

Short abstract
The malignancy of human colorectal carcinoma is correlated with a unique expression pattern of a specific group of genes, allowing the classification of tumor tissues into two clinically distinct groups.
Abstract
Background
Individual human carcinomas have distinct biological and clinical properties: gene-expression profiling is expected to unveil the underlying molecular features. Particular interest has been focused on potential diagnostic and therapeutic applications. Solid tumors, such as colorectal carcinoma, present additional obstacles for experimental and data analysis.
Results
We analyzed the expression levels of 1,536 genes in 100 colorectal cancer and 11 normal tissues using adaptor-tagged competitive PCR, a high-throughput reverse transcription-PCR technique. A parametric clustering method using the Gaussian mixture model and the Bayes inference revealed three groups of expressed genes. Two contained large numbers of genes. One of these groups correlated well with both the differences between tumor and normal tissues and the presence or absence of distant metastasis, whereas the other correlated only with the tumor/normal difference. The third group comprised a small number of genes. Approximately half showed an identical expression pattern, and cancer tissues were classified into two groups by their expression levels. The high-expression group had strong correlation with distant metastasis, and a poorer survival rate than the low-expression group, indicating possible clinical applications of these genes. In addition to c-yes, a homolog of a viral oncogene, prognostic indicators included genes specific to glial cells, which gives a new link between malignancy and ectopic gene expression.
Conclusions
The malignancy of human colorectal carcinoma is correlated with a unique expression pattern of a specific group of genes, allowing the classification of tumor tissues into two clinically distinct groups.
Background
Gene-expression profiling is a powerful tool with which to elucidate the molecular features underlying variations in individual cancer tissues. Diagnostic and therapeutic applications are the most obvious goals, and have been the main focus of analytical efforts. For example, gene-expression analysis was used successfully to discover a new classification of diffuse B-cell lymphoma, dividing this disease into groups with different prognoses [1].
In spite of its increasing popularity, many technical and analytical aspects of gene-expression profiling are still unresolved. Solid tumors such as gastrointestinal or breast cancers are actually mixtures of cancerous and non-cancerous tissues. Changes in gene expression in the cancer cells have to be detected through RNAs mixed with those from normal tissues, requiring more accurate measurements. To get round this problem, some studies have restricted their analysis to samples that contain a minimum of non-cancerous cells [2,3]. However, this type of analysis ignores a large population of tumors, and thus requires additional evaluation to confirm its diagnostic applications.
The vast complexity of some gene-expression data also makes data analysis quite difficult. Currently, the most popular method for unveiling underlying features of gene-expression profiles is hierarchical cluster analysis [4]. Because of a lack of valid statistical evaluation methods, however, clusters are often subject to interpretation by the investigator.
In this report, we applied unique approaches to characterize colorectal cancer gene expression. Colorectal carcinoma is one of the most prevalent and well characterized human cancers, and, in spite of recent advances in diagnosis and therapeutics, is still a leading cause of death [5-8]. We report here the measurement of gene-expression levels using a high-throughput quantitative PCR system [9,10] based on adaptor-tagged competitive PCR (ATAC-PCR) [11]. ATAC-PCR utilizes unique adaptors for different cDNAs. As reverse transcription-PCR (RT-PCR) can detect alterations in gene expression with sensitivity unattainable with hybridization-based techniques [12], ATAC-PCR should allow the discovery of new molecular features of human colorectal cancers. In addition, we applied a parametric clustering method to identify clusters of expressed genes, and by this method we have identified genes linked to malignancy.
Results
Survey of expressed genes in colorectal cancer and ATAC-PCR assay
We first surveyed the genes expressed in colorectal cancer tissues using expressed sequence tag (EST) sequencing [13]. A 3' end-directed cDNA library was produced from RNAs purified from six colorectal cancers and 5,465 EST clones were sequenced; from these, we selected 1,344 genes for analysis, which were also deposited in the RefSeq database. We then designed PCR primers for 1,536 genes, including 192 other genes either known to be involved in colorectal cancer or identified as tumor-specific in previous microarray experiments [14]. Thus, this set includes only those genes that are expressed in colorectal cancers, an advantage over more universal sets, such as UniGene, which include many nonspecific genes. The expression levels of these genes in sample RNAs derived from 100 cancer and 11 normal tissues were then assayed by ATAC-PCR. In this assay, using seven adaptors, five RNA samples and two controls with different inoculated amounts are processed in a single reaction. A typical electropherogram from the DNA analyzer is shown in Figure 1. The data matrix resulting from this analysis consists of 1,536 genes × 111 tissue samples. All expression data are available as additional files (see Additional data files).
Figure 1.
