

Abstract
The rate of oxygen consumption by certain tissues is impaired when mice or rats are injected with lipopolysaccharide. A similar change in the rate of oxygen consumption is observed when Caco-2 human enterocyte-like cells are incubated in vitro with cytomix, a cocktail of cytokines containing tumor necrosis factor, IL-1β, and IFN-γ. The decrease in the rate of oxygen consumption is not due to a change in oxygen delivery (e.g. on the basis of diminished microvascular perfusion), but rather to an acquired intrinsic defect in cellular respiration, a phenomenon that we have termed 'cytopathic hypoxia'. A number of different biochemical mechanisms have been postulated to account for cytopathic hypoxia in sepsis, including reversible inhibition of cytochrome a,a3 by nitric oxide, and irreversible inhibition of one or more mitochondrial respiratory complexes by peroxynitrite. Recently, however, our laboratory has obtained data to suggest that the most important mechanism underlying the development of cytopathic hypoxia is depletion of cellular stores of nicotinamide adenine dinucleotide (NAD+/NADH) as a result of activation of the enzyme, poly(ADP-ribose) polymerase-1. If cytopathic hypoxia is important in the pathophysiology of established sepsis and multiorgan dysfunction syndrome, then efforts in the future will need to focus on pharmacological interventions designed to preserve normal mitochondrial function and energy production in sepsis.
Keywords: ATP, LPS, NAD, PARP, sepsis
A cellular energetics primer
The First Law of Thermodynamics states that the total amount of energy in a system remains constant before and after any sort of transforming event. The Second Law of Thermodynamics states that, even though the total amount of energy does not change after a transforming event, the total amount of usable energy (the Gibbs free energy) is always decreased. In accordance with the Second Law of Thermodynamics, all reversible chemical reactions proceed in the direction that results in a net decrease in the Gibbs free energy for the system; in other words, the change in the Gibbs free energy (ΔG) is always less than zero.
When cells in living systems need to carry out a reaction for which ΔG is positive, they couple the reaction to another reaction that is energetically favorable (i.e. characterized by ΔG < 0). If the algebraic sum of the ΔG values for the two coupled reactions is negative then the formation of the desired product can proceed. For example, the amidation of glutamate by an ammonium ion to form glutamine is an 'endergonic' reaction (ΔG > 0). To drive this reaction toward the formation of glutamate, cells couple it to another reaction, the hydrolysis of ATP to form ADP and inorganic phosphate, which is 'exergonic' (ΔG is negative). The algebraic sum of ΔG for the two reactions is negative, and the coupled reactions proceed yielding the products glutamine, ADP, inorganic phosphate, and a hydrogen ion.
Hundreds of reactions within cells would not proceed without this sort of coupling. Also, in most cases, the exergonic reaction that drives the formation of the desired product is hydrolysis of the terminal pyrophosphate ester linkage of ATP to yield ADP and inorganic phosphate. The hydrolysis of ATP also drives other energy-requiring processes in cells, such as the active pumping of solutes against a concentration gradient across a membrane barrier. Therefore, for proper functioning, all cells need a steady supply of ATP. Stated another way, ATP is the energy currency of the cell.
ATP can be generated in cells as a result of both aerobic and anaerobic processes. Anaerobic generation of ATP, or the energetically equivalent compound guanosine triphosphate, occurs in both the cytosol and mitochondria as a result of the phosphorylation reactions that are catalyzed by the enzymes phosphoglycerate kinase, pyruvate kinase, and succinyl coenzyme A synthase (Fig. 1). Aerobic generation of ATP occurs in the mitochondria as a result of a carefully orchestrated series of reactions that effectively couple the oxidation of substrates by molecular oxygen (O2), on the one hand, to the phosphorylation of ADP to form ATP, on the other.
Figure 1.
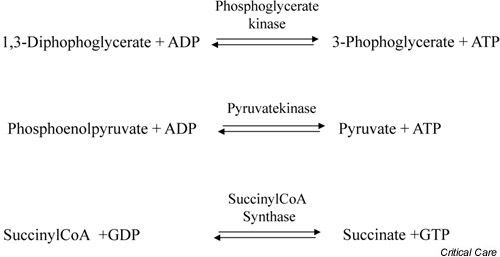
Biochemical reactions that result in substrate level phosphorylation of either ADP or guanosine diphosphate (GDP). The products of these reactions are ATP and guanosine triphosphate (GTP), respectively. CoA, coenzyme A.
To provide a somewhat more detailed account of oxidative phosphorylation, it is will be useful to briefly review the basic principles of reduction–oxidation (redox) chemistry. Good reducing agents are elements or compounds that have a strong propensity to donate electrons to another element or compound. Conversely, good oxidizing agents are elements or compounds that avidly accept electrons. Molecular oxygen (dioxygen, O2) is a very potent oxidizing agent. Two strong reducing agents, namely the reduced form of nicotinamide adenine dinucleotide (NADH) and the reduced form of flavin adenine dinucleotide, are produced in cells during glycolysis and the citric acid cycle. These two reducing agents are oxidized by O2 in mitochondria, and the energy released during this process is used to drive the formation of ATP.
The reaction of a strong reducing agent such as NADH with a powerful oxidizing agent such as O2 releases a large amount of energy. To take optimal advantage of this highly exergonic redox reaction and to capture as much of the energy released as possible in a usable form (i.e. the high-energy terminal pyrophosphate bond in ATP), mitochondria 'step-down' the reducing potential of NADH (and the reduced form of flavin adenine dinucleotide) in stages. The electrons are thus not transferred from NADH to O2 all at once, but rather are transferred through a series of intermediate compounds, called electron carriers, that have progressively lower reducing potentials. Several of the electron carriers involved in the mitochondrial respiratory chain are organized as complexes located within the inner mitochondrial membrane. These complexes use the energy released during electron transfer to actively pump protons from the mitochondrial matrix into the intermembrane space, thereby generating an electrochemical gradient across the inner mitochondrial membrane. The presence of this gradient drives hydrogen ions through a mitochondrial enzyme, the FoF1ATPase, that catalyzes the formation of ATP from ADP and inorganic phosphate.
For each mole of glucose metabolized to carbon dioxide and water, the net yield of ATP from substrate level (anaerobic) phosphorylation reactions is 4 moles of ATP, whereas the net yield of ATP from oxidative phosphorylation reactions is 32 moles of ATP. The oxidative metabolism in normally functioning mitochondria is thus far more efficient at producing ATP than is the anaerobic metabolism, and many cell types (such as hepatocytes, neurons, and cardiac myocytes) are dependent on a steady supply of O2.
