

Abstract
Background
Using suitable error models for gene expression measurements is essential in the statistical analysis of microarray data. However, the true probabilistic model underlying gene expression intensity readings is generally not known. Instead, in currently used approaches some simple parametric model is assumed (usually a transformed normal distribution) or the empirical distribution is estimated. However, both these strategies may not be optimal for gene expression data, as the non-parametric approach ignores known structural information whereas the fully parametric models run the risk of misspecification. A further related problem is the choice of a suitable scale for the model (e.g. observed vs. log-scale).
Results
Here a simple semi-parametric model for gene expression measurement error is presented. In this approach inference is based an approximate likelihood function (the extended quasi-likelihood). Only partial knowledge about the unknown true distribution is required to construct this function. In case of gene expression this information is available in the form of the postulated (e.g. quadratic) variance structure of the data.
As the quasi-likelihood behaves (almost) like a proper likelihood, it allows for the estimation of calibration and variance parameters, and it is also straightforward to obtain corresponding approximate confidence intervals. Unlike most other frameworks, it also allows analysis on any preferred scale, i.e. both on the original linear scale as well as on a transformed scale. It can also be employed in regression approaches to model systematic (e.g. array or dye) effects.
Conclusions
The quasi-likelihood framework provides a simple and versatile approach to analyze gene expression data that does not make any strong distributional assumptions about the underlying error model. For several simulated as well as real data sets it provides a better fit to the data than competing models. In an example it also improved the power of tests to identify differential expression.
Background
An analysis of gene expression data typically includes the application of some multivariate statistical techniques such as clustering, classification, PCA etc. These high-level procedures all require the assumption of a low-level error model for the data. In practice, this model is often only specified implicitly rather than explicitely. Nevertheless, its choice has a great impact on subsequent statistical considerations.
In the case of array data, the error model characterizes the variability of gene expression intensity measurements [1]. It is essential, e.g., to get a measure of precision of an estimated expression level, to statistically evaluate the difference between treatment and control samples, to calibrate and normalize data sets using regression techniques, to increase prediction accuracy in classification, and to assess confidence in high-level analysis (e.g. clustering). Some of these applications may be robust against a misspecified low-level model but most will not. Thus, a careful choice of a suitable error model is warranted.
The observed intensity IP at a microarray probe may be decomposed into
IP = IT + IS + error, (1)
where IT denotes the true signal (foreground, due to the transcript) and IS is the stray signal (background, not due to the transcript) [2]. On this original scale IT is approximately linearly proportional to the true transcript concentration [2-4]. Distributional assumptions for the error term in Equation 1 used in the literature include
• a normal foreground and background (e.g. [5]),
• a Gamma foreground with unspecified background [6],
• a log-normal foreground and normal background, [1]
• a log-normal foreground and background [7], and
• an asinh-normal model [8,9].
At present, as there currently is no generally accepted mechanistic or empirical model for gene expression measurements, there is no agreement which of these suggested error models comes closest to the truth (except perhaps that a normal model on the observed scale can be ruled out). Moreover, all of the models are fairly difficult to distinguish statistically for the small sample size present in microarray data.
Because of this difficulty to choose among simple parametric error models for the observed probe intensities two other alternative ways to describe the error term in Equation 1 have been explored in the literature. First, it sometimes is possible to obtain a fully non-parametric estimate of the underlying error distribution (e.g. [10]). However, the drawback of completely empirical error models is that they generally require quite a lot of data, i.e. many measurements per gene and condition, and more than are usually available. In addition, non-parametric approaches are prone to overfitting and ignore known prior structural information on the data.