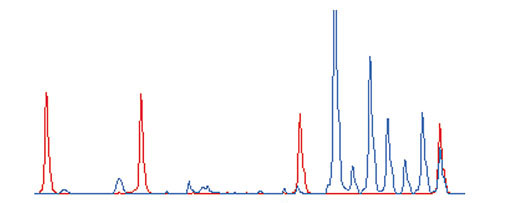
An example of an ATAC-PCR electropherogram. The height of each peak represents fluorescence intensity. Blue, PCR product signals; red, size marker signals. Starting from the left, the marker sizes are 35, 50, 75 and 100 bases. The seven blue peaks on the right correspond to ATAC-PCR products. From the left, the first peak corresponds to 10 equivalents of control cDNA, the second corresponds to two equivalents of control cDNA, and the last five peaks correspond to five equivalents of sample cDNA.
Parametric clustering of gene-expression data
To classify genes expressed in human colorectal cancers into statistically significant groups, we applied a parametric cluster analysis based on the Gaussian mixture model [15-17] and Bayes inference. This analysis enables a rigorous clustering approach, making possible the detection of global structures hidden within data matrices. The main problem faced during clustering was that of missing and saturated values included in the data matrix. Quantitative assays, such as RT-PCR and DNA microarrays, have limited dynamic ranges: values outside these ranges are not accurate. The replacement of these values with saturation values and the presence of missing values did not allow fitting of expression data to the Gaussian mixture model. Therefore, we selected 341 genes which displayed less than six saturated and less than five missing values, assuming that the data outside the measurable range in these samples would not significantly affect the statistical outcome. We assumed that the gene-expression data did not carry information requiring analysis of full dimension, and multivariate characters of the gene-expression vectors were extracted by principal component analysis.
The distribution of the first factor scores differed from normal distributions, and seemingly possessing spatial characteristics, while those of the fourth and later factor scores showed high similarity to normal distributions with little or no spatial character (Figure 2). We estimated that the multivariate nature of the gene-expression vectors could be represented by the first, second and third factor scores. In addition, for revealing a group structure, the between-cluster variation should not be dominated by within-cluster variation. Inclusion of fourth and later factor scores would blur the group structure because of dominant within-cluster variation. Parametric clustering using the variational Bayes method [18] was therefore carried out using the first three components. The analysis revealed a clear categorization of three groups, with two groups containing a large number of genes (GM-A and GM-B) and a third group containing only a small number (GM-C) (Figure 3).
Figure 2.
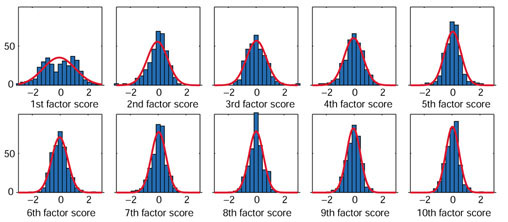
Distributions of the first to tenth factor scores obtained by principal component analysis. Vertical axis, number of genes; horizontal axis, factor scores. The interval of each column is 0.3, and the range of the central column is between -0.15 and 0.15. Red curves are normal distributions fitted to the columns. Percent variance explained by each component is as follows, from the first to the tenth: 12.33, 4.96, 4.76, 4.22, 3.28, 3.08, 2.57, 2.51, 2.27, 2.17.
Figure 3.
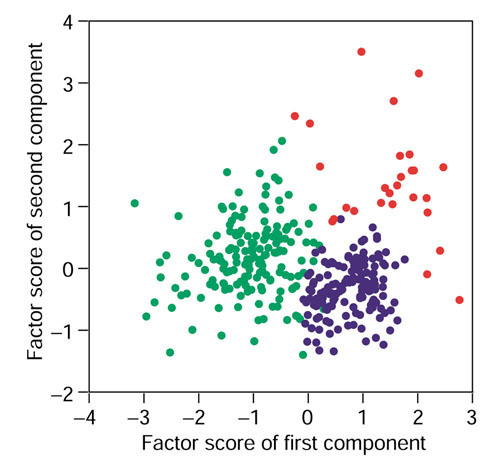
Parametric clustering of genes using the Gaussian mixture model. The dots represent a two-dimensional matrix of genes generated by principal component analysis of the gene-expression data. Horizontal axis, factor score of the first component extracted from the expression patterns of each gene; vertical axis, factor score of the second component. Green, group GM-A; blue, group GM-B; red, group GM-C.
