increasing LVEDVI and preserved CI die of the classic distributive shock.
The value of right ventricular performance parameters in predicting outcome is less clear. Multiple studies [24,25,26,27] have shown that, in sepsis, the right ventricle behaves similar to the left ventricle, exhibiting acute dilation and decreased RVEF. Persistence of right ventricular systolic dysfunction is associated with poor outcome [26,27]. Opinions diverge, however, on whether initial right ventricular dilation and decreased RVEF portend poor prognosis. Vincent et al. [27] suggested that survival correlates with higher initial RVEF, whereas Parker et al. [26] observed lower RVEF in survivors of septic shock than in nonsurvivors. The question requires further study.
Patients with sepsis and septic shock can show resistance to the vasopressor and inotropic effects of catecholamines. Nonsurvivors of septic shock have been shown to have an attenuated inotropic response to a dobutamine stress test compared with survivors [30]. Conversely, increased SVI, increased mixed venous oxygen saturation, ventricular dilation and drop in diastolic blood pressure in response to a dobutamine stress test all predict survival in septic patients [31].
Etiology of myocardial depression in sepsis and septic shock
Myocardial hypoperfusion
The possibility of myocardial dysfunction in sepsis was originally proposed and described in the 1960s. Its etiology, however, remained a mystery. For many years, the leading theory was that sepsis was associated with a globally decreased myocardial perfusion, leading to ischemic injury and myocardial depression. Two studies disproved that view. Cunnion et al. [32] performed serial measurements of coronary blood flow and metabolism using thermodilution coronary sinus catheters in septic patients (Fig. 5). They found normal or elevated coronary blood flow in septic patients compared with normal controls with comparable heart rates, and found no difference in blood flow between patients who developed myocardial dysfunction and those who did not. There was no net myocardial lactate production. Dhainaut et al. [33], using the same technique, confirmed these findings.
Figure 5.
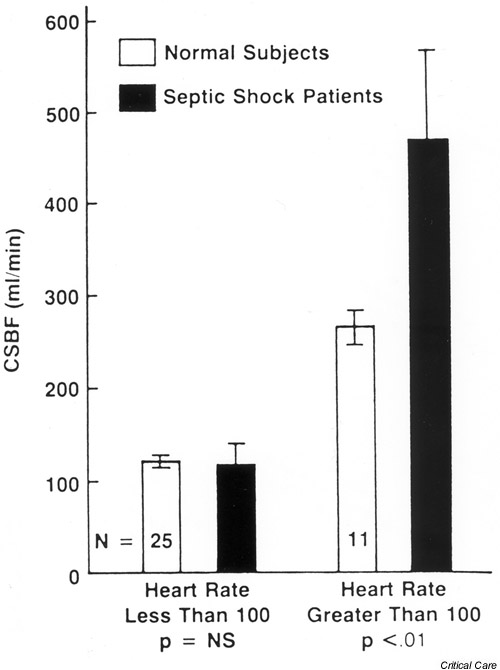
Mean coronary sinus blood flow (CSBF) in seven patients with septic shock compared with normal subjects. Flow measurements were stratified into heart rates above and below 100 beats/min. Coronary blood flow in septic shock patients equaled (heart rate <100 beats/min) or exceeded (heart rate >100 beats/min) coronary blood flow in control patients. Reproduced with permission from [32].
Furthermore, studies on animal models of sepsis [34] demonstrated that myocardial oxygen metabolism and high-energy phosphates were preserved in septic shock, neither of which is compatible with myocardial ischemia. However, Turner et al. [35] recently measured increased troponin I levels in patients with septic shock, demonstrating some degree of myocardial cell injury in the course of septic shock. It remains unclear from their study whether direct cardiac injury plays a role in sepsis-induced myocardial dysfunction or is the result of other factors, including a myocardial depressant substance (MDS) or exogenous catecholamine administration.