A second widely pursued alternative is to try to find a transformation of the data to a different scale where the error term follows a normal distribution and where the variance is constant and intensity independent. For gene expression data, this can often be achieved, at least approximately, using the log-transform or some other related function [8,9,11-13]. Thus, in this perspective the problem of finding a suitable error model is equivalent to the problem of choosing an appropriate transformation. Note, however, that an ideal scale combines ease of interpretation, constancy of variance variance, normal errors, and additivity of systematic effects. Unfortunately, these properties cannot in general be achieved simultaneously [14]. In particular, if a non-linear transformation such as the log-function is applied to the data, the expectation of the transformed intensity is not anymore a linear measure of the transcript concentration.
Approximate Error Models
In this paper, the use of approximate semi-parametric error models, rather than parametric or or nonparametric models, is advocated for gene expression data. In particular, the quasi-likelihood framework is considered that allows statistical analysis even when the knowledge of the underlying error distribution is incomplete. Applied to gene expression analysis, this approach allows to model the data while at the same time avoiding strong assumptions about the underlying distribution and the optimal scale.
In the next sections the utility of these approximate error models for gene expression data is explored. First, the general quasi-likelihood theory is introduced. Subsequently, a suitable quasi-likelihood function for gene expression data is derived. Then simulated and real data are analyzed. Finally, some conclusions concerning low-level models and transformations for gene expression data are drawn.
Results and Discussion
Quasi-Likelihood Framework
Quasi-likelihood (QL) is a framework for statistical modeling that employs an approximate likelihood function rather using than a fully specified likelihood. The advantage of this approach is that no probability structure has to be specified, as the estimating function is constructed from the first two moments only. This is a useful strategy for dealing with non-normal multivariate data (e.g. [15], chap. 14). Note that microarray data are non-normal and multivariate.
The original quasi-likelihood idea goes back to Wedderburn [16] who employed it in a regression setting. He introduced
![]()
as the quasi-log-likelihood function for independent observations yi with expectations E(yi) = μi and variances var(yi) = V(μi) where V is some known function. In a regression problem μi usually depends on a linear predictor xB via a link function g by μi = g-1 (xB). Wedderburn showed that with respect to μi and the regression coefficients B the function Q(μi;yi) has properties similar to those of the log-likelihood [16].
However, the original quasi-likelihood as in Equation 2 cannot be used to infer free parameters θ contained in a variance function Vθ(u). To fix this, Nelder and Pregibon [17] suggested to use the extended quasi-log-likelihood function
![]()
Mean parameters that maximize the original QL function also maximize Q+(μi,θ;yi) but variance parameters θ, as well as parameters in the link function g, can now also be estimated by maximizing the extended version.
QL and extended QL estimators have desirable finite sample and asymptotic statistical properties [18-21]. They are closely related to saddlepoint approximations from exponential families [22,23]. As a result, maximum quasi-likelihood estimates coincide frequently with inferences from an exact likelihood. For example, for V(μ) = σ2 the extended quasi-log-likelihood Q+(μi,σ2;yi) is exactly the normal log-likelihood (see Table 1).
Table 1.
Examples for extended quasi-log-likelihood functions
| V(μ) | Q+ | Comment |
| σ2 | normal distribution | |
| μσ2 | Poisson distribution (σ2 = 1) | |
| μ2σ2 | approx. Gamma distribution (α = 1/σ2, β =μσ2) |
|
| (μ - β)2σ2 + ρ2 | 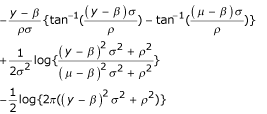 |
this paper |
Gene Expression Variance Structure and Quasi-Likelihood Function
A model in the quasi-likelihood framework requires only knowledge of the relationship between the mean and the variance, i.e. the function V in Equation 3.