We also carried out hierarchical cluster analysis (Figure 4) and compared the results with those of the parametric clustering. Clustering was truncated at the 88-cluster level. Groups GM-A and GM-B corresponded to two major branches of the hierarchical clustering, showing that both techniques detected a similar overarching organization. The genes from group GM-C were found to reside mainly in cluster 43. Regions lacking bottom dots were not characterized using parametric clustering. Although additional clusters in these regions correlated with differences between tumor and normal samples, these differences were not statistically evaluated, as they required further verification with additional samples.
Figure 4.
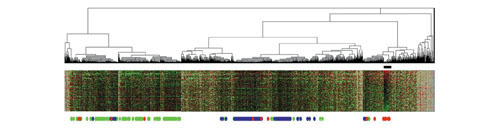
Hierarchical cluster analysis of the genes on the basis of expression patterns. A total of 1,536 genes are aligned horizontally. One hundred cancer and 11 normal tissue samples are vertically aligned in the same order as in Figure 6a. The bottom dots indicate genes grouped by parametric clustering. Green, group GM-A; blue, group GM-B; red, group GM-C. The black bar corresponds to cluster 43.
Characteristics of groups GM-A and -B
Genes selected by supervised methods, such as selection based on correlation with a clinical parameter, include those universally correlated with the parameter and those correlating only within the analyzed sample set. To avoid the uncertainty inherent in supervised methods, we examined the correlations between clinical phenotypes and gene groups instead of individual genes. We devised a correlation ratio (CR) to serve as an indicator for correlation with clinical parameters. Genes were first sorted by CR value order, and then the CRs of the original total dataset were compared with those of permuted data (Figure 5). In group GM-A, the CRs were significantly higher than those of the total dataset for both the differences between tumor and normal tissues and the presence or absence of distant metastases (Figure 5a,c). In contrast, the GM-B group possessed a high CR only for the difference between tumor and normal tissues (Figure 5b,b). For these parameters, the CR values of the total dataset were consistently higher throughout the full range of CRs, suggesting the correlation was not restricted to a small number of genes, but was a global character of each group (Figure 5a,b,c). We could not identify significant correlations for other parameters, including lymph-node metastases (Figure 5e,f) and histological type (data not shown).
Figure 5.
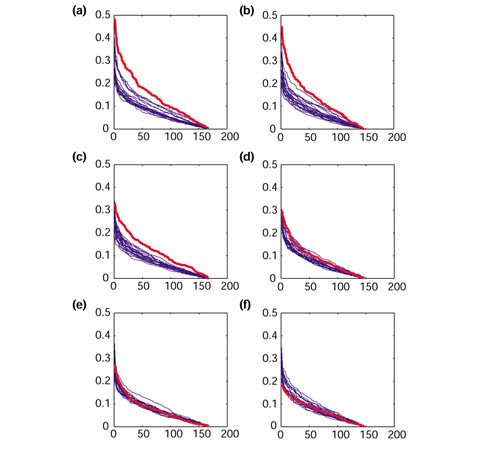
Correlation of gene expression with cancer phenotype. The vertical axis represents the correlation ratio (CR) of the differences between tumor and normal tissues in (a) group GM-A and (b) GM-B; or the presence or absence of distant metastasis in (c) GM-A and (d) GM-B; or lymph node metastasis in (e) GM-A and (f) GM-B. The horizontal axis represents the genes sorted by CR. Red, original data; blue, trials of permuted data.
The GM-C group contains genes linked to malignancy
With the GM-C group, which contained only a small number of genes, the correlations were analyzed differently. Approximately half of these members, named TCL (tumor-classifier) genes, had identical expression patterns. The average expression levels of 12 representative genes (listed in Table 1) were used as a guideline for the separation of the samples into two subpopulations, with group 1 displaying high expression and group 2 low expression.
Table 1.