Circulatory depressant substances
Wiggers' landmark report [36] in 1947 postulating the presence of a circulating myocardial depressant factor in hemorrhagic shock provided the basis for the accepted current theory of myocardial dysfunction in septic shock. The presence of a myocardial depressant factor in sepsis was later confirmed experimentally by Lefer [37] in the late 60 s. Clinical studies performed at the same time associated death from septic shock with a hypodynamic circulatory state marked by a decreased CO [6,7,8]. Although these earlier studies depended on the measurement of CVP to assess preload, a factor now recognized as unreliable in the critically ill, Lefer's early contributions furthered research into MDS in sepsis [37].
The first study to actually show a link between septic shock-associated myocardial depression in humans with the myocyte depressant effects of a patient's own septic serum was carried out by Parrillo et al. in 1985 [38]. Serum from patients with septic shock generated a significant concentration-dependent depression of in vitro myocyte contractility (Fig. 6). The investigators were also able to demonstrate a strong correlation between the timing and degree of septic shock-associated decrease in LVEF in vivo and cardiac myocyte depression in vitro induced by exposure to serum from the same patients.
Figure 6.
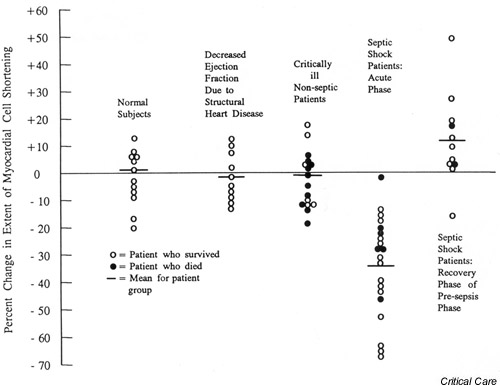
The effect of serum from septic shock patients and control groups on the extent of myocardial cell shortening of spontaneously beating rat heart cells in vitro. Septic shock patients during the acute phase demonstrated a statistically significant lower extent of shortening (P < 0.001) compared with any other group. Reproduced with permission from [38].
Subsequent work focused on identifying the presence of a MDS. In a study of 34 patients with septic shock [39], researchers noted high levels of myocardial depressant activity that correlated with higher peak serum lactate, with increased ventricular filling pressures and with increased LVEDVI. In addition, a trend towards higher mortality was present in patients with high levels of myocardial depressant activity compared with patients with lower or absent activity (36% versus 10%) [39]. These studies would argue in favor of a circulating substance rather than hypoperfusion as the causative factor in septic shock-associated myocardial depression.
Myocardial depression in sepsis: cytokines
Although the existence of MDS was demonstrated by the previous studies [36,37,38,39], the identity of the molecules remained in question. Potential circulating inflammatory mediators that could cause septic myocardial depression include the prostaglandin group, leukotrienes, platelet activating factor, histamine and endorphins. However, the substance was found to be heat labile, soluble in water, and its activity in filtration studies was present in the >10 kDa fraction [39]. Although full molecular characterization was not possible with the available data, the characteristics were most consistent with either a polypeptide or protein. The list of potential cytokine mediators of myocardial depression is exhaustive; however, TNF-α and IL-1β play a central role and deserve further consideration.
TNF-α shares a similar biochemical profile with MDS, making it a plausible mediator of the myocardial effects of sepsis and septic shock [38,40,41]. Experimentally, increased levels of TNF-α produce fever, lactic acidosis, disseminated intravascular coagulation, acute lung injury and death. The cardiovascular effects are similar to clinical septic shock; namely, hypotension, increased CO and low SVR [42,43]. Human volunteers given TNF-α infusions demonstrate similar responses [44,45].
A number of studies have shown that, when TNF-α is administered to human and animal myocardial tissue in vitro or ex vivo, the result is a concentration-dependent depression of contractility [46,47,48,49]. Furthermore, removal of TNF-α from the serum of patients with septic shock partially eliminates its myocardial depressant effect [47]. Although larger phase III clinical trials have shown no overall survival benefit when anti-TNF-α monoclonal antibody has been administered to patients with septic shock, left ventricular function did improve in this patient group [50].