Evidence for impaired mitochondrial respiration in sepsis
Many factors can compromise the delivery of O2 to tissues in patients with sepsis. Among these factors are acute lung injury, which of course can cause arterial hypoxemia. In addition, dilation of capacitance vessels combined with microvascular hyperpermeability can lead to an acute decrease in the left ventricular preload and can thereby compromise cardiac output. Left ventricular performance can be further compromised by the intrinsic decrease in myocardial contractility that occurs in many patients with sepsis. Perfusion through the microvasculature can also be embarrassed by abnormalities in arteriolar tone as well as plugging of capillaries by sequestered platelets and leukocytes. In addition, the deformability of erythrocytes is impaired in patients with sepsis, and this pathological phenomenon might further contribute to derangements in microvascular blood flow.
Given the importance of impaired perfusion in sepsis, it is not surprising that intensivists have focused enormous effort on developing better ways to monitor blood flow and O2 delivery. Furthermore, convincing data are now available to support the view that early aggressive efforts to improve systemic O2 delivery by administering intravenous fluids, packed red blood cells, and inotropic agents can improve the outcome for patients with septic shock [1]. By the same token, however, efforts to improve systemic O2 delivery later in the course of sepsis are at best ineffective [2,3], and at worst deleterious [4].
If improving perfusion and O2 delivery in patients with established sepsis fails to improve survival or prevent organ system dysfunction, then one might wonder whether alterations in energy metabolism are important at all in the pathogenesis of the syndrome [5]. Alternatively, one could hypothesize that cellular energetics are deranged in sepsis not just because O2 delivery is impaired, but also, and perhaps even more importantly, because the ability of cells to utilize available O2 is compromised.
Some years ago, we coined the term 'cytopathic hypoxia' to describe just such an acquired intrinsic derangement in cellular respiration [6,7]. While the clinical significance of this phenomenon remains to be established with certainty, a considerable body of evidence has accumulated to support the notion that cytopathic hypoxia occurs in experimental animals with sepsis or endotoxemia or cells exposed in vitro to proinflammatory cytokines. Moreover, it is becoming increasingly apparent that activation of a key enzyme, poly(ADP-ribose) polymerase-1 (PARP-1), plays a central role in the pathogenesis of cytopathic hypoxia. The purpose of the present article is to review some of these data.
Measurements of tissue oxygen partial pressure and the cytochrome a,a3 redox state
Tissue hypoxia is an expected correlate of any of the three classical causes of impaired cellular aerobic metabolism identified by Barcroft more than 75 years ago [8]. Therefore, when the delivery of O2 decreases on the basis of low arterial oxygen tension (pO2), anemia, and/or hypoperfusion, cells extract a greater fraction of the available O2 in an effort to defend aerobic ATP production. As a consequence, the distribution of tissue pO2 values shifts to the left (i.e. toward values closer to zero). In contrast, when ATP production is impaired as a result of an intrinsic derangement in cellular respiration, cells extract less O2 per unit time from the available supply. The expected consequence of this change in O2 extraction is a rightward shift of the tissue pO2 distribution (i.e. toward higher values).
One line of evidence supporting the concept of cytopathic hypoxia comes from studies making measurements of tissue pO2 in patients or experimental animals. If tissue hypoperfusion were a major factor contributing to cellular dysfunction in sepsis, septic shock, or endotoxemia, then we would predict that abnormally low values for tissue pO2 would be detected in these conditions. But, if impaired O2 utilization by cells were a major factor, then we would predict that tissue pO2 values would be normal or even higher than normal. Indeed, observations of this sort have been reported. For example, Astiz and colleagues used cecal ligation and puncture in rats as a model of sepsis. They showed that the mean skeletal muscle pO2 was similar in septic animals and in normal controls provided that the rats with peritonitis were infused with albumin solution to expand the intravascular volume [9].
In a conceptually similar study, Hotchkiss et al. used a novel approach to determine whether tissue hypoxia occurs following the induction of sepsis in rats [10]. The tissue pO2 was not measured directly, but rather was estimated by measuring the retention of [18F]fluoroisonidazole, a lipophilic 2-nitroimidazole derivative that is irreversibly bound to intracellular macromolecules under hypoxic, but not normoxic, conditions. As a positive control, these investigators showed that retention of [18F]fluoroisonidazole was increased in gastrocnemius muscle rendered ischemic by proximal application of a rubber tourniquet. When the retention of [18F]fluoroisonidazole in a variety of tissues, such as skeletal muscle and the liver, was compared in groups of septic rats and of nonseptic controls, however, no differences were apparent. These data provided very strong evidence that sepsis in rats is not associated with tissue hypoxia.
Some data support the even more remarkable conclusion that the tissue pO2 actually increases in sepsis relative to normal values. For example, VanderMeer et al. used a porcine model to investigate the effects of endotoxemia on intestinal mucosal pO2 [11]. When anesthetized pigs were infused with lipopolysaccharide (LPS) and simultaneously resuscitated to maintain normal cardiac output, the mean mucosal pO2 increased significantly (Fig. 2). Rosser et al. similarly reported that bladder mucosal pO2 increased in rats challenged with LPS [12]. The very same pattern has been observed in humans. Boekstegers and colleagues documented that the pO2 distribution in skeletal muscle was shifted toward values less than normal in patients with cardiogenic shock, as expected, but was shifted to the right (i.e. to supranormal values) in patients with septic shock [13]. Sair et al. also recently reported that skeletal muscle tissue pO2 levels are higher than normal in patients with severe sepsis [14]. These data showing that the tissue pO2 actually increases during sepsis are consistent with the view that cellular utilization of O2 is impaired under these conditions.
Figure 2.
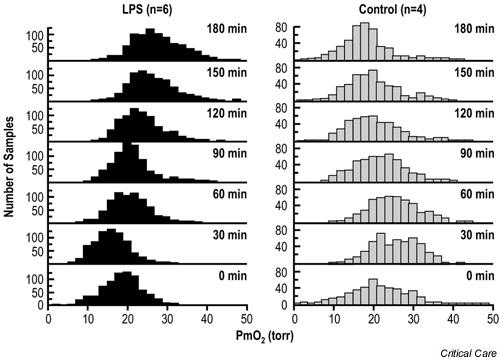
Ileal mucosal oxygen tension in pigs infused with lipopolysaccharide (LPS) (150 μg/kg; n = 6) or normal saline (n = 4). The animals were resuscitated with lactated Ringer's solution and dextran-70 solution to maintain a cardiac output of 90–110% of the baseline value. Mucosal oxygen tension (pO2) histograms were obtained with an array of Clark-type O2-sensitive electrodes driving an amplifier and a computerized data acquisition system. Adapted from [11] with permission.