List of TCL (tumor-classifier) genes
| GS number | Accession number | Symbol | Annotation |
| GS2892 | NM_004368 | CNN2* | Homo sapiens calponin 2 (CNN2), mRNA |
| GS3019 | NM_003348 | UBE2N* | Homo sapiens ubiquitin-conjugating enzyme E2N (homologous to yeast UBC13) (UBE2N) |
| GS3386 | NM_003337 | UBE2B* | Homo sapiens ubiquitin-conjugating enzyme E2B (RAD6 homolog) (UBE2B), mRNA |
| GS3387 | NM_013317 | hT1a-1* | Homo sapiens hT1a-1 (hT1a-1), mRNA |
| GS3588 | AF131848 | * | Homo sapiens clone 24922 mRNA sequence, complete coding sequence |
| GS4015 | NM_005433 | YES1* | Homo sapiens v-yes-1 Yamaguchi sarcoma viral oncogene homolog 1 (YES1), mRNA |
| GS4022 | NM_002433 | MOG* | Homo sapiens myelin oligodendrocyte glycoprotein (MOG), mRNA |
| GS4163 | AC007565 | * | Homo sapiens chromosome 19, cosmid R27656, complete sequence |
| GS4780 | AD001530 | * | Homo sapiens XAP-5 mRNA, complete coding sequence |
| GS4941 | NM_016380 | * | Homo sapiens differentiation-related protein dif13 (LOC51212), mRNA |
| GS4945 | NM_016343 | CENPF* | Homo sapiens centromere protein F (350/400 kD, mitosin) (CENPF), mRNA |
| GS4946 | NM_002439 | MSH3* | Homo sapiens mutS (E. coli) homolog 3 (MSH3), mRNA |
| GS3170 | L35240 | † | Human enigma gene, complete coding sequence |
| GS715 | AL096800 | † | Human DNA sequence from clone RP1-303A1 on chromosome 6 |
| GS3002 | AL023806 | STM2† | Human DNA sequence from clone 466P17 on chromosome 6q24 |
| GS1102 | Y18000 | NF2† | Homo sapiens NF2 gene |
| GS5239 | AL139229 | † | Human DNA sequence from clone RP4-540A13 on chromosomeXq22.1-22.3 |
| GS4947 | NM_018520 | Homo sapiens hypothetical protein PRO2268 (PRO2268), mRNA | |
| GS1341 | AC006165 | Homo sapiens clone UWGC:y54c125 from 6p21, complete sequence | |
| GS4512 | NM_005768 | C3F | Homo sapiens putative protein similar to nessy (Drosophila) (C3F), mRNA |
| GS4501 | AF261689 | Homo sapiens DNA polymerase epsilon p17 subunit gene, complete coding sequence | |
| GS6969 | AL022316 | Human DNA sequence from clone CTA-126B4 on chromosome 22q13.2-13.31 | |
| GS6493 | AF113695 | Homo sapiens clone FLB5224 PRO1365 mRNA, complete coding sequence | |
| M15990 | M15990 | yes | yes |
*Group GM-C genes used in the experiment detailed in Figure 3. †Additional genes in group C.
We then examined these gene-expression patterns for possible correlations with distant metastases. Group 1 consisted of 21 metastatic cases and 27 non-metastatic cases, whereas group 2 consisted of 7 metastatic cases and 44 non-metastatic cases (Figure 6a). Thus, distant metastasis was significantly correlated with elevated expression of the TCL genes (Fisher exact test, p < 0.01). Normal tissues, like the group 2 samples, displayed low levels of TCL gene expression.
Figure 6.
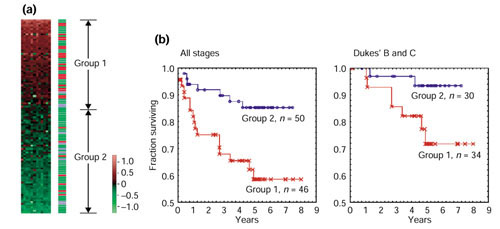
Expression of TCL (tumor-classifier) genes was correlated with the malignant potential of colorectal cancer. Results used the 100 cancer and 11 normal tissues. (a) Correlation with distant metastasis. We aligned the 12 genes listed at the top of Table 1 from left to right horizontally. All samples were then sorted vertically by the average of the gene-expression levels of the 12 genes. The border of groups 1 and 2 is set at 0. The right bar indicates remote metastasis status, with red, green and blue representing remote metastasis positive, remote metastasis negative, and normal tissues, respectively. (b) Kaplan-Meier plot analysis of groups 1 and 2. Vertical axis, fraction of survival; horizontal axis, survival time in years. The groups of patients analyzed consisted of either all stages (left) or Dukes' B and C stages only (right). All the expression data and relevant clinical information are available as additional data files.
Kaplan-Meier plotting revealed significant differences in survival rates between group 1 and group 2, with a 5-year survival rate of 57.6% for group 1 and 85.7% for group 2 (p < 0.01) (Figure 6b). Currently, cancer stage classification is the most diagnostically informative practice in clinical medicine. Patients classified as Dukes' A have good prognosis, whereas those ranked as Dukes' D have poor prognosis. Dukes' B and C are intermediate stages for which risk assessment is more difficult [19]. The Kaplan-Meier analysis was carried out on the Dukes' B and C patients, revealing a significant difference in the 5-year survival rates between patients in groups 1 and 2, which were 69.9 and 93.5%, respectively (p < 0.05) (Figure 6c).
Additional TCL genes were recovered from cluster number 43 from the hierarchical cluster analysis, including some absent from the GM-C group. These genes are listed in Table 1.