Despite the uncertainty with regard to the exact form of the error distribution for gene expression measurements there is some agreement about the variance structure V. In the two-component model for the measured intensity (see Equation 1) let the expectations be E(IP) = E(IT) + E(IS) or μ = E(IT) + β. If the variance of the stray signal is assumed to be constant (var(IS) = ρ2) and the true signal assumed to exhibit a constant coefficient of variation σ (so that var(IT) = E(IT)2σ2), then the total overall variance structure is
var(IP) = (μ - β)2σ2 + ρ2 := V(μ; β, σ, ρ). (4)
Such a quadratic variance-mean relationship is observed in a lot of microarray data (e.g. [1,8,9,12,13]) and references therein), and therefore also assumed in the following. However, any other appropriate variance function could be used equally well in the quasi-likelihood framework.
From the variance function Equation 4 the extended quasi-log-likelihood function can be computed using Equation 3, resulting in
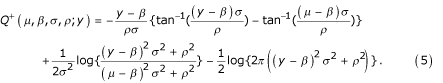
This function constitutes the approximate error model used in this paper. Note again that it is derived solely from the putative variance structure of gene expression data (Equation 4) with no further assumptions. Point estimates of the mean and variance parameters (μ, β, σ, ρ) are obtained by maximizing this function.
Ideally one would like to estimate one set of these parameters for each gene and condition separately. However, this is feasable only if there are a lot of replications, otherwise a parameter reduction is advised. Typically, the estimate of the coefficient of variation σ can be shared across all genes and conditions [24]. Similar parameter reduction may be applied to the estimates of ρ and β.
The quasi-likelihood approach also allows the estimation of approximate confidence intervals for the estimated parameters (for example θ). One way is to employ the profile quasi-likelihood Q+(θ) = Q+(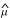 i, θ; yi), where i is optimized for fixed θ, to construct the interval
i, θ; yi), where i is optimized for fixed θ, to construct the interval
{θ, 2Q+ (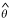 ) - 2Q+ (θ) ≤ d}.
) - 2Q+ (θ) ≤ d}.
The estimate maximizes Q+(θ) and the threshold d may be chosen as some percentage point of a χ2 distribution (for a one-parameter approximate 95% confidence interval d = 3.84). Alternatively, a variety of standard bootstrap procedures are applicable to construct confidence intervals for the quasi-likelihood point estimates [17]. However, the bootstrap intervals tend to be more conservative (i.e. wider) than the above likelihood-based interval.
Effect of Calibration
Prior to any analysis the raw microarray data generally need to be calibrated (or normalized). This also affects the error model. If linear location-scale transformation y' = a + by is assumed for each chip, see e.g. [25-27], then the transformed variance structure is
E(I'P) = E(a + bIP) = a + bμ = μ'
var(I'P) = (μ' - a - bβ)2σ2 + (bρ)2
Thus, for uncalibrated data the background parameter β is confounded with the shift and scale parameters a and b, while the background error ρ is confounded with scale parameter b. The coefficient of variation a is not affected by the transformation.
It may be desired to estimate calibration parameters a and b in addition to the error model itself. In this case, however, it is necessary to assume that β and ρ are shared parameters across all chips or channels. It is unclear, however, whether this is a realistic assumption for real data. Note, however, that this is the basis for estimating a and b in the transformation-based approach by Huber et al. [8].
Simulation Study
To explore the adequacy of the quasi-likelihood approximate error model for gene expression data a simulation study was performed.
Data were generated according to three different schemes. First, as true error model a convolution of normal and log-normal distribution was assumed [1]. As a second model an asinh-normal (ANL) distribution was assumed [8,9]. Note that in these two cases the approximate error model provided by quasi-likelihood is misspecified as both distributions are not part of the exponential family. As third true error model a Gamma distribution was considered [6].
The simulated whole-chip data consisted of 7000 genes, with the coefficient of variation set to σ = 0.25, the background parameters set to β = 25000 and ρ = 5000. The 7000 true expression levels μi - β were drawn randomly from a log-normal distribution (with log-mean 8 and standard deviation 2). These values were chosen to match the molecular data analyzed in [1]. Data y from the convolution model and the Gamma model were generated directly on the observed scale. To generate data y from the asinh-normal distribution, data x were drawn from a normal distribution N(u, s2) and subsequently transformed to the observed scale via y = asinh(a + bx). Note that this is possible because there exists a one-to-one mapping of the parameters on the normal scale (u, s2, a, b) and those of the transformed scale (μ, σ, β, ρ), see Table 2. For each variant 4, 10 and 20 replicates per gene were drawn. Figure 1 shows the observed mean-variance relationship of an example with 10 replicates per gene.