This same idea is also supported by another related study. Simonsen et al. used near-infrared spectroscopy to monitor the redox state of the terminal element of the mitochondrial respiratory chain, cytochrome a,a3, in skeletal muscle cells of baboons rendered septic by an infusion of viable Escherichia coli [15]. The functional status of cytochrome a,a3 was monitored by periodically causing temporary skeletal muscle ischemia using a proximally placed tourniquet. Inflating the tourniquet caused a decrease in the signal from oxidized cytochrome a,a3, whereas deflating the tourniquet resulted in an increase in the near-infrared signal from the reoxidized enzyme. Early in the sepsis protocol (i.e. at 6 hours), the rate of cytochrome a,a3 reduction following tourniquet ischemia was the same as that at baseline, although the rate of reoxidation following the release of ischemia was slowed. These data were interpreted as being consistent with decreased delivery of O2 to the tissue. Later in the sepsis protocol (e.g. at 18 hours), the rate of cytochrome a,a3 reduction during tourniquet ischemia was markedly slowed, a finding that was felt to be consistent with the presence of either a defect in the ability of the enzyme to accept electrons from O2 or a limitation in the availability of reducing equivalents (i.e. NADH and/or the reduced form of flavin adenine dinucleotide). These data are particularly interesting because they suggest that cytopathic hypoxia is not present early in sepsis, but that it develops after the septic process has evolved for many hours. These temporal considerations might explain the positive results obtained in a clinical trial of 'early goal-directed' hemodynamic support by Rivers et al. [1] and the negative results obtained in a similar trial but including patients with more established disease [4].
It is important to point out that not all studies of sepsis have obtained data showing that tissue pO2 values are normal or increased. Indeed, contrary findings have been reported by a number of investigators. For example, two well-performed studies showed that the intestinal mucosal pO2 decreased when experimental animals were infused with LPS to a sepsis-like state [16,17]. Sair and colleagues similarly reported that the skeletal muscle pO2 decreased markedly in a rat model of endotoxemia [18]. Interestingly, in the study by Sair et al. there was no difference between endotoxemic animals and controls with respect to skeletal muscle perfusion. The development of tissue hypoxia following the injection of LPS was therefore attributed to impaired microvascular control of nutritive flow.
Indirect and direct measurements of mitochondrial respiration
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) is a colorless compound that is reduced by functioning mitochondria to a blue dye (MTT-formazan). The concentration of the blue product can be determined spectrophotometrically. The reduction of MTT is thus a convenient, albeit indirect, way to assess mitochondrial function.
Bankey et al. used this approach in 1974 to assess mitochondrial function in cocultures of rat hepatoctyes and rat liver macrophages [19]. Sequential stimulation of the cultures with a proinflammatory cytokine (IL-6) and then LPS decreased MTT reduction by about 50%. Similar results were reported more recently by Szabó and colleagues, who showed that MTT reduction was decreased in cultured macrophages and vascular smooth muscle cells following incubation with a proinflammatory cytokine (IFN-γ) plus LPS [20].
Unno et al. used the reduction of MTT to MTT-formazan to assess mitochondrial function in vivo [21]. In that study, rats were injected with saline or a low dose of LPS (5 mg/kg) that caused neither hypotension nor mortality. Twenty-four hours later, the lumen of the intestine was loaded with a solution of MTT. After a 30 min incubation period, the epithelial layer was scraped off the intestine and the concentration of MTT-formazan in enterocytes was determined spectrophotometrically. The MTT reduction was shown to be significantly lower in enterocytes from endotoxemic rats as compared with enterocytes from normal controls (Fig. 3). In the study by Unno et al., treatment of the endotoxemic rats with aminoguanidine, a drug that blocks inducible nitric oxide synthase (iNOS), restored MTT reduction back to normal levels. This latter finding suggests that impaired mitochondrial respiration in sepsis is mediated, at least partially, by a mechanism that depends on increased production of nitric oxide free radical (NO·) via the iNOS pathway.
Figure 3.
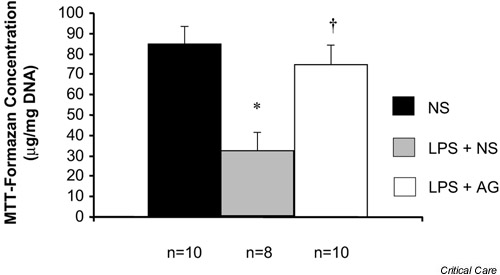
Effect of lipopolysaccharide (LPS) on the ileal mucosal mitochondrial function in rats. Rats were injected with normal saline (NS), LPS (5 mg/kg) or the same dose of LPS plus aminoguanidine (AG). The mitochondrial function was measured 24 hours later using MTT (see text). *P < 0.05 versus NS, †P < 0.05 versus LPS. Adapted from [21] with permission.
The MTT assay reflects the activity of a number of different dehydrogenases, particularly succinate dehydrogenase [22], and is not a direct measure of mitochondrial O2 consumption per se. Several studies have obtained more direct evidence that cellular or mitochondrial respiration is impaired in animals with sepsis or endotoxemia. For example, Kantrow et al. showed that hepatocytes isolated from septic rats consumed significantly less O2 than did hepatocytes from nonseptic control rats [23]. King et al. subsequently sought to determine whether ileal mucosal O2 consumption is impaired in endotoxemic rats [24]. Rats were injected with either LPS or a similar volume of the saline vehicle. Eight hours later, a strip of ileal mucosa was harvested from the animals and mounted in a polaragraphic chamber, and the rate of O2 consumption determined using standard methods. The rate of O2 consumption was significantly lower for mucosal samples from endotoxemic rats as compared with control rats (Fig. 4). If the endotoxemic rats were treated with aminoguanidine to block iNOS activity, then normal ileal mucosal O2 consumption was preserved. These findings thus provide further support for the notion that the development of cytopathic hypoxia in LPS-challenged rats requires iNOS-dependent NO· production.