Discussion
A recent study indicated that RT-PCR could detect changes in gene expression that were missed by microarrays [12]. The difficulty of constructing calibration curves, however, has prevented the implementation of quantitative PCR for high-throughput analysis. ATAC-PCR solves this problem by including control samples within a single reaction tube [20]. In addition, because we used native RNA as a control, quantitation is most accurate around physiological concentrations. Thus, the technique is ideal for detecting small differences between samples, as is necessary for the analysis of solid tumors. In addition, ATAC-PCR requires much smaller amounts of RNA than microarray analysis, a crucial advantage when dealing with clinical samples.
The main focus of this study was to discover genes whose expression in colon cancer samples correlated with clinical parameters. There are two major approaches to this problem - supervised [21-23] and unsupervised [1,24] - and both have weaknesses. In the supervised approach, genes are collected that might display correlations between their expression and various clinical parameters. Diagnostic methods are then constructed using the collected genes to confirm or deny these hypothetical correlations, and answers are only obtained after validation with an external dataset. In addition, such diagnostic systems are usually too complicated to be easily accessible to clinicians.
By unsupervised methods, clusters are first found that contain groups of genes or samples which share common gene-expression patterns in a given gene-expression data matrix. Then, correlations with clinical parameters are explored for each cluster. This approach is similar to that of pathology, where the aim is to determine morphological classifications and then determine how those classifications correlate with clinical data. The main difficulty of the unsupervised approach to gene-expression analysis lies in the identification of statistically valid clusters.
For example, hierarchical cluster analysis allows the branches of a dendrogram to be swapped at any level of stratification. Consequently, 2n-1 alignments can describe a clustering of n cases. As there are no established methods to determine either the optimal alignment or the optimal cluster number, statistical evaluation of proposed cluster models can be very difficult. This is a problem common to other methods, such as k-means clustering and self-organizing mapping (SOM). Models are therefore usually subject to the interpretation of individual scientists.
Parametric cluster analysis based on probability models offers a solution to the above problem. With the underlying probability model, determining the number of clusters and their structures becomes a statistical problem, and we have solved this problem by a method based on the Gaussian mixture model and Bayes inference, using the variational Bayes method [18]. The main features of our method are as follows.
First, conventional clustering methods, including hierarchical, k-means, SOM and probabilistic mixture models trained by the EM algorithm [14] often produce unstable results depending on initial parameters and conditions. The variational Bayes method produces stable clustering, partly because the algorithm does not have a strong dependence on initial parameters.
Second, the free energy (see Materials and methods) approximates the log marginal likelihood, which represents the fitness (likelihood) of the model structure to the given data. On this basis we can reliably produce a model with an appropriate number of clusters.
Third, clusters with non-hyperspherical shapes can be represented. Conventional methods assume spherical shapes for clusters, but this assumption may not be appropriate. As we define a mixture of full-covariance Gaussian distributions, our method can accommodate clusters with hyperelliptical shapes and oblique axes (for example, GM-C in Figure 3).
By this method, we have successfully identified groups of genes whose expression is correlated with clinical parameters, an important step towards the molecular classification of cancer. Heuristic methods may be suited for biological problems, because the identification of a large number of possible clusters is advantageous for hypothesis generation. In contrast, identification of statistically valid clusters is of the highest priority for cancer classification, because the intention is to apply these classifications to future clinical samples. In particular, Bayes inference may have an advantage, because the generalization ability of Bayesian predictive distributions tends to exceed that of the maximum likelihood [25].
Because parametric cluster analysis is based on the assumption that the data are distributed according to a mixture of Gaussian distributions, we excluded genes which possibly violate this assumption. The excluded genes would contain additional members of the three groups identified by the parametric clustering, and some of the genes may constitute small clusters of possible biological and clinical interest. Expanding the dynamic range of the ATAC-PCR assay may help to settle this problem. The dynamic range depends on the number and inoculated amounts of the control cDNAs, and additional assays with different amounts of control cDNA should serve to expand the dynamic range.
We prioritized clustering of genes, not tissues, because biological interpretation of gene clusters is easier than that of tissue clusters. The TCL genes include several genes possessing clear relationships with malignancy, c-yes [26], the human homolog of v-yes, a Yamaguchi sarcoma virus oncogene, is one such example. Although c-yes, a member of the tyrosine kinase oncogene family, exhibits elevated expression in a subpopulation of colorectal carcinomas, its relationship with prognosis has not been well characterized [27]. Overexpression of c-yes maybe associated with active proliferation of cancer cells. In addition, two ubiquitin-conjugating enzymes discovered during this analysis may be involved in protein degradation during anoxia-induced cancer-cell death stemming from rapid growth of peripheral cells. Ectopic gene expression in cancer tissues, such as the frequently observed expression of adrenocorticotropic hormone (ACTH) in lung cancer cells [28], can occasionally evoke serious symptoms in cancer patients. In the absence of symptoms, however, ectopic gene expression is rarely identified. Genes for myelin oligodendrocyte glycoprotein [29] and NF2 [30], which are specific to oligodendroglia and Schwann cells, were identified as TCL genes in our screen. This observation suggests that there may be a unique link between malignancy and ectopic gene expression. Further studies, however, will be required to clarify whether additional genes with the TCL expression pattern or the ectopically expressed genes themselves are responsible for the malignant phenotype.
