

Abstract
Background
A limiting factor of cDNA microarray technology is the need for a substantial amount of RNA per labeling reaction. Thus, 20–200 micro-grams total RNA or 0.5–2 micro-grams poly (A) RNA is typically required for monitoring gene expression. In addition, gene expression profiles from large, heterogeneous cell populations provide complex patterns from which biological data for the target cells may be difficult to extract. In this study, we chose to investigate a widely used mRNA amplification protocol that allows gene expression studies to be performed on samples with limited starting material. We present a quantitative study of the variation and noise present in our data set obtained from experiments with either amplified or non-amplified material.
Results
Using analysis of variance (ANOVA) and multiple hypothesis testing, we estimated the impact of amplification on the preservation of gene expression ratios. Both methods showed that the gene expression ratios were not completely preserved between amplified and non-amplified material. We also compared the expression ratios between the two cell lines for the amplified material with expression ratios between the two cell lines for the non-amplified material for each gene. With the aid of multiple t-testing with a false discovery rate of 5%, we found that 10% of the genes investigated showed significantly different expression ratios.
Conclusion
Although the ratios were not fully preserved, amplification may prove to be extremely useful with respect to characterizing low expressing genes.
Keywords: mRNA amplification, microarray, gene expression, multiple hypothesis testing, linear mixed effects model
Background
The substantial amount of RNA required for expression analysis is a limiting factor for the cDNA microarray technology in a number of potentially important applications. Two main approaches, signal amplification and global mRNA amplification, have been developed to overcome this obstacle. Signal amplification, such as dendrimer technology [1] and tyramide signal amplification (TSA) [2] aim to increase the fluorescent signal emitted per mRNA molecule. Global mRNA amplification has the purpose of increasing the number of available transcript equivalents for sufficient labeling from a limited starting amount. In current implementations, mRNA amplification techniques require less RNA than those based on signal amplification.
Van Gelder et al. [3] devised a multistep strategy to amplify mRNA from limited quantities of cDNA in studies of gene expression. Their method is commonly referred to in the literature as the Eberwine method. The general steps involve reverse transcription of mRNA with an oligo dT-primer bearing a T7-promoter site. After synthesis of double stranded cDNA, antisense RNA was transcribed by the T7-polymerase in an in vitro transcription reaction. Two recent reports have described detailed protocols for mRNA amplification used with microarray gene expression profiling [4,5]. We have followed the optimized protocol presented by Baugh et al. [5] which has retained the basis of the Eberwine strategy, but where they have introduced some modifications in order to reduce undesired products. The protocol involves two rounds of amplification, which we found to be suitable for further studies as the output of aRNA obtained from 0.2 μg total RNA was sufficient for several hybridizations. Although both reports demonstrate the potential of mRNA amplification, it is clear that the challenge lies in a detailed comparison of data sets obtained from amplified samples versus non-amplified samples to estimate the consistency of the results. This is a critical step in order to validate the use of microarray data obtained from amplified material quantitatively.
Global mRNA amplification has been employed in a range of applications, including more recent studies where microarrays have been utilized for expression profiling [6-9]. An optimal upscaling of mRNA that introduced minimal systematic biases, would maintain the relative transcript abundance present in the initial mRNA sample. Examinations of the transcript level preservation through mRNA amplification procedures have so far involved Northern blot confirmation using a limited number of genes [10], dot blot differential screening of selected cDNAs using probes synthesized from poly(A) RNA or aRNA [11], the use of internal RNA standards [12], hierarchical clustering to compare consistency of outliers [4], validation by quantitative RT-PCR [13] or comparisons of ratio/intensity distribution of the entire gene set, and subgroups determined by gene abundance [8].
Based on microarray analysis, estimates of ratio preservation through mRNA amplification have been assessed from correlation between ratio profiles from amplified aRNA and the total RNA [4,8,13-15]. The studies referred to here have applied 1–3 rounds of amplification. Wang et al. [4] presented an informative study, although limited, as they only focused on the maintenance between two RNA sources of differentially expressed genes or "outliers". A relatively stringent filtering criterion was used and outliers determined by total RNA hybridization experiments were used as controls. By applying hierarchical clustering, they identified reproducible outliers across arrays of variable experimental conditions. When aRNA was generated from 0.25–3.0 μg of total RNA, 85–92% of the outliers from the control experiments were also identified as differentially expressed genes after aRNA hybridization analysis. Scheidl et al. [8] presented all genes in common between aRNA and total RNA, and not only the outliers. Through a series of plots of pre-processed data, they indicated the degree of preservation that could be expected under the experimental conditions applied. Although the intensity levels were not preserved, the relative abundances of transcripts were maintained, giving rise to a relatively high correlation factor (0.84). Pearson correlation coefficients of ratio-to-ratio measurements were calculated according to mean intensities of the corresponding genes, showing decreased coefficients as intensities approached background levels. Zhao et al. [15] also obtained a similar average correlation coefficient (0.82), when using an optimized protocol as did Feldman et al. [16], who obtained a correlation coefficient of 0.8. Hu et al. [14] and Puskás et al. [13] took the outcome of amplification one step further and pointed out the observation that additional genes were detected in arrays hybridized with amplified material. Absolute intensity levels were elevated on arrays hybridized with labeled aRNA and as a consequence more genes were registered. Hu et al. [14] selected a few of these genes. They confirmed by other molecular techniques that these genes were in fact expressed in the cells and not the result of unspecific binding or artifacts. This indicated that amplified RNA was more sensitive to low abundance transcripts than the standard method using total RNA. Due to improved signal to background levels using aRNA, Feldman et al. [16] not only scored more genes on arrays with amplified material, but also observed a doubling in the number of outliers. Hence, within the limits investigated so far, the general conclusion is that microarray data from amplified material is comparable to non-amplified material, but there is a slight decrease in correlation coefficients reflecting changes in transcript ratios.
In this study, we refer to total RNA as the "true" standard. However, this is not necessarily correct. Using ANOVA analysis, Smith et al. [17] found that the variation between total RNA replicates was highly significant. Other molecular techniques could be used to verify the reliability of our total RNA standard, but for the moment few reasonable approaches are available for high throughput verification. Quantitative real time PCR is not suited to query thousands of genes. Similarly, SAGE appears not to lend itself to query the number of experiments performed in this report. Also, the publicly available SAGE data may not correspond to the samples used in microarray experiments, with respect to e.g. transcript representation and priming method.
Quantitative studies of transcript ratio variations between amplified RNA and total RNA are few, and no consensus with respect to the sources of the observed results have been obtained. The high degree of correlation between independent amplifications from the same RNA indicates that repetition of the amplification procedure does not appear to be the source of variation [5,13,15,18]. Nevertheless, it is clear that microarray hybridizations are affected by substantial variability even under controlled conditions. Previously reported correlation coefficients between total RNA and aRNA profiles represent the total amount of variation, including noise, without quantification of the variation due to the amplification procedure itself. Our aim therefore, was to perform further comparisons of microarray data sets obtained from amplified and non-amplified material by introducing an ANOVA based statistical design where the goal was to isolate the variation in gene expression data due to amplification from other sources of variation present in the data set. In addition we used a multiple t-test to identify the genes that displayed consistent ratios (referred to as the "conservative set") from those that contributed to ratio variation due to the amplification. To find the overall effect of amplification on the ratios, we compared Signal to Noise Ratios (SNRs) including variations due to amplification or not. This was done both for the total and "conservative set".
Results and Discussion
The goal of this study is to evaluate if ratios are preserved by the amplification protocol under study. We assess this question by examining data of observed intensities and ratios between two cell lines in cDNA microarray experiments using non-amplified material and amplified material. Two different cell lines (MT-1 and U2OS) are considered since it is impossible to hybridize non-amplified and amplified material on the same array. The experimental design is presented in Figure 1, and explained in the Methods Section. The design consists of four hybridizations of non-amplified material and eight hybridizations of amplified material.
Figure 1.

Experimental design. For a particular cell line, the material was divided into six batches; two batches of non-amplified RNA were directly labeled (white shapes) and four batches of diluted total RNA were amplified before labeling (grey shapes). These batches were split in two and labeled Cy5 (red) or Cy3 (green). In total, four array experiments were based on non-amplified material, and eight experiments were based on amplified material.
Preservation of ratios can be addressed by performing multiple hypothesis testing by comparing ratios between the two cell lines for the amplified material with ratios between the two cell lines for the non-amplified material for each gene. This strategy can lead to a division of the total number of genes under study into a set of genes where the ratio is clearly preserved (called the "conservative set"), a set of genes where the ratio is clearly not preserved (called the "rejected set"), and a set of genes where the conclusion about ratio preservation is unclear (called the "undetermined set"). This strategy has been investigated, but since the size of the undetermined set can be substantial the strategy is not completely satisfactory.
We have chosen to address the question of preservation of ratios on an overall level with the aid of a statistical analysis of variance model. An additional motivation for using such a model is that we are also interested in investigating if the variability caused by the amplification protocol is large compared with the total variability in the experiments. We found many sources of variation present in the cDNA experiments; variation between amplified and non-amplified material (protocol, P), between the two different cell lines (C), between different batches of each cell line, (B) between different arrays (A), between the two different florescent dyes (D), between different spots (AG), between different genes (G) and combinations thereof. Based on this, an experimental design was developed and an ANOVA-based statistical model was constructed. From estimates of the parameters present in the ANOVA-model, a Signal to Noise Ratio (SNR) evaluation criterion was constructed. The ANOVA-model is based on modelling normalized log2-intensities, but SNR evaluations are calculated with focus on ratios. The details are found in the Methods Section.
Results on the total data set
Data from the cDNA experiments were analysed and preprocessed as described in the Methods section. See Table 1 and Table 2 for details on filtered genes. The experiments using aRNA produced superior hybridization quality. This was reflected in the lower number of filtered genes per array compared with the total RNA hybridizations. On average, 35% of the genes were filtered from arrays hybridized with amplified material, versus 63% of the genes filtered from arrays hybridized with non-amplified material (Table 2). As a measurement of array quality, Pearson correlations were calculated between the different arrays. For the amplified arrays, 28 pairs of arrays can be formed, and the average correlation coefficient was 0.90 (ranging from 0.82 to 0.96). For the non-amplified arrays, six pairs of arrays can be formed, and the average correlation coefficient was 0.87 (ranging from 0.82–0.95 for the six pairs). The average correlation between amplified and non-amplified arrays was 0.79 (ranging from 0.71 to 0.84).
Table 1.
Overview of filtered genes.
| Description | Number of genes |
| 1Genes on an array | 12497 (100%) |
| 2Genes filtered out from all arrays | 1738 (14%) |
| 3Total number of genes after filtering | 10759 (86%) |
| 4Genes only detected in non-amplified material (at least one array) | 64 (0.5%) |
| 5Genes only detected in amplified material (at least one array) | 4377 (35%) |
| 6Genes detected for both groups | 6318 (50.5%) |
1Number of genes printed on an array. 2Genes not detected on any of the twelve arrays. 3Genes with signals on at least on array. 4Genes that were filtered out from all arrays with amplified material, but present on at least one array with non-amplified material. 5Genes that were filtered out from all arrays with non-amplified material, but present on at least one array with amplified material. 6Number of genes that were detected both in arrays with non-amplified material and in arrays with amplified material. These genes are again split into two groups; one with genes fulfilling the criterion of 3 observations in each group (4331 genes); and one with those not fulfilling this criterion (1987 genes).
Table 2.
Filtering per array.
| Arrays hybridized with amplified material | Arrays hybridized with non-amplified material | |||||||||||
| Array number | 1 | 2 | 5 | 6 | 7 | 9 | 10 | 11 | 3 | 4 | 8 | 12 |
| Genes flagged by Genepix | 5192 | 2155 | 1007 | 2152 | 3151 | 2209 | 3212 | 1073 | 4750 | 4909 | 2903 | 4374 |
| 41.55% | 17025% | 8.06% | 17.22% | 25.21% | 17.68% | 25.70% | 8.59% | 38.01% | 39.28% | 23.23% | 35.00% | |
| Genes flagged manually | 15 | 1 | 3 | 5 | 2 | 6 | 10 | 1 | 116 | 15 | 4223 | 16 |
| 0.12% | 0.008% | 0.024% | 0.04% | 0.016% | 0.048% | 0.08% | 0.008% | 0.93% | 0.12% | 33.79% | 0.13% | |
| Additionally Filtered by (spot-background) / background > 1 | 2078 | 1621 | 1055 | 2081 | 2081 | 2033 | 1680 | 2596 | 3226 | 2223 | 2117 | 2521 |
| 16.63% | 12.97% | 8.44% | 16.65% | 16.15% | 16.27% | 13.44% | 20.77% | 25.81% | 17.79% | 16.94% | 20.17% | |
| Sum | 7285 | 3777 | 2065 | 4238 | 5171 | 4238 | 4902 | 3670 | 8092 | 7147 | 9243 | 6911 |
| 58.30% | 30.22% | 16.52% | 33.91% | 41.38% | 33.40% | 39.22% | 29.37% | 64.75% | 57.19% | 73.96% | 55.30% | |
The ANOVA-model was fitted to the filtered data set of 10759 genes. Parameter estimates in the ANOVA-model are found in Table 3. To sum up, the main findings of Table 3 is a comparison of the different sources of variability. The estimates of the variability in batch, 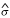 B, and in interaction between array and dye, AD, were small. This meant that there was a low variability between samples from the same cell line and that the different dyes had about the same effect for different arrays. The estimated variability in the interaction between gene and dye, DG, was small, which meant that there was no prominent dye effect. The unexplained modeling and measurement error, denoted σε, was estimated to be moderate, and the estimated variability in the cell line, gene, and protocol interaction, CGP, was comparable to ε. This implies that the amplification did not preserve the ratios perfectly. The estimate of the variability of the arrays (hybridization effect), A, was larger than the estimated cell line, gene, and protocol interaction. The estimated variability due to the spot effect, AG, was as expected quite large. The estimated variability of the interaction between cell line and gene, CG, was large, and this was reasonable, since each cell line had a unique gene expression profile. Also the estimated variability in the gene-protocol interaction, GP, was large, indicating that the magnitude of the intensities was not amplified equally for all genes. The estimated variability in gene, G, was as expected very large, since the genes obviously had different expression levels. The ranking of size of the estimated random effects are AD <B <DG <ε <CGP <A <AG <CG <GP <G.
B, and in interaction between array and dye, AD, were small. This meant that there was a low variability between samples from the same cell line and that the different dyes had about the same effect for different arrays. The estimated variability in the interaction between gene and dye, DG, was small, which meant that there was no prominent dye effect. The unexplained modeling and measurement error, denoted σε, was estimated to be moderate, and the estimated variability in the cell line, gene, and protocol interaction, CGP, was comparable to ε. This implies that the amplification did not preserve the ratios perfectly. The estimate of the variability of the arrays (hybridization effect), A, was larger than the estimated cell line, gene, and protocol interaction. The estimated variability due to the spot effect, AG, was as expected quite large. The estimated variability of the interaction between cell line and gene, CG, was large, and this was reasonable, since each cell line had a unique gene expression profile. Also the estimated variability in the gene-protocol interaction, GP, was large, indicating that the magnitude of the intensities was not amplified equally for all genes. The estimated variability in gene, G, was as expected very large, since the genes obviously had different expression levels. The ranking of size of the estimated random effects are AD <B <DG <ε <CGP <A <AG <CG <GP <G.
Table 3.
Parameter estimates for the total data set consisting of 10759 genes and for the "conservative set" of 2721 genes.
| Parameter | Type of effect1 | Estimate Total set | Estimate Conserved set |
| μ | Fixed | 10.56 | 12.18 |
| C (cell line 2; U2OS) | Fixed | 0.086 | -0.028 |
| D (Cy3) | Fixed | -0.026 | -0.022 |
| P (non-amplified) | Fixed | -0.74 | -1.14 |
| CP22 (cell line 2 and non-amplified) | Fixed | -0.0043 | 0.0089 |
| σA (array) | Random | 0.28 | 0.30 |
| σB (batch) | Random | 0.041 | 0.041 |
| σC (gene) | Random | 1.36 | 0.98 |
| σAD | Random | 0.039 | 0.040 |
| σGP | Random | 0.85 | 0.79 |
| σCG | Random | 0.74 | 0.54 |
| σAG | Random | 0.44 | 0.44 |
| σDG | Random | 0.048 | 0.050 |
| σCGP | Random | 0.22 | 0.028 |
| σε | Random | 0.22 | 0.22 |
Annotations: μ = overall mean signal intensity, C = cell line type, D = labeled dye, P = protocol. 1 Of the fixed effects, C, D, and P have two levels while CP is an interaction between two fixed effects. The random effects were modeled as independently normally distributed random effects with zero mean and constant standard deviation. σCGP is the cell line specific GP effect and is the effect of primary interest.
Results on the evaluation of the SNR-criterion are found in Table 4. The SNR was reduced from 9.91 to 5.22 by the inclusion of the cell line, gene, and protocol interaction. Thus, the overall implication of the amplification was a doubling of relative noise in the experiments. From this we can conclude that not all the ratios are preserved when looking at the total data set of 10759 genes. 6318 of the 10759 genes are present both on arrays with amplified and on arrays with unamplified material. Only these 6318 genes contributed directly in the estimation of the parameters measuring the variability caused by amplification (e.g. the variability in the cell line, gene, and protocol interaction). The other 10759-6318 = 4441 genes were useful because they contributed to the estimation of the other parameters of the model.
Table 4.
Signal to Noise Ratio (SNR) for the two data sets.
| Data set | SNR without σCGP | SNR with σCGP |
| Total set1 | 9.91 | 5.22 |
| Conservative set2 | 5.38 | 5.30 |
1The total data set included 10579 genes detected on at least one of the twelve arrays. 2The conservative data set included genes found to have equal ratio expression between amplified and non-amplified material. σCGP is the cell line specific GP effect and is the effect of primary interest.
Results on the "conservative set"
Multiple hypothesis testing was performed to divide the total data set into a "conservative set", a "rejected set" and an "undetermined set". To be able to assess the variability in the gene expression ratios, we only considered genes where the number of observations in each group (amplified versus non-amplified) was at least three. This was true for 4331 of the 10759 genes.
1) We defined a "conservative set" of genes for which we did not reject the hypothesis