IL-1β induces similar pathophysiologic responses to TNF-α. IL-1β is increased in both human and animal models of sepsis and septic shock [51]. When exposed to IL-1β, in vitro as well as ex vivo myocardial contractility is depressed [49,52,53]. Immunoabsorption of IL-1β partially neutralizes cardiac myocyte depressant activity of human septic serum [47]. Administration of IL-1β antagonist attenuates the hemodynamic and metabolic manifestations of septic shock [54,55]. As with TNF-α, overall mortality has not been improved in randomized trials with IL-1β [54,55]
A number of studies have postulated that cytokine synergy plays a key role in septic myocardial depression. When considering IL-1β and TNF-α independently, substantially higher concentrations of each cytokine are required to depress contractility in rat cardiac myocytes than when these two cytokines are combined [47]. These findings have also been validated in ex vivo studies of isolated human atrial trabeculae [56]. The combination of TNF-α and IL-1β may cause myocyte depression at concentrations 50–100 times lower than would be required if applied individually. Such concentrations are well within those found in blood during human septic shock. Available data supports a causative role for TNF-α and IL-1β acting synergistically in septic myocardial depression.
Cellular mechanisms of septic myocardial depression
Depression of myocardial contractility by TNF-α, IL-1β and septic serum in vitro occurs in two distinct time frames. The early phase of cardiac myocyte depression occurs within minutes of exposure either to TNF-α, to IL-1β, to TNF-α and IL-1β given together or to septic serum [47,48]. In vivo canine studies also demonstrate the ability of TNF-α to induce rapid myocardial depression [43,57]. Furthermore, there is a convincing relationship between the degree of in vitro early cardiac myocyte depression produced by human septic serum and the decrease in LVEF from those same patients during acute septic shock [38]. Other studies also document a delayed depressant effect of TNF-α, IL-1β and supernatants of activated macrophages on in vitro myocardial tissue [52,53,57,58]. This effect begins hours after exposure and may persist for days. This late phase of myocardial depression appears to occur by a distinctly different biochemical pathway than the early depression, and may involve de novo protein synthesis.
The generation of NO may be central to both early and late depression of myocytes in in vitro and in vivo models of septic shock. The role of NO in cardiac contractility is still under study. NO is produced by the conversion of L-arginine to L-citrulline by the nitric oxide synthase, which exists in inducible nitric oxide synthase (iNOS) and constitutive nitric oxide synthase (cNOS) forms. Studies support the role of NO generated by cNOS in the physiologic regulation of cardiac contractility [59,60,61]. In vitro studies of cardiac myocytes superperfused with either NO, nitroprusside (NO donor) or the NO donor SIN-1 demonstrate reductions in myocardial contractility [62]. In human studies, nitroprusside infusion into coronary arteries depresses intraventricular pressure while improving diastolic relaxation and distensibility [63].
In sepsis and septic shock, the pathophysiologic production of NO may contribute to cardiovascular dysfunction. The initial process is thought to occur from sequential NO and cGMP generation via cNOS activation rather than by de novo synthesis of iNOS. This is consistent with the short time frame to the onset of myocardial depression [48,56,64]. These studies cannot, however, account for the late-onset of cytokine-driven myocardial depression discussed earlier. Further studies have shown, in in vitro myocardial preparations, that the generation of iNOS, NO and cGMP may be responsible for late onset myocardial depression [48,53,65,66].
Studies thus suggest that NO-mediated depression of myocardial contractility appears in two separate time frames by two distinct pathways. Prior to Kinugawa et al.'s study [67], it might have been thought these mechanisms were mutually exclusive. Kinugawa et al. demonstrated, using an avian cardiac myocyte model, a biphasic response to IL-6 administration. IL-6 given in high concentration produced both early (<30 min) and late (24 hours) cardiac myocyte depression, associated with a decrease in intracellular calcium. Early depression could be blocked by pretreatment with a calcium chelator, implicating that the calcium/calmod-ulin-dependent cNOS is involved. However, late depression could not be blocked by the chelator, supporting the role for the calcium/calmodulin-independent iNOS.