Unlike other diagnosis-oriented studies using supervised approaches [2,22,23], TCL genes were not selected for metastatic or prognostic properties. The diagnostic conclusions obtained by such studies using such properties were optimized for the cancer populations selected for the analysis, and thus may not be as effective with future samples [31]. In addition, gene-expression profiles reveal only the intrinsic properties of cancer tissues; treatment decisions in clinical practice are made using many factors surrounding individual patients. Stage classification, an integration of clinical parameters, has the most important role in therapeutic decisions. New methods that improve the current diagnostic schemes based on stage classifications should continually be evaluated. Thus, this novel molecular classification might serve as a potential candidate for advancing diagnostic determinations.
Although it is not common to describe 'diseases of gene expression', it is not inappropriate to define diseases by gene-expression patterns, as these reflect the intrinsic properties of tissues better than other parameters. It may be too early to discuss whether the molecular grouping should be treated as either one of the clinical parameters or a crucial factor defining disease entities. It is crucial to identify additional TCL genes and clarify their functional relationships with tumor characteristics. It is also important to explore correlation with chromosomal abnormalities. We expect, however, that TCL genes will eventually define a new categorization scheme for colorectal cancer, facilitating better understanding of disease etiology and development of therapies.
Materials and methods
Samples
A total of 100 Japanese primary colorectal cancer specimens and corresponding normal colonic epithelial specimens were obtained from surgical resections during a period from April 1994 to September 2000. All tumor patients were diagnosed at advanced stages. All normal tissues were histopathologically confirmed to be free of cancer cells. None of the patients was treated pre-operatively with either chemotherapy or radiotherapy. Patient prognosis was followed for a median of 41.2 months. Additional specimens were snap frozen in liquid nitrogen and stored at -80°C until use. These resected samples were used appropriately according to the guidelines of the ethics committee of Osaka University.
ATAC-PCR assay
Total RNA was purified from clinical samples using Trizol reagent (Invitrogen). A 3'-end cDNA library was constructed using a mixture of RNA from six malignant samples. Utilizing software developed in our laboratory, we designed PCR primers for ATAC-PCR. Reactions were carried out as described previously [20]. Briefly, seven adaptors were used, two of which were assigned to a mixture of 10 malignant tissues, including those used to create the library. Each reaction mixture contained 10 equivalents of control cDNA with the smallest adaptor, two equivalents of control cDNA with the second smallest adaptor, and five equivalents of each sample, where one equivalent is the amount of cDNA template corresponding to 1.2 ng total RNA. Amplified products were separated using an ABI 3700 DNA analyzer. We then calculated the relative expression levels as compared to the control. The accuracy of this technique, comparable to other RT-PCR methods such as real-time PCR, has been established through studies at the Nara Institute of Science and Technology, often with confirmation by real-time PCR [10].
Data preprocessing
The data matrix was first normalized by the median of the cases. Values greater than 20 and less than 0.05 were truncated to 20 and 0.05 respectively, because reliable quantitation is only obtained within this range. All data were then converted to logarithmic scale, base 10. After selection of genes for parametric clustering, the missing values were estimated by the k-nearest neighbor method [32], where k = 15. This k-value was empirically determined by trials to obtain optimum estimation of missing values artificially introduced into the dataset of the 785 genes having no missing values.
Parametric cluster analysis
A probabilistic generative model for an L-dimensional gene factor vector y is defined as a Gaussian mixture model:
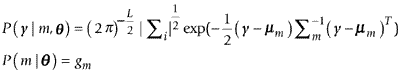
P(y | m, θ) is the probability distribution of the mth component, and is an L-dimensional Gaussian distribution with a mean μm and a covariance matrix Σm. gm is the mixing rate parameter, satisfying gm ≥ 0 and 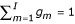 . θ is a parameter of the Gaussian mixture model, and is defined as θ = {(gm, μm, Σm) | m = 1,..., I}. The index of the components, m, is a hidden variable. I is the number of clusters.
. θ is a parameter of the Gaussian mixture model, and is defined as θ = {(gm, μm, Σm) | m = 1,..., I}. The index of the components, m, is a hidden variable. I is the number of clusters.