Table 2.
Parameter mapping in the simulation study
| observed scale y | normal scale x |
| μ = ( |
|
| σ2 = |
s2 = log(1 + σ2) |
| E(y)= μ | E(x) = u |
| var(y) = (μ - β)2σ2 + ρ2 | var(x) = s2 |
| y = asinh(a + by) | x ~ N(u, s2) |
Figure 1.

Variance-mean relationship for simulated data: true value (red), maximum-likelihood estimate β (green), maximum-quasi-likelihood estimate (blue)
Subsequently, the extended quasi-likelihood (EQL) model (Equation 5) and the ANL model were fitted to the simulated data by maximizing the extended quasi-likelihood function and the likelihood function derived from the asinh-normal distribution, respectively. The results are shown in Table 3 for the convolution model as the underlying true distribution, in Table 4 for the asinh-normal distribution, and in Table 5 for the Gamma distribution as the true error model.
Table 3.
Parameter estimates (true model: convolution of normal and log-normal distribution)
| EQL | ANL | true value | |||||
| Replicates | 4 | 10 | 20 | 4 | 10 | 20 | |
| err( |
0.0008 | 0.0007 | 0.0006 | 0.0010 | 0.0014 | 0.0009 | 0 |
| 20605 | 20012 | 19332 | 22979 | 24178 | 25161 | 25000 | |
| 0.1906 | 0.2035 | 0.2032 | 0.2340 | 0.2395 | 0.2427 | 0.25 | |
| 4150.1 | 4559.8 | 4669.6 | 4995.4 | 5053.6 | 5082.3 | 5000 | |
| -log L | 280822 | 708618 | 1420480 | 280205 | 708308 | 1419534 | |
err(![]() i) = avg((
i) = avg((![]() i - μi)/μi) EQL: all parameters estimated via EQL ANL: all parameters estimated using asinh-normal assumption
i - μi)/μi) EQL: all parameters estimated via EQL ANL: all parameters estimated using asinh-normal assumption
Table 4.
Parameter estimates (true model: ANL)
| EQL | ANL | true value | |||||
| Replicates | 4 | 10 | 20 | 4 | 10 | 20 | |
| err( |
0.0013 | 0.0007 | 0.0003 | 0.0013 | 0.0007 | 0.0003 | 0 |
| 20646 | 20686 | 19102 | 24848 | 25206 | 24999 | 25000 | |
| 0.1907 | 0.2073 | 0.2065 | 0.2176 | 0.2376 | 0.2427 | 0.25 | |
| 4132.2 | 4539.3 | 4591.8 | 4293.4 | 4727.8 | 4876.3 | 5000 | |
| -log L | 280546 | 708608 | 1419646 | 280362 | 707998 | 1418279 | |
See Table 3 for abbreviations.
Table 5.
Parameter estimates (true model: Gamma distribution)
| EQL | ANL | true value | |||||
| Replicates | 4 | 10 | 20 | 4 | 10 | 20 | |
| err( |
0.0001 | 0.0006 | 0.0001 | 0.0001 | 0.0006 | 0.0001 | 0 |
| -647.63 | 973.56 | 988.59 | 7616.7 | 5663.5 | 4821.5 | 0 | |
| 0.2142 | 0.2458 | 0.2538 | 0.2308 | 0.2479 | 0.2519 | 0.25 | |
| 34.713 | 7.7921 | 5.3068 | 3991.5 | 3846.1 | 3818.3 | 0 | |
| -log L | 289207 | 728069 | 1462227 | 289469 | 728668 | 1463451 | |
See Table 3 for abbreviations.
The colored lines in Figure 1 indicate the true variance-mean relationships and the maximum-likelihood and maximum quasi-likelihood estimates, respectively.
If the true error model was the asinh-normal distribution (Table 4) or the similar convolution model (Table 3) then – as expected – the fit of the correctly specified model (ANL) was always better than that of the EQL model, though in terms of the log-likelihood only by a small amount. Parameter estimation using this correct probability model was more efficient than with EQL. In both cases parameters μi were well estimated. Variance parameters β, σ and ρ were underestimated by the approximate error model. The sample size (4, 10, 20 replicates per gene) did not greatly impact EQL estimates.
On the other hand, if the true model followed a Gamma distribution (Table 5) the EQL model fitted the data consistently better than the ANL model. However, the difference in log-likelihood between the two candidate models was again comparatively small. As the Gamma model does not contain background signal the true values for β and ρ are zero. In this case the parameter estimates based on the ANL model are highly biased upwards, whereas estimates from the EQL approach were almost unbiased.
Thus, while the EQL model was based only on the postulated variance structure with no additional information on higher distributional moments, it nevertheless provided a reasonable fit to the ANL and convolution generated data and a very good fit to the Gamma-generated data.
Leukemia Data
Next, the EQL and ANL model was fitted to the Leukemia data from Golub et al. [28]. After preprocessing and filtering as in [28] 3051 genes and 38 samples remained. The estimation results based on the EQL and ANL error models are shown in Table 6. The data available from the Golub et al. website were already calibrated and background-corrected, hence the parameter β was set close to zero both for the approximate error model EQL as well as the ANL model. For this data set, the fit of the approximate error model EQL is better than of the parametric ANL model, i.e. the EQL models achieves a much higher (quasi) log-likelihood. The estimated EQL and ANL parameter values are similar, with EQL estimates being slightly smaller than the corresponding ANL parameter values.
Table 6.
Fit to Leukemia data
| -log L | ||||
| EQL | 0.0010 | 0.6895 | 0.0054 | 825827 |
| ANL | 0.0010 | 0.7957 | 0.0216 | 828054 |
See Table 3 for abbreviations.
The Leukemia data set contains subsets of samples from two tumor classes, AML and ALL [28]. A statistical test can then be employed to reveal which genes are differentially expressed between the two groups. One approach that explicitely takes account of the underlying error model is based on the likelihood ratio test [29]. For normal errors this approach is (asymptotically) equivalent to the standard t-test, but unlike the t-test it can also be applied for any other assumed error model. To determine p-values the test distribution for the likelihood ratio was assumed to be χ2 (a more accurate distribution may be obtained, e.g., using a bootstrap approach). Figure 2 shows the number of differentially expressed genes given a nominal α value (type I error) for the individual pairwise tests, estimated using the approximate error model (open triangles) and the ANL model (filled triangles). In the data set there are a large number of differentially expressed genes. As the type I error is controlled by α the percentage of statistically significant differentially expressed genes shows the power of the test in dependence of the chosen error model. The approximate error model EQL fits the data better than the parametric model ANL, and Figure 2 shows that this leads to a subsequent improved power to select differentially expressed genes.
Figure 2.

Number of differentially expressed genes in dependence of the nominal α value (type I error), computed using the approximate error model (open triangles) and the ANL model (filled triangles).
Conclusions
Error models play an important though often implicit part in the analysis of gene expression data. There are a lot of possibilities to model the error of intensity measurements, and these are mirrored by the wide choice of parametric models and corresponding data transformations. As non-parametric approaches are not always applicable due to lack of sufficient replicate data, in this paper the use of approximate error models based on quasi-likelihood is suggested as a further alternative.
Quasi-likelihood is a versatile and simple framework for semi-parametric modeling that requires only a partial specification of the underlying probability structure. This is ideal for microarray data where there is agreement on the variance versus mean relationship of the measured intensities but no suitable mechanistic model available to guide the search for the true underlying error distribution. A further advantage of quasi-likelihood is also that it is scale-neutral, i.e. it can be used to analyze data on any preferred scale. Thus, using an approximate error model within the quasi-likelihood framework allows to analyze data on the original observed scale, where the expected intensity corresponds directly to transcript concentration, without the need for a complicated transformation. Third, quasi-likelihood can be viewed as a compromise between traditional parametric and non-parametric approaches.
In this paper both for simulated and molecular data the approximate error model fitted the data as good or better than a competing parametric model derived from an transformation-based approach. Moreover, in a model-based test for differential expression the approximate error model had more power on the same level of type I error than the parametric model. It is expected that the favorable properties of quasi-likelihood also hold for other data sets.
Employing an approximate error model in a statistical analysis comprises a tradeoff between the a priori available information on the true model and the efficacy of an inference from the data. If the true underlying model is fully known, using an approximate model such as quasi-likelihood inevitably entails loss of efficiency and leads to bias in parameter estimation. However, if a suitable error model is not readily available and if multiple unknown sources of error have to be taken into account, then the quasi-likelihood approach is advantageous as it provides an optimal estimating equation under very general conditions, and may thus outperform other ad-hoc parametric models.
While in this paper quasi-likelihood was used for modeling and inference purposes, it is generally applicable also in a regression setting [14]. This points towards further possible applications of the quasi-likelihood framework in gene expression analysis. For instance, normalization procedures may benefit from using an approximate error model (e.g. [30]). Systematic effects in the data such as those due to different arrays, dyes, etc. can also be inferred by regression and ANOVA techniques [27,31] and hence are amenable to analysis by quasi-likelihood, too. In a related line, the affinity of probes on a chips may thus also be estimated by using quasi-likelihood, rather than assuming a normal error as in [5]. Finally, high-level analysis such as classification can incorporate quasi-likelihood models.
In summary, approximate error models such as provided by the quasi-likelihood framework enable the analysis of gene expression data despite our ignorance of the true underlying low-level processes generating the observed data.
Acknowledgments
Acknowledgments
This work was supported by an Emmy Noether research grant (STR 624/1-2) from the Deutsche Forschungsgemeinschaft. I thank Anja von Heydebreck and Wolfgang Huber for discussion.
References
- Rocke DM, Durbin B. A model for measurement error for gene expression analysis. J Comp Biol. 2001;8:557–570. doi: 10.1089/106652701753307485. [DOI] [PubMed] [Google Scholar]
- Hubbell E, Liu W-M, Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- Yue H, Eastman PS, Wang BB, Minor J, Doctolero MH, Nuttall RL, Stack R, Becker JW, Montgomery JR, Vainer M, Johnston R. An evaluation of the performance of cDNA microarrays for detecting changes in global mRNA expression. Nucleic Acids Res. 2001;29:e41. doi: 10.1093/nar/29.8.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdas L, Coombes KR, Baggerly K, Abruzzo L, Highsmith WE, Krogmann T, Hamilton SR, Zhang W. Sources of nonlinearity in cDNA microarray expression measurements. Genome Biology. 2001;2:research0047.1–7. doi: 10.1186/gb-2001-2-11-research0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui WK. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J Comp Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Explorations, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. [DOI] [PubMed]
- Huber W, von Heydebreck A, Sültmann H, Postka A, Vingron M. Variance-stabilizing applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18:96S–104S. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- Durbin B, Hardin J, Hawkins D, Rocke DM. A variance-stabilizing transformation for gene expression microarray data. Bioinformatics. 2002;18:105S–110S. doi: 10.1093/bioinformatics/18.suppl_1.s105. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani R, Storey JD, Tusher V. Empirical Bayes analysis of a microarray experiment. J Amer Statist Assoc. 2001;96:1151–1160. doi: 10.1198/016214501753382129. [DOI] [Google Scholar]
- Yeung KY, Fraley C, Murua A, Raftery AE, Ruzzo WL. Model-based clustering and data transformations for gene expression data. Bioinformatics. 2001;17:977–987. doi: 10.1093/bioinformatics/17.10.977. [DOI] [PubMed] [Google Scholar]
- Geller SC, Gregg JP, Hagerman P, Rocke DM. Transformation and normalization of oligonucleotide microarray data. Technical report. 2002. [DOI] [PubMed]
- Cui X, Kerr MK, Churchill GA. Data transformation for cDNA microarray data. Technical report. 2002.
- McCullagh P, Nelder JA. Generalized Linear Models. 2. Chapman and Hall, London; 1989. [Google Scholar]
- Pawitan Y. In All Likelihood: Statistical Modelling and Inference Using Likelihood. Clarendon Press, Oxford; 2001. [Google Scholar]
- Wedderburn RWM. Quasi-likelihood functions, generalized linear models, and the Gauss-Newton method. Biometrika. 1974;61:439–447. [Google Scholar]
- Nelder JA, Pregibon D. An extended quasi-likelihood function. Biometrika. 1987;74:221–232. [Google Scholar]
- McCullagh P. Quasi-likelihood functions. Ann Statist. 1983;11:59–67. [Google Scholar]
- Godambe VP, Heyde CC. Quasi-likelihood and optimal estimation. Intl Statist Review. 1987;55:231–244. [Google Scholar]
- Firth D. On the efficiency of quasi-likelihood estimation. Biometrika. 1987;74:233–245. [Google Scholar]
- Nelder JA, Lee Y. Likelihood, quasi-likelihood and pseudolikelihood: some comparisons. J R Statist Soc B. 1992;54:273–284. [Google Scholar]
- Barndorff-Nielsen O, Cox DR. Edgeworth and saddlepoint approximations with statistical applications (with discussion). J R Statist Soc B. 1979;41:279–312. [Google Scholar]
- Efron B. Double exponential families and their use in generalized linear regression. J Amer Statist Assoc. 1986;81:709–721. [Google Scholar]
- Chen Y, Dougherty ER, Bittner ML. Ratio-based decisions and the quantitative analysis of cDNA microarray images. J Biomed Optics. 1997;2:364–374. doi: 10.1117/1.429838. [DOI] [PubMed] [Google Scholar]
- Beißbarth T, Fellenberg K, Brors B, Arribas-Prat R, Boer JM, Hauser NC, Scheideler M, Hoheisel JD, Schütz G, Poustka A, Vingron M. Processing and quality control of DNA array hybridization data. Bioinformatics. 2000;16:1014–1022. doi: 10.1093/bioinformatics/16.11.1014. [DOI] [PubMed] [Google Scholar]
- Sapir M, Churchill GA. Estimating the posterior probability of differential gene expression from microarray data. Poster, The Jackson Laboratory. 2000.
- Kerr MK, Afshari CA, Bennett L, Bushel P, Martinez J, Walker NJ, Churchill GA. Statistical analysis of a gene expression microarray experiment with replication. Statistica Sinica. 2002;12:203–217. [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Ideker TE, Thorsson V, Siegel AF, Hood LE. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J Comp Biol. 2000;7:805–817. doi: 10.1089/10665270050514945. [DOI] [PubMed] [Google Scholar]
- Kepler TB, Crosby L, Morgan KT. Normalization and analysis of DNA microarray data by self-consistency and local regression. Genome Biology. 2002;3:research0037.1–research0037.12. doi: 10.1186/gb-2002-3-7-research0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MK, Churchill GA. Statistical design and the analysis of gene expression microarray data. Genet Res. 2001;77:123–128. doi: 10.1017/S0016672301005055. [DOI] [PubMed] [Google Scholar]