The study of Kinugawa et al. clearly suggests that cytokines (or sepsis) can stimulate the heart to sequentially produce NO by both cNOS and iNOS. The data would suggest that TNF-α, IL-1β and possibly other cytokines involved in septic shock could mediate their cardiodepressant effects by at least two distinct mechanisms. Early cardiodepressant activity may involve both a NO-dependent but β-adrenoreceptor-independent mechanism and a NO-independent defect of β-adrenoreceptor signal transduction [68]. Late depression involves an iNOS-dependent defect of β1-adrenergic signal transduction with further addition of an undefined defect of myocardial contractility. While NO generation may contribute to the cardiodepressant effects seen in sepsis, its precise role and the role of other mechanisms underlying septic myocardial dysfunction continue to be defined.
Conclusion
Myocardial dysfunction is an important variable in sepsis and septic shock. The impairment in cardiac function results from a combination of systolic and diastolic dysfunction. The presence of a circulating MDS, which probably represents different cytokines, including TNF-α and IL-1β, acting synergistically is clear. The myocardial depressant effect of these cytokines is at least partly mediated through mechanisms of NO generation. While the current standard of therapy in patients with septic shock is still directed towards re-establishing organ and tissue perfusion and oxygen delivery by fluid resuscitation and inotropic support, ongoing research aimed at understanding the cellular mechanisms of cardiac dysfunction may lead to exciting new therapeutic options.
Competing interests
None declared.
Abbreviations
CI = cardiac index; cNOS = constitutive nitric oxide synthase; CO = cardiac output; CVP = central venous pressure; IL = interleukin; iNOS = inducible nitric oxide synthase; LVEDVI = left ventricular end-diastolic volume index; LVEF = left ventricular ejection fraction; MDS = myocardial depressant substance; NO = nitric oxide; PAC = pulmonary artery catheter; PAWP = pulmonary artery wedge pressure; RVEF = right ventricular ejection fraction; SVR = systemic vascular resistance; TNF-α = tumor necrosis factor alpha.
References
- Center for Diseases Control and Prevention National Center for Health Statistics: mortality patterns – United States, 1990. Monthly Vital Stat Rep. 1993;41:5. [Google Scholar]
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- Van der Poll T, Van Deventer JH. Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am. 1999;13:413–426. doi: 10.1016/s0891-5520(05)70083-0. [DOI] [PubMed] [Google Scholar]
- MacLean LD, Mulligan WG, McLean APH, Duff JH. Patterns of septic shock in man: a detailed study of 56 patients. Ann Surg. 1967;166:543–562. doi: 10.1097/00000658-196710000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowes GHA, Vucinic M, Weidner MG. Circulatory and metabolic alterations associated with survival or death in peritonitis. Ann Surg. 1966;163:844–866. doi: 10.1097/00000658-196606000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima H, Weil MH, Shubin H, Cavanilles J. Hemodynamic and metabolic studies on shock associated with gram-negative bacteremia. Medicine (Baltimore) 1973;52:287–294. doi: 10.1097/00005792-197307000-00007. [DOI] [PubMed] [Google Scholar]
- Weil MH, Nishijima H. Cardiac output in bacterial shock. Am J Med. 1978;64:920–922. doi: 10.1016/0002-9343(78)90444-8. [DOI] [PubMed] [Google Scholar]
- Blain CM, Anderson TO, Pietras RJ, Gunnar RM. Immediate hemodynamic effects of gram-negative vs gram-positive bacteremia in man. Arch Intern Med. 1970;126:260–265. [PubMed] [Google Scholar]