Our parametric clustering method is based on determination of the posterior distribution of parameters and hidden variables by the Bayes inference. With a certain value of I, the posterior distribution P(H, θ | Y) may be estimated according to the Bayes theorem:
![]()
where P0(θ), Y and H are the prior distribution of parameters, a set of gene-factor vectors defined as Y = {y1, ..., yN}, and a set of hidden variables, respectively. The normalization term, P(Y), is the marginal likelihood.
Because of the need to integrate over the parameters, computations of the Bayes inference can seldom be performed exactly, and approximation is necessary. We applied the variational Bayes method [18]. To approximate the posterior distribution, a trial posterior distribution, Q(H, θ), is prepared, and a free energy is defined as follows:
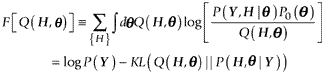
where KL(· || ·) is the Kullback-Leibler (KL) divergence between two probability distributions. In the variational Bayes method, iterative modulation of the trial posterior distribution to yield the true posterior distribution is achieved by maximizing the free energy, F [Q(H, θ)]. It should be noted that maximizing the free energy is equivalent to minimizing the KL divergence, because log P(Y) does not depend on Q(H, θ).
We assumed independence in the trial posterior distribution: Q(H, θ) = Q(H)Q(θ), and used a conjugate prior distribution P0(θ). Using various values of I, the maximum free energy was obtained by an efficient iteration algorithm similar to the expectation-maximization (EM) algorithm used in the maximum likelihood inference. The I value with the largest maximum free energy was then selected. After obtaining the posterior distributions, Q(H) and Q(θ), clustering was performed to classify the ith gene by
![]()
In our case, L = 3 and I = 3.
Correlation analysis
The correlation ratio of gene i is defined by the following equation.
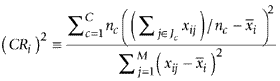
where nc is the number of genes in a particular class Jc; xi,j is the expression level of gene i in sample j; and 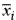 is the average expression level of gene i. A more detailed description of the parametric cluster and correlation analyses may be obtained from S.I. upon request.
is the average expression level of gene i. A more detailed description of the parametric cluster and correlation analyses may be obtained from S.I. upon request.
Other statistical analysis
For hierachical cluster analysis and survival analysis, the data matrix was normalized to the median of the cases and then to the median of the individual genes. Subsequently, data preprocessing was conducted in the same way as for the parametric cluster analysis. Hierarchical cluster analysis was performed using Ward's method with ClustanGraphics software [33]. The significant cluster level was determined by bootstrap validation. Survival analysis was performed using STATISTICA 6.1J software (StatSoft). No clinical parameter biases, other than distant metastasis status, were detected for molecular groups 1 and 2.
Additional data files
All the gene-expression data (1536 genes × 111 samples) and the annotation of the assayed genes are available as an Excel file and an accompanying Word file. An Excel data file for Figure 6 lists the gene-expression data of the 12 GM-C genes. Gene-expression data are those normalized for hierarchical cluster analysis as described in the Materials and methods section.
Supplementary Material
Gene-expression data and the annotation of assayed genes
An accompanying Word file to the gene-expression data and annotation of assayed genes
An Excel data file for Figure 6 listing the gene-expression data of the 12 GM-C genes
Acknowledgments
Acknowledgements
We thank Satoko Maki and Keiko Miyaoka for technical assistance and Kenta Mitsuhara for database management. This work was supported by a Grant-in-Aid for the Development of Innovative Technology from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–535. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Fahy B, Bold RJ. Epidemiology and molecular genetics of colorectal cancer. Surg Oncol. 1998;7:115–123. doi: 10.1016/s0960-7404(99)00021-3. [DOI] [PubMed] [Google Scholar]
- Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci USA. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterman DA, Alon U, Sierk AJ, Levine AJ. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001;61:3124–3130. [PubMed] [Google Scholar]
- Matoba R, Saito S, Ueno N, Maruyama C, Matsubara K, Kato K. Gene expression profiling of mouse postnatal cerebellar development. Physiol Genomics. 2000;4:155–164. doi: 10.1152/physiolgenomics.2000.4.2.155. [DOI] [PubMed] [Google Scholar]