Figure 4.
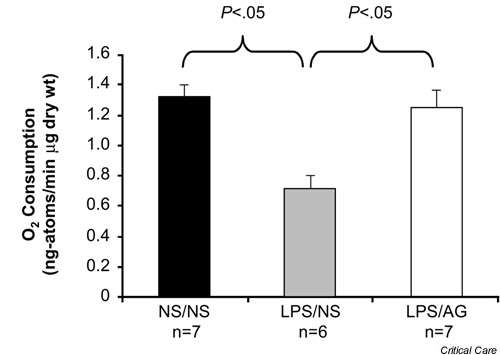
Effect of lipopolysaccharide (LPS) on ileal mucosal O2 consumption. Rats in the NS/NS group were injected at T = 0 hours with normal saline (NS) and were treated with NS. Rats in the LPS/NS group were injected with LPS (5 mg/kg) at T = 0 hours and were treated with NS. Rats in the LPS/AG group were challenged with the same dose of LPS and treated with aminoguanidine (30 mg/kg per dose at T = 1, 3 and 6 hours). Ex vivo O2 consumption was measured at T = 8 hours. Adapted from [24] with permission.
Potential mechanisms to explain cytopathic hypoxia in sepsis
A number of different but mutually compatible mechanisms might foster the development of cytopathic hypoxia under pathological conditions. These mechanisms include the diminished delivery of pyruvate into the mitochondrial tricarboxylic acid (TCA) cycle, the inhibition of key mitochondrial enzymes that are involved in either the TCA cycle or the electron transport chain, and the activation of the enzyme PARP-1.
Inhibition of pyruvate dehydrogenase
The end-product of glycolysis is pyruvic acid, a three-carbon alpha-keto acid. Pyruvic acid can either be reduced to lactic acid or enter the TCA, ultimately to be oxidized to water and carbon dioxide. Pyruvate dehydrogenase (PDH) catalyzes the reaction whereby pyruvate, in the presence of NAD+ and coenzyme A, is converted to acetyl-coenzyme A. Because of its pivotal role in the regulation of intermediary metabolism, the activity of PDH is tightly regulated by both end-product inhibition and reversible phosphorylation. A group of isoenzymes, the PDH kinase family, catalyzes the phosphorylation of PDH to its inactive form. A PDH phosphatase catalyzes the dephosphorylation of the inactive form to the active form of the enzyme complex. Vary and coworkers showed that the PHH inactive form:PHA active form ratio in skeletal muscle tissue increases during chronic sepsis in rats [25,26]. The mechanism responsible for this effect is increased PDH kinase activity rather than decreased PDH phosphatase activity [27,28]. Because inactivation of PDH limits the flux of the substrate through the TCA cycle, excess pyruvate accumulates in cells and leads to increased production of lactate. Therefore, according to the data obtained by Vary's laboratory, hyperlactatemia in sepsis is not necessarily evidence of impaired O2 delivery, but can be evidence of the combined effects of PDH inhibition and accelerated glucose transport into cells [26].
Nitric oxide-mediated inhibition of cytochrome a,a3
As already noted, sepsis is associated with iNOS induction and increased production of the pluripotent signaling and effector molecule NO·. At physiologically relevant concentrations (~1 μM), NO· rapidly but reversibly inhibits the enzymatic activity of cytochrome a,a3, the terminal complex of the mitochondrial electron transport chain [29,30,31,32]. NO·-mediated inhibition of mitochondrial O2 consumption is the result of competition by the two gases (i.e. O2 and NO·) for the same binding site on the enzyme complex [33,34]. Accordingly, the inhibitory effect of NO· tends to be more pronounced when the pO2 is relatively low [30,31,32]. Although much of the work related to this mechanism has been carried out using NO· derived from an exogenous source (i.e. authentic NO· gas or a chemical compound that releases NO· in solution), data are available to show that endogenously produced NO· is also capable of causing reversible inhibition of the terminal element in the mitochondrial respiratory chain (cytochrome a,a3), leading to reduced cellular respiration [35].
Peroxynitrite-mediated inhibition of mitochondrial enzymes
Although NO· reversibly inhibits cytochrome a,a3, the compound reacts with only a limited range of intracellular targets and should not be regarded as toxic. Under the right conditions, however, NO· reacts rapidly with O2-· to form peroxynitrite (ONOO-), which is a potent oxidizing and nitrosating agent [36,37,38,39]. Appropriate conditions for the production of ONOO- are present during a variety of acute inflammatory conditions, including ischemia/reperfusion injury and sepsis. In addition, small quantities of O2-· are continually being produced by mitochondria at the level of the three enzymatic complexes interacting with Q10. Under certain conditions, such as when O2 availability is limited [40] or cytochrome a,a3 is inhibited by NO· [41], mitochondria generate increased quantities of O2-· by this mechanism. Moreover, a calcium-dependent nitric oxide synthase isoform that is either identical to or is very similar to neuronal nitric oxide synthase is present in mitochondria [42,43,44,45]. Mitochondria are thus capable, within the confines of the organelle itself, of generating both NO· and O2-·, and hence, under appropriate conditions, large quantities of the potentially toxic moiety ONOO- [46].
In the laboratory, exposing mitochondria to ONOO- from an exogenous source causes irreversible inhibition of mitochondrial respiration. Several mechanisms have been implicated as being important in this phenomenon. Specifically, ONOO- has been shown to inhibit the mitochondrial FoF1ATPase that carries out phosphorylation of ADP to form ATP [47]. In addition, ONOO- also inhibits two of the mitochondrial enzyme complexes, namely Complex I [47] and Complex II [47], that are involved in electron transport. {Note added in proof: Brealey et al. recently reported that Complex I activity in skeletal muscle biopsies from patients with sepsis is inversely correlated with plasma nitrite/nitrate levels (marker for NO· production) and shock severity [48].} Finally, ONOO- inhibits the activity of aconitase, the TCA cycle enzyme that converts citrate into isocitrate [49]. Endogenous production of ONOO- secondary to iNOS induction plus O2-· generation has been implicated as the major factor leading to impaired mitochondrial respiration in some tissues, such as the rat diaphragm, following in vivo challenge with LPS [50].
The poly(ADP-ribose) polymerase hypothesis
PARP-1 is a nuclear enzyme that participates in a variety of cellular functions, including the repair of single-strand breaks in nuclear DNA [51,52], DNA replication [53], and apoptosis [53]. PARP-1 is activated by single-strand breaks in nuclear DNA, and then catalyzes the cleavage of NAD+ into ADP-ribose and nicotinamide and the polymerization of the resultant ADP-ribose units into branching poly(ADP-ribose) homopolymers [54,55]. Simultaneously, poly-ADP ribose is degraded by various nuclear enzymes, especially poly(ADP-ribose) glycohydrolase [55,56]. The concurrent actions of PARP-1 and poly(ADP-ribose) glycohydrolase constitute the functional equivalent of a nicotinamide adenine dinucleotidase. In states of acute inflammation, reactive oxygen species including ONOO- (and related oxidants) can induced single-strand breaks in nuclear DNA, and thereby can activate PARP-1. As a consequence, the NAD+/NADH content of cells can be depleted. Since NADH is the main reducing equivalent used to support oxidative phosphorylation, activation of PARP-1 can lead to a marked impairment in the ability of cells to utilize O2 to support ATP synthesis; in other words, cytopathic hypoxia.
The notion that redox stress can lead to PARP-1 activation and metabolic inhibition on this basis was first articulated by Schraufstatter et al. about 15 years ago [57]. More recently, Szabó et al. showed that exposure of cultured cells to physiologically relevant concentrations of ONOO- activated PARP-1 and, on this basis, resulted in impaired mitochondrial respiration [58]. Szabó and coworkers further showed that endogenously generated ONOO- was capable of activating PARP-1 and thereby of inhibiting mitochondrial respiration in cultured immunostimulated macrophages [59] and vascular smooth muscle cells [60]. Of note, in these studies, alterations in cellular respiration were not detected by directly measuring O2 consumption, but were inferred by quantitating the reduction of MTT [59].
Recent in vitro studies performed by Khan et al. provide further support for the importance of PARP-1-dependent NAD+/NADH depletion as a mechanism for cytopathic hypoxia caused by inflammatory mediators [61]. Human Caco-2 enterocytic cells growing on microcarrier beads were used as a 'reductionist' model of the intestinal epithelium. The consumption of O2 by these cells was measured directly using an O2-sensitive optode. Incubation of the cells with cytomix (a cocktail of three proinflammatory cytokines: TNF-α, IL-1β, and IFN-γ) decreased cellular O2 consumption by more than 50% (Fig. 5a). This phenomenon was entirely reversible; if the cells were washed free of the cytokine cocktail and then incubated for a short period in normal culture medium, the normal rate of O2 consumption was restored (Fig. 5b). The cytokine-induced decrease in O2 consumption was thus not caused by cell death, but by some sublethal process that impaired normal cellular respiration.
Figure 5.
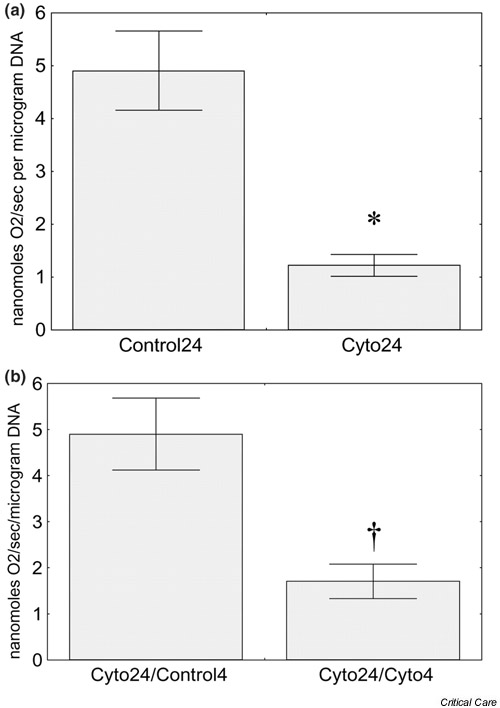
Effect of incubation with cytomix on O2 consumption by Caco-2 enterocytes. (a) In some studies, O2 consumption was measured after cells growing on microcarrier beads were incubated for 24 hours with either control medium (Control24; n = 11) or medium containing cytomix (Cyto24; n = 12). (b) In additional experiments, enterocytes that had been exposed to cytomix for 24 hours were washed extensively, and then incubated for a further 4 hours with either cytokine-free medium (Cyto24/Control4; n = 9) or cytomix-containing medium (Cyto24/Cyto4; n = 4). Data were analyzed using Student's t test. *P < 0.05 versus Control24, †P < 0.05 versus Cyto24/Control4. Reprinted from [61] with permission.
The decrease in O2 consumption induced by incubation with cytomix was significantly ameliorated if any one of the following pharmacological agents was also present during the period of exposure to the cytokine cocktail: 4,5-dihydroxy-1,3-benzene disulfonic acid, an O2-· scavenger; 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, a NO· scavenger; 5,10,15,20-tetrakis-[4-sulfonatophenyl]-porphyrinato-iron [III], a ONOO- decomposition catalyst; urate, a ONOO- scavenger; 3-aminobenzamide, a PARP-1 inhibitor; or N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethyl-acetamide HCl, a chemically dissimilar and more potent PARP-1 inhibitor [61]. The decrease in O2 uptake induced by cytomix was associated with significantly decreased cellular levels of NAD+/NADH.
Khan et al. reasoned that if NAD+/NADH depletion is responsible for the decrease in cellular respiration induced by cytomix, then replenishing cellular levels of this catalytically essential nucleotide should tend to restore normal rates of O2 consumption [61]. NAD+, however, is a bulky and highly charged molecule that would not be expected to diffuse across the cytosolic membrane. Furthermore, extracellular NAD+/NADH is a substrate for a cell-surface enzyme, nicotinamide adenine dinucleotide glycohydrolase, which converts NAD+/NADH into cyclic ADP-ribose [62]. Simply adding NAD+ or NADH to the incubation medium would thus not be expected to have much of an effect on cellular levels of NAD+/NADH. In an effort to circumvent this problem, the investigators incubated cytomix-stimulated Caco-2 cells with NAD+ encapsulated in liposomes. This strategy worked; when cytomix-stimulated Caco-2 cells were coincubated with liposome-encapsulated NAD+, the cellular NAD+/NADH level was increased to about 85% of the control value (Fig. 6). Furthermore, coincubating cytomix-stimulated Caco-2 cells with liposome-encapsulated NAD+ prevented the development of cytopathic hypoxia (Fig. 7).
Figure 6.
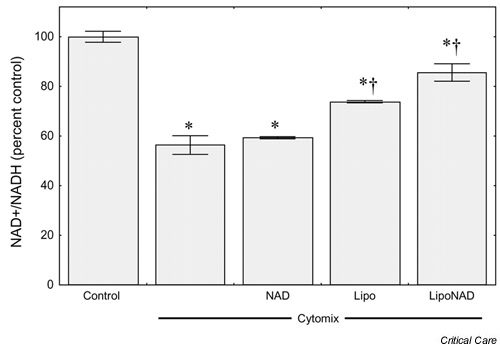
Effect of liposomal nicotinamide adenine dinucleotide (NAD+) on NAD+/NADH levels in Caco-2 enterocytes. Caco-2 cells growing as monolayers in six-well chambers were incubated for 24 hours under cytokine-free conditions alone (Control) or with cytomix in the presence or absence of free NAD+ (NAD), of empty liposomes (Lipo), or of NAD+-containing liposomes (LipoNAD). Results are means ± SEM (n = 4 per condition). Data were analyzed by analysis of variance followed by Duncan's multiple range test. *P < 0.05 versus control, †P < 0.05 versus Cyto. Reprinted from [61] with permission.
Figure 7.
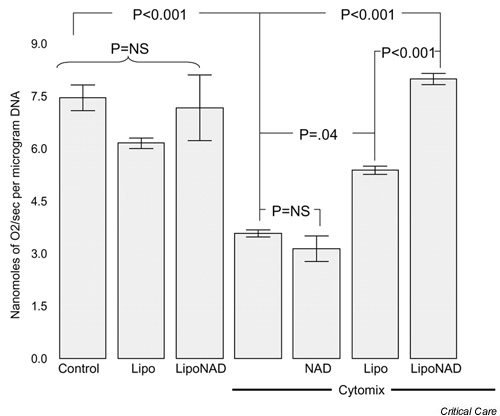
Effect of liposomal nicotinamide adenine dinucleotide (NAD+) on O2 consumption by Caco-2 cells growing on microcarrier beads. Control cells growing on microcarrier beads in six-well dishes were incubated for 24 hours under cytokine-free conditions alone (Control), with empty liposomes (Lipo), or with NAD+-containing liposomes (LipoNAD). Cytokine-stimulated cells were incubated for 24 hours with cytomix alone or with free NAD+ (NAD), with empty liposomes (Lipo), or with NAD+-containing liposomes (LipoNAD). Results are means ± SEM (n = 6–8 per condition). Data were analyzed by analysis of variance followed by Duncan's multiple range test. NS, not significant. Reprinted from [61] with permission.
In addition to the findings just described, other data support the view that PARP-1 activation is a major factor contributing to the pathogenesis of sepsis. For example, using pharmacological agents to block PARP-1 can prevent LPS-induced vascular contractile dysfunction in rodents [63,64]. Moreover, in comparison with wild-type controls, mice with a genetic defect in the PARP-1 enzyme (i.e. PARP-1 knockout mice) are relatively resistant to the lethal of effects LPS [65,66]. Goldfarb et al. recently showed that treatment with a potent PARP-1 inhibitor (N-[6-oxo-5,6-dihydro-phenanthridin-2-yl]-N,N-dimethylacetamide HCl) significantly improved survival in a porcine model of lethal bacterial peritonitis [67].
Conclusion
Several lines of evidence support the notion that cellular energetics are deranged in established sepsis not on the basis of inadequate tissue perfusion, but rather on the basis of impaired mitochondrial respiration. If this concept is correct, then a promising approach will be to develop pharmacological strategies, notably potent and selective PARP-1 inhibitors, to restore normal mitochondrial function and cellular energetics.
Competing interests
None declared.
Abbreviations
ΔG = change in the Gibbs free energy; IFN = interferon; IL = interleukin; iNOS = inducible nitric oxide synthase; LPS = lipopolysaccharide; MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; NAD+ = nicotinamide adenine dinucleotide; NADH = reduced form of nicotinamide adenine dinucleotide; NO· = nitric oxide free radical; O2 = molecular oxygen; ONOO- = peroxynitrite; PARP-1 = poly(ADP-ribose) polymerase-1; PDH = pyruvate dehydrogenase; pO2 = partial pressure of oxygen; redox = reduction–oxidation; TCA = tricarboxylic acid.
Acknowledgments
Acknowledgement
This work was supported by NIH grants GM 37631 and GM58484.
References
- Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- Tuchschmidt J, Fried J, Astiz M, Rackow E. Elevation of cardiac output and oxygen improves outcome in septic shock. Chest. 1992;102:216–220. doi: 10.1378/chest.102.1.216. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Brazzi L, Pelosi P, Latini R, Tognoni G, Pesenti A, Fumagalli R. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med. 1995;333:1025–1032. doi: 10.1056/NEJM199510193331601. [DOI] [PubMed] [Google Scholar]
- Hayes MA, Timmins AC, Yau EHS, Palazzo M, Hinds CJ, Watson D. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717–1722. doi: 10.1056/NEJM199406163302404. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis. JAMA. 1992;267:1503–1509. [PubMed] [Google Scholar]
- Fink MP. Cytopathic hypoxia in sepsis. Acta Anaesthesiol Scand. 1997;41(suppl 100):87–95. doi: 10.1111/j.1399-6576.1997.tb05514.x. [DOI] [PubMed] [Google Scholar]
- Fink MP. Cytopathic hypoxia: mitochondrial dysfunction as a mechanism contributing to organ dysfunction in sepsis. Crit Care Clin North Am. 2001;17:219–237. doi: 10.1016/s0749-0704(05)70161-5. [DOI] [PubMed] [Google Scholar]
- Barcroft J. On anoxaemia. Lancet. 1920;ii:485. [Google Scholar]
- Astiz M, Rackow EC, Weil MH, Schumer W. Early impairment of oxidative metabolism and energy production in severe sepsis. Circ Shock. 1988;26:311–320. [PubMed] [Google Scholar]
- Hotchkiss RS, Rust RS, Dence CS, Wasserman TH, Song S-K, Hwang D-R, Karl IE, Welch MJ. Evaluation of the role of cellular hypoxia in sepsis by the hypoxic marker [18F]fluoroisonidazole. Am J Physiol. 1991;261:R965–R972. doi: 10.1152/ajpregu.1991.261.4.R965. [DOI] [PubMed] [Google Scholar]
- VanderMeer TJ, Wang H, Fink MP. Endotoxemia causes ileal mucosal acidosis in the absence of mucosal hypoxia in a normodynamic porcine model of septic shock. Crit Care Med. 1995;23:1217–1226. doi: 10.1097/00003246-199507000-00011. [DOI] [PubMed] [Google Scholar]
- Rosser DM, Stidwill RP, Jacobson D, Singer M. Oxygen tension in the bladder epithelium rises in both high and low cardiac output endotoxemic sepsis. J Appl Physiol. 1995;79:1878–1882. doi: 10.1152/jappl.1995.79.6.1878. [DOI] [PubMed] [Google Scholar]
- Boekstegers P, Weidenhofer S, Pilz G, Werdan K. Peripheral oxygen availability within skeletal muscle in sepsis and septic shock: comparison to limited infection and cardiogenic shock. Infection. 1991;19:317–323. doi: 10.1007/BF01645355. [DOI] [PubMed] [Google Scholar]
- Sair M, Etherington PJ, Winlove CP, Evans TW. Tissue oxygenation and perfusion in patients with systemic sepsis. Crit Care Med. 2001;29:1343–1349. doi: 10.1097/00003246-200107000-00008. [DOI] [PubMed] [Google Scholar]
- Simonson SG, Welty-Wolf K, Huang Y-CT, Griebel JA, Caplan MS, Fracica PJ, Piantadosi CA. Altered mitochondrial redox responses in Gram negative septic shock in primates. Circ Shock. 1994;43:34–43. [PubMed] [Google Scholar]
- Vallet B, Lund N, Curtis SE, Kelly D, Cain SM. Gut and muscle tissue PO2 in endotoxemic dogs during shock and resuscitation. J Appl Physiol. 1994;76:793–800. doi: 10.1152/jappl.1994.76.2.793. [DOI] [PubMed] [Google Scholar]
- Hasibeder W, Germann R, Wolf HJ, Haisjackl M, Hausdorfer H, Riedmann B, Bonatti J, Gruber E, Schwarz B, Waldenberger P, Friesenecker B, Furtner B. Effects of short-term endotoxemia and dopamine on mucosal oxygenation in porcine jejunum. Am J Physiol. 1996;270:G667–G675. doi: 10.1152/ajpgi.1996.270.4.G667. [DOI] [PubMed] [Google Scholar]
- Sair M, Etherington PJ, Curzen NP, Winlove CP, Evans TW. Tissue oxygenation and perfusion in endotoxemia. Am J Physiol. 1996;271:H1620–H1625. doi: 10.1152/ajpheart.1996.271.4.H1620. [DOI] [PubMed] [Google Scholar]
- Bankey PE, Hill S, Geldon D. Sequential insult enhances liver macrophage-signaled hepatocyte dysfunction. J Surg Res. 1994;57:185–191. doi: 10.1006/jsre.1994.1129. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Day BJ, Crapo JD, Salzman AL, Szabó C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br J Pharmacol. 1997;120:259–267. doi: 10.1038/sj.bjp.0700872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno N, Wang H, Menconi MJ, Tytgat SHAJ, Larkin V, Smith M, Morin MJ, Chavez A, Hodin RA, Fink MP. Inhibition of inducible nitric oxide synthase ameliorates lipopolysaccharide-induced gut mucosal barrier dysfunction in rats. Gastroenterology. 1997;113:1246–1257. doi: 10.1053/gast.1997.v113.pm9322519. [DOI] [PubMed] [Google Scholar]
- Gross SS, Levi R. Tetrahydrobiopterin synthesis. An absolute requirement for cytokine-induced nitric oxide generation by vascular smooth muscle. J Biol Chem. 1992;267:25722–25729. [PubMed] [Google Scholar]
- Kantrow SP, Taylor DE, Carraway MS, Piantadosi CA. Oxidative metabolism in rat hepatocytes and mitochondria during sepsis. Arch Biochem Biophys. 1997;345:278–288. doi: 10.1006/abbi.1997.0264. [DOI] [PubMed] [Google Scholar]
- King CJ, Tytgat S, Delude RL, Fink MP. Ileal mucosal oxygen consumption is decreased in endotoxemic rats but is restored toward normal by treatment with aminoguanidine. Crit Care Med. 1999;27:2518–2524. doi: 10.1097/00003246-199911000-00032. [DOI] [PubMed] [Google Scholar]
- Vary TC, Siegel JH, Nakatani T, Sato T, Aoyama H. Effect of sepsis on activity of pyruvate dehydrogenase complex in skeletal muscle and liver. Am J Physiol. 1986;250:E634–E640. doi: 10.1152/ajpendo.1986.250.6.E634. [DOI] [PubMed] [Google Scholar]
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock. 1996;6:89–94. doi: 10.1097/00024382-199608000-00002. [DOI] [PubMed] [Google Scholar]
- Vary TC, Hazen S. Sepsis alters pyruvate dehydrogenase kinase activity in skeletal muscle. Mol Cell Biochem. 1999;198:113–118. doi: 10.1023/a:1006993910781. [DOI] [PubMed] [Google Scholar]
- Vary TC. Increased pyruvate dehydrogenase kinase activity in response to sepsis. Am J Physiol. 1991;250:E669–E674. doi: 10.1152/ajpendo.1991.260.5.E669. [DOI] [PubMed] [Google Scholar]
- Borutaité V, Brown GC. Rapid reduction of nitric oxide by mitochondria, and reversible inhibition of mitochondrial respiration by nitric oxide. Biochem J. 1996;315:295–299. doi: 10.1042/bj3150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- Takehara Y, Kanno T, Yoshioka T, Inoue M, Utsumi K. Oxygen-dependent regulation of mitochondrial energy metabolism by nitric oxide. Arch Biochem Biophys. 1995;323:27–32. doi: 10.1006/abbi.1995.0005. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Sato EF, Kuroki T, Inoue M. Role of glutathione and nitric oxide in the energy metabolism of rat liver mitochondria. Febs Lett. 1997;415:341–345. doi: 10.1016/s0014-5793(97)01156-3. [DOI] [PubMed] [Google Scholar]
- Torres J, Darley-Usmer V, Wilson MT. Inhibition of cytochrome c oxidase in turnover by nitric oxide: mechanism and implications for control of respiration. Biochem J. 1995;312:169–173. doi: 10.1042/bj3120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffre A, Sarti P, D'Itri E, Buse G, Soulimane T, Brunori M. On the mechanism of inhibition of cytochrome c oxidase by nitric oxide. J Biol Chem. 2000;271:33404–33408. doi: 10.1074/jbc.271.52.33404. [DOI] [PubMed] [Google Scholar]
- Brown GC, Bolanos JP, Heales SJ, Clark JB. Nitric oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci Lett. 1995;193:201–204. doi: 10.1016/0304-3940(95)11703-y. [DOI] [PubMed] [Google Scholar]
- Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic Biol Med. 2001;25:392–403. doi: 10.1016/s0891-5849(98)00095-1. [DOI] [PubMed] [Google Scholar]
- Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med. 2001;30:463–488. doi: 10.1016/s0891-5849(00)00373-7. [DOI] [PubMed] [Google Scholar]
- Du G, Mouithys-Mickalad A, Sluse FE. Generation of superoxide anion by mitochondria and impairment of their functions during anoxia and reoxygenation in vitro. Free Radic Biol Med. 1988;25:1066–1074. doi: 10.1016/s0891-5849(98)00148-8. [DOI] [PubMed] [Google Scholar]
- Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci USA. 2001;98:14126–14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafourifar P, Schenk U, Klein SD, Richter C. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J Biol Chem. 1999;27:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- Lacza Z, Puskar M, Figueroa JP, Zhang J, Rajapakse N, Busija DW. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic Biol Med. 2001;31:1609–1615. doi: 10.1016/s0891-5849(01)00754-7. [DOI] [PubMed] [Google Scholar]
- Packer MA, Porteous CM, Murphy MP. Superoxide production by mitochondria in the presence of nitric oxide forms peroxynitrite. Biochem Mol Biol Int. 1996;40:527–534. doi: 10.1080/15216549600201103. [DOI] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Bioch Biophys. 1994;308:96–102. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- Brealey D, Brand M, Hargreaves I, Heales S, Land S, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondiral dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not its precursor, nitric oxide. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- Boczkowski J, Lisdero C, Lanone S, Samb A, Carreras MC, Boveris A, Aubier M, Poderoso JJ. Endogenous peroxynitrite mediates mitochondrial dysfunction in rat diaphragm during endotoxemia. FASEB J. 1999;13:1637–1646. doi: 10.1096/fasebj.13.12.1637. [DOI] [PubMed] [Google Scholar]
- Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283:593–596. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- Saitoh MS, Poirier GG, Lindahl T. Dual function for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry. 1994;33:7099–7106. doi: 10.1021/bi00189a012. [DOI] [PubMed] [Google Scholar]
- Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares H, Smulson ME. Involvement of PARP and poly(ADP-ribosyl)ation in the early stages of apoptosis and DNA replication. Mol Cell Biochem. 1999;193:137–148. [PubMed] [Google Scholar]
- Lautier D, Lageux J, Thibodeau J, Ménard L, Poirier GG. Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol Cell Biochem. 1993;122:171–193. doi: 10.1007/BF01076101. [DOI] [PubMed] [Google Scholar]
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Jacobson EL, Jacobson MK. Molecular heterogeneity and regulation of poly(ADP-ribose) glycohydrolase. Mol Cell Biochem. 1999;193:75–81. [PubMed] [Google Scholar]
- Schraufstatter IU, Hinshaw DB, Hyslop PA, Spragg RG, Cochrane CG. Oxidant injury of cells. DNA strand-breaks activate polyadenosine diphosphate-ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide. J Clin Invest. 1986;77:1312–1320. doi: 10.1172/JCI112436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C, Zingarelli B, O'Connor M, Salzman AL. DNA strand breakage, activation of poly-ADP ribosyl synthetase, and cellular energy depletion are involved in the cytotoxicity in macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci USA. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B, O'Connor M, Wong H, Salzman AL, Szabó C. Peroxynitrite-mediated DNA strand breakage activates poly-ADP ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J Immunol. 1996;156:350–358. [PubMed] [Google Scholar]
- Szabó C, Zingarelli B, Salzman AL. Role of poly-ADP ribosyltransferase activation in the vascular contractile and energetic failure elicited by exogenous and endogenous nitric oxide and peroxynitrite. Circ Res. 1996;78:1051–1063. doi: 10.1161/01.res.78.6.1051. [DOI] [PubMed] [Google Scholar]
- Khan AU, Delude RL, Han YH, Sappington PL, Han X, Carcillo JA, Fink MP. Liposomal NAD+ prevents diminished O2 consumption by immunostimulated Caco-2 cells. Am J Physiol. 2002;282:L1082–L1091. doi: 10.1152/ajplung.00358.2001. [DOI] [PubMed] [Google Scholar]
- Kim H, Jacobson EL, Jacobson MK. Synthesis and degredation of cyclic ADP-ribose by NAD glycohydrolases. Science. 1993;261:1330–1333. doi: 10.1126/science.8395705. [DOI] [PubMed] [Google Scholar]
- Pulido EJ, Shames BD, Selzman CH, Barton HA, Banerjee A, Bensard DD, McIntyre RC., Jr Inhibition of PARS attenuates endotoxin-induced dysfunction of pulmonary vasorelaxation. Am J Physiol. 1999;277:L769–L776. doi: 10.1152/ajplung.1999.277.4.L769. [DOI] [PubMed] [Google Scholar]
- Szabó A, Salzman AL, Szabó C. Poly (ADP-ribose) synthetase activation mediates pulmonary microvascular and intestinal mucosal dysfunction in endotoxin shock. Life Sci. 1998;63:2133–2139. doi: 10.1016/s0024-3205(99)80010-1. [DOI] [PubMed] [Google Scholar]
- Kuhnle S, Nicotera P, Wendel A, Leist M. Prevention of endotoxin-induced lethality, but not of liver apoptosis in poly(ADP-ribose) polymerase-deficient mice. Biochem Biophys Res Commun. 1999;263:433–438. doi: 10.1006/bbrc.1999.1393. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet J-C, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb RD, Marton A, Szabó E, Virag L, Salzman AL, Glock D, Akhter I, McCarthy R, Parrillo JE, Szabó C. Protective effect of a novel, potent inhibitor of poly(adenosine 5'-diphosphate-ribose) synthetase in a porcine model of severe bacterial sepsis. Crit Care Med. 2002;30:974–980. doi: 10.1097/00003246-200205000-00004. [DOI] [PubMed] [Google Scholar]