- Packman MI, Rackow EC. Optimum left heart filling pressure during fluid resuscitation of patients with hypovolemic and septic shock. Crit Care Med. 1983;11:165–169. doi: 10.1097/00003246-198303000-00003. [DOI] [PubMed] [Google Scholar]
- Winslow EJ, Loeb HS, Rahimtoola SH, Kamath S, Gunnar RM. Hemodynamic studies and results of therapy in 50 patients with bacteremic shock. Am J Med. 1973;54:421–432. doi: 10.1016/0002-9343(73)90038-7. [DOI] [PubMed] [Google Scholar]
- Krausz MM, Perel A, Eimerl D, Cotev S. Cardiopulmonary effects of volume loading in patients with septic shock. Ann Surg. 1977;185:429–434. doi: 10.1097/00000658-197704000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SM, Shelhamer JH, Natanson C, Alling DW, Parrillo JE. Serial cardiovascular variables in survivors and non-survivors of human septic shock: heart rate as an early predictor of prognosis. Crit Care Med. 1987;15:923–929. doi: 10.1097/00003246-198710000-00006. [DOI] [PubMed] [Google Scholar]
- Parker MM, Suffredini AF, Natanson C, Ognibene FP, Shelhamer JH, Parrillo JE. Responses of left ventricular function in survivors and non-survivors of septic shock. J Crit Care. 1989;4:19–25. [Google Scholar]
- Weisel RD, Vito L, Dennis RC, Hechtman HB. Myocardial depression during sepsis. Am J Surg. 1977;133:512–521. doi: 10.1016/0002-9610(77)90141-6. [DOI] [PubMed] [Google Scholar]
- Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, Damske BA, Parrillo JE. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- Ognibene FP, Parker MM, Natanson C, Shelhamer JH, Parrillo JE. Depressed left ventricular performance. Response to volume infusion in patients with sepsis and septic shock. Chest. 1988;93:903–910. doi: 10.1378/chest.93.5.903. [DOI] [PubMed] [Google Scholar]
- Ellrodt AG, Riedinger MS, Kimchi A, Berman DS, Maddahi J, Swan HJC, Murata GH. Left ventricular performance in septic shock: reversible segmental and global abnormalities. Am Heart J. 1985;110:402–409. doi: 10.1016/0002-8703(85)90163-2. [DOI] [PubMed] [Google Scholar]
- Raper RF, Sibbald WJ, Driedger AA, Gerow K. Relative myocardial depression in normotensive sepsis. J Crit Care. 1989;4:9–18. [Google Scholar]
- Jafri SM, Lavine S, Field BE, Thill-Baharozian MC, Carlson RW. Left ventricular diastolic function in sepsis. Crit Care Med. 1991;18:709–714. doi: 10.1097/00003246-199007000-00005. [DOI] [PubMed] [Google Scholar]
- Munt B, Jue J, Gin K, Fenwick J, Tweeddale M. Diastolic filling in human severe sepsis: an echocardiographic study. Crit Care Med. 1998;26:1829–1833. doi: 10.1097/00003246-199811000-00023. [DOI] [PubMed] [Google Scholar]
- Poelaert J, Declerck C, Vogelaers D, Colardyn F, Visser CA. Left ventricular systolic and diastolic function in septic shock. Intensive Care Med. 1997;23:553–560. doi: 10.1007/s001340050372. [DOI] [PubMed] [Google Scholar]
- Sibbald WJ, Paterson NAM, Holliday RL, Anderson RA, Lobb TR, Duff JH. Pulmonary hypertension in sepsis: measurement by the pulmonary artery diastolic-pulmonary wedge pressure gradient and the influence of passive and active factors. Chest. 1978;73:583–591. doi: 10.1378/chest.73.5.583. [DOI] [PubMed] [Google Scholar]
- Kimchi A, Ellrodt GA, Berman DS, Murata GH, Riedinger MS, Swan HJC, Murata GH. Right ventricular performance in septic shock: a combined radionuclide and hemodynamic study. J Am Coll Cardiol. 1984;4:945–951. doi: 10.1016/s0735-1097(84)80055-8. [DOI] [PubMed] [Google Scholar]
- Schneider AJ, Teule GJJ, Groenveld ABJ, Nauta J, Heidendal GAK, Thijs LG. Biventricular performance during volume loading in patients with early septic shock, with emphasis on the right ventricle: a combined hemodynamic and radionuclide study. Am Heart J. 1988;116:103–112. doi: 10.1016/0002-8703(88)90256-6. [DOI] [PubMed] [Google Scholar]
- Parker MM, McCarthy KE, Ognibene FP, Parrillo JE. Right ventricular dysfunction and dilatation, similar to left ventricular changes, characterize the cardiac depression of septic shock in humans. Chest. 1990;97:126–131. doi: 10.1378/chest.97.1.126. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Reuse C, Frank N, Contempre B, Kahn RJ. Right ventricular dysfunction in septic shock: assessment by measurements of right ventricular ejection fraction using the thermodilution technique. Acta Anaesthesiol Scand. 1989;33:34–38. doi: 10.1111/j.1399-6576.1989.tb02856.x. [DOI] [PubMed] [Google Scholar]
- Baumgartner J, Vaney C, Perret C. An extreme form of hyperdynamic syndrome in septic shock. Intensive Care Med. 1984;10:245–249. doi: 10.1007/BF00256261. [DOI] [PubMed] [Google Scholar]
- Groeneveld ABJ, Nauta JJ, Thijs L. Peripheral vascular resistance in septic shock: its relation to outcome. Intensive Care Med. 1988;14:141–147. doi: 10.1007/BF00257468. [DOI] [PubMed] [Google Scholar]
- Rhodes A, Lamb FJ, Malagon R, Newman PJ, Grounds M, Bennett D. A prospective study of the use of a dobutamine stress test to identify outcome in patients with sepsis, severe sepsis or septic shock. Crit Care Med. 1999;27:2361–2366. doi: 10.1097/00003246-199911000-00007. [DOI] [PubMed] [Google Scholar]
- Schupp E, Kumar A, Bunnell E, Uretz E, Calvin J, Ali A, Parrillo JE. The cardiovascular response to incremental doses of dobutamine in septic shock: prediction of survival [abstract]. Crit Care Med. 1994;22:A109. [Google Scholar]
- Cunnion RE, Schaer GL, Parker MM, Natanson C, Parrillo JE. The coronary circulation in human septic shock. Circulation. 1986;73:637–644. doi: 10.1161/01.cir.73.4.637. [DOI] [PubMed] [Google Scholar]
- Dhainaut JF, Huyghebaert MF, Monsallier JF, Lefevre G, Dall'Ava-Santucci J, Brunet F, Villemant D, Carli A, Raichvarg D. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose and ketones in patients with septic shock. Circulation. 1987;75:533–541. doi: 10.1161/01.cir.75.3.533. [DOI] [PubMed] [Google Scholar]
- Solomon MA, Correa R, Alexander HR, Koev LA, Cobb JP, Kim DK, Roberts WC, Quezado ZMN, Schotz TD, Cunnion RE, Hoffman WD, Bacher J, Yatsiv I, Danner RL, Banks SM, Ferrans VJ, Balaban RS, Natanson C. Myocardial energy metabolism and morphology in a canine model of sepsis. Am J Physiol. 1994;266:H757–H768. doi: 10.1152/ajpheart.1994.266.2.H757. [DOI] [PubMed] [Google Scholar]
- Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Crit Care Med. 1999;27:1775–1780. doi: 10.1097/00003246-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Wiggers CJ. Myocardial depression in shock. A survey of cardiodynamic studies. Am Heart J. 1947;33:633–650. doi: 10.1016/0002-8703(47)90079-3. [DOI] [PubMed] [Google Scholar]
- Lefer AM. Mechanisms of cardiodepression in endotoxin shock. Circ Shock. 1979;1(suppl):1–8. [PubMed] [Google Scholar]
- Parrillo JE, Burch C, Shelhamer JH, Parker MM, Natanson C, Schuette W. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. J Clin Invest. 1985;76:1539–1553. doi: 10.1172/JCI112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JM, Cunnion RE, Burch-Whitman C, Parker MM, Shelhamer JH, Parrillo JE. A circulating myocardial depressant substance is associated with cardiac dysfunction and peripheral hypoperfusion (lactic acidemia) in patients with septic shock. Chest. 1989;95:1072–1080. doi: 10.1378/chest.95.5.1072. [DOI] [PubMed] [Google Scholar]
- Cunnion RE, Parrillo JE. Myocardial dysfunction in sepsis. Recent insights. Chest. 1989;95:941–945. doi: 10.1378/chest.95.5.941. [DOI] [PubMed] [Google Scholar]
- Seckinger P, Vey E, Turcatti G, Wingfield P, Dayer J. Tumor necrosis factor inhibitor: purification, NH2-terminal amino acid sequence and evidence for anti-inflammatory and immunomodulatory activities. Eur J Immunol. 1990;20:1167–1174. doi: 10.1002/eji.1830200533. [DOI] [PubMed] [Google Scholar]
- Eichacker PQ, Hoffman WD, Farese A, Banks SM, Kuo GC, MacVittie TJ, Natanson C. TNF but not IL-1 in dogs causes lethal lung injury and multiple organ dysfunction similar to human sepsis. J Appl Physiol. 1991;71:1979–1989. doi: 10.1152/jappl.1991.71.5.1979. [DOI] [PubMed] [Google Scholar]
- Eichenholz PW, Eichacker PQ, Hoffman WD, Banks SM, Parrillo JE, Danner RL, Natanson C. Tumor necrosis factor challenges in canines: patterns of cardiovascular dysfunction. Am J Physiol. 1992;263:H668–H675. doi: 10.1152/ajpheart.1992.263.3.H668. [DOI] [PubMed] [Google Scholar]
- van der Poll T, van Deventer SJ, Hack CE, Wolbink GJ, Aarden LA, Buller HR, ten Cate H. Effects on leukocytes following injection of tumor necrosis factor into healthy humans. Blood. 1992;79:693–698. [PubMed] [Google Scholar]
- van der Poll T, Romjin JA, Endert E, Borm JJ, Buller HR, Sauerwein HP. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am J Physiol. 1991;261:E457–E465. doi: 10.1152/ajpendo.1991.261.4.E457. [DOI] [PubMed] [Google Scholar]
- Gu M, Bose R, Bose D, Yang J, Li X, Light RB, Mink S. Tumor necrosis factor-alpha but not septic plasma depresses cardiac myofilament contraction. Can J Anesth. 1998;45:280–286. doi: 10.1007/BF03012028. [DOI] [PubMed] [Google Scholar]
- Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor-alpha and interleukin1-beta are responsible for depression of in vitro myocardial cell contractility induced by serum from humans with septic shock. J Exp Med. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel MS, Oddis CV, Jacobs TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines mediated by nitric oxide. Science. 1992;257:387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- Weisensee D, Bereiter-Hahn J, Low-Friedrich I. Effects of cytokines on the contractility of cultured cardiac myocytes. Int J Immunopharmacol. 1993;15:581–587. doi: 10.1016/0192-0561(93)90075-a. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Bakker J, Marecaux G, Schandene L, Kahn RJ, Dupont E. Administration of anti-TNF antibody improves left ventricular function in septic shock patients: results of a pilot study. Chest. 1992;101:810–815. doi: 10.1378/chest.101.3.810. [DOI] [PubMed] [Google Scholar]
- Hesse DG, Tracey KJ, Fong Y, Manogue KR, Palladinao MA, Cerami A, Shines GT, Lowry SF. Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet. 1988;166:147–153. [PubMed] [Google Scholar]
- Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. IL-1 and TNF inhibit cardiac myocyte adrenergic responsiveness. Proc Natl Acad Sci. 1989;86:6753–6757. doi: 10.1073/pnas.86.17.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosenpud JD, Campbell SM, Mendelson DJ. Interleukin-1 induced myocardial depression in an isolated beating heart preparation. J Heart Transplant. 1989;8:460–464. [PubMed] [Google Scholar]
- Fisher CJJr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, Reines HD, Shelly MP, Thompson BW, LaBreque JF, Catalano MA, Knaus WA, Saddoff JC. Recombinant human IL-1 receptor antagonist in the treatment of patients with the sepsis syndrome: results from a randomized, double blind placebo controlled trial. JAMA. 1994;271:1836–1843. [PubMed] [Google Scholar]
- Fisher CJ, Jr, Slotner GJ, Opal SM, Pribble JP, Bone RC, Emmanuel G, Ng D, Bloedow DC, Catalano MA. Initial evaluation of human recombinant IL-1 receptor antagonist in the treatment of the sepsis syndrome; a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. 1994;22:12–21. doi: 10.1097/00003246-199401000-00008. [DOI] [PubMed] [Google Scholar]
- Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, Harken AH. Tumor necrosis factor-α and interleukin-1β synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–1318. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- Walley KR, Hebert PC, Wakai Y, Wilcox PG, Road JD, Cooper DJ. Decrease in left ventricular contractility after tumor necrosis factor-α in infusion in dogs. J Appl Physiol. 1994;76:1060–1067. doi: 10.1152/jappl.1994.76.3.1060. [DOI] [PubMed] [Google Scholar]
- DeMeules JE, Pigula FA, Mueller M, Raymond SJ, Gamelli RL. Tumor necrosis factor and cardiac function. J Trauma. 1992;32:686–692. doi: 10.1097/00005373-199206000-00003. [DOI] [PubMed] [Google Scholar]
- Hare JM, Keaney JF, Balligand JL, Loscalzo J, Smith TW, Colucci WS. Role of nitric oxide in parasympathetic modulation of beta-adrenergic myocardial contractility in normal dogs. J Clin Invest. 1995;95:360–366. doi: 10.1172/JCI117664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer DB, Colucci WS. Nitric oxide in the failing myocardium. Cardiol Clin North Am. 1998;16:657–664. doi: 10.1016/s0733-8651(05)70042-4. [DOI] [PubMed] [Google Scholar]
- Balligand JL, Kobzik L, Han X, Kaye DM, Belhassen L, O'Hara DS, Kelly RA, Smith TW, Michel T. NO-dependent parasympathetic signalling is due to the activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J Biol Chem. 1995;270:14582–14586. doi: 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- Brady AJ, Warren JB, Poole-Wilson PA, Williams TJ, Harding SE. Nitric oxide attenuates cardiac myocyte contraction. Am J Physiol. 1993;265:H176–H182. doi: 10.1152/ajpheart.1993.265.1.H176. [DOI] [PubMed] [Google Scholar]
- Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprussside infusion. Circulation. 1994;89:2070–2078. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- Kumar A, Brar R, Wang P, Dee L, Skorupa G, Khadour F, Schulz R, Parillo JE. The role of nitric oxide and cyclic GMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999;276:R265–R276. doi: 10.1152/ajpregu.1999.276.1.R265. [DOI] [PubMed] [Google Scholar]
- Rozanski GJ, Witt RC. IL-1 inhibits beta-adrenergic control of the cardiac calcium current: role of L-arginine/nitric oxide pathway. Am J Physiol. 1994;267:H1753–H1758. doi: 10.1152/ajpheart.1994.267.5.H1753. [DOI] [PubMed] [Google Scholar]
- Smith JA, Radomski MW, Schulz R, Moncada S, Lewis MJ. Porcine ventricular endocardial cells in culture express the inducible form of nitric oxide synthase. Br J Pharmacol. 1993;108:1107–1110. doi: 10.1111/j.1476-5381.1993.tb13512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinugawa K, Takahashi T, Kohmoto O, Yao A, Aoyagi T, Monomura S, Hirata Y, Serizawa T. Nitric oxide-mediated effects of IL-6 on [Ca2+]i and cell contraction in cultured chick ventricular myocytes. Circ Res. 1994;75:285–295. doi: 10.1161/01.res.75.2.285. [DOI] [PubMed] [Google Scholar]
- Kumar A, Krieger A, Symeoneides S, Kumar A, Parrillo JE. Myocardial dysfunction in septic shock: Part II. Role of cytokines and nitric oxide. J Cardiothorac Vasc Anesth. 2001;15:485–511. doi: 10.1053/jcan.2001.25003. [DOI] [PubMed] [Google Scholar]
- Parillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction and therapy. Ann Intern Med. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]