- Iwao K, Matoba R, Ueno N, Ando A, Miyoshi Y, Matsubara K, Noguchi S, Kato K. Molecular classification of primary breast tumors possessing distinct prognostic properties. Hum Mol Genet. 2002;11:199–206. doi: 10.1093/hmg/11.2.199. [DOI] [PubMed] [Google Scholar]
- Kato K. Adaptor-tagged competitive PCR: a novel method for measuring relative gene expression. Nucleic Acids Res. 1997;25:4694–4696. doi: 10.1093/nar/25.22.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland MJ. Transcript abundance in yeast varies over six orders of magnitude. J Biol Chem. 2002;277:14363–14366. doi: 10.1074/jbc.C200101200. [DOI] [PubMed] [Google Scholar]
- Matoba R, Kato K, Saito S, Kurooka C, Maruyama C, Sakakibara Y, Matsubara K. Gene expression in mouse cerebellum during its development. Gene. 2000;241:125–131. doi: 10.1016/s0378-1119(99)00457-6. [DOI] [PubMed] [Google Scholar]
- Takemasa I, Higuchi H, Yamamoto H, Sekimoto M, Tomita N, Nakamori S, Matoba R, Monden M, Matsubara K. Construction of preferential cDNA microarray specialized for human colorectal carcinoma: molecular sketch of colorectal cancer. Biochem Biophys Res Commun. 2001;285:1244–1249. doi: 10.1006/bbrc.2001.5277. [DOI] [PubMed] [Google Scholar]
- McLachlan GJ, Peel D. Finite Mixture Models. New York: Wiley; 2000. [Google Scholar]
- Yeung K-Y, Fraley C, Murua A, Raftery AE, Ruzzo WL. Model-based clustering and data transformations for gene expression data. Bioinformatics. 2001;17:977–987. doi: 10.1093/bioinformatics/17.10.977. [DOI] [PubMed] [Google Scholar]
- McLachlan GJ, Bean RW, Peel D. A mixture model-based approach to the clustering of microarray expression data. Bioinformatics. 2002;18:413–422. doi: 10.1093/bioinformatics/18.3.413. [DOI] [PubMed] [Google Scholar]
- Attias H. A variational Bayesian framework for graphical models. In: Solla SA, Leen TK, Müller K-R, editor. In Advances in Neural Information Processing Systems 12. Cambridge: MIT Press; 2000. pp. 206–212. [Google Scholar]
- McLeod HL, Murray GI. Tumor markers of prognosis in colorectal cancer. Brit J Cancer. 1999;79:191–203. doi: 10.1038/sj.bjc.6690033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba R, Kato K, Kurooka C, Maruyama C, Sakakibara Y, Matsubara K. Correlation between gene functions and developmental expression patterns in the mouse cerebellum. Eur J Neurosci. 2000;12:1357–1371. doi: 10.1046/j.1460-9568.2000.01034.x. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumors. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Oba S, Sato M, Takemasa I, Monden M, Matsubara K, Ishii S. Missing value estimation using mixture of PCAs. In: Dorronsoro JR, editor. In Artificial Neural Networks - ICANN LNC2415. New York: Springer; 2002. pp. 492–497. [Google Scholar]
- Sukegawa J, Semba K, Yamanashi Y, Nishizawa M, Miyajima N, Yamamoto T, Toyoshima K. Characterization of cDNA clones for the human c-yes gene. Mol Cell Biol. 1987;7:41–47. doi: 10.1128/mcb.7.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licato LL, Brenner DA. Analysis of signaling protein kinases in human colon or colorectal carcinomas. Dig Dis Sci. 1998;43:1454–1464. doi: 10.1023/a:1018894227169. [DOI] [PubMed] [Google Scholar]
- Terzolo M, Reimondo G, Ali A, Bovio S, Daffara F, Paccotti P, Angeli A. Ectopic ACTH syndrome: molecular bases and clinical heterogeneity. Ann Oncol. 2001;12 Suppl 2:S83–S87. doi: 10.1093/annonc/12.suppl_2.s83. [DOI] [PubMed] [Google Scholar]
- Pham-Dinh D, Allinquant B, Ruberg M, della Gaspera B, Nussbaum JL, Dautigny A. Characterization and expression of the cDNA coding for the human myelin/oligodendrocyte glycoprotein. J Neurochem. 1994;63:2353–2356. doi: 10.1046/j.1471-4159.1994.63062353.x. [DOI] [PubMed] [Google Scholar]
- Trofatter JA, Maccollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DN, Eldridge R, Kley N, Menon AG, Pulaski K, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- Ambroise C, McLachlan GJ. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc Natl Acad Sci USA. 2002;99:6562–6566. doi: 10.1073/pnas.102102699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanskaya O, Cantor M, Sherlock G, Brown PO, Hastie T, Tibshirani R, Botstein D, Altman RB. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- Clustan http://www.clustan.com
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene-expression data and the annotation of assayed genes
An accompanying Word file to the gene-expression data and annotation of assayed genes
An Excel data file for Figure 6 listing the gene-expression data of the 12 GM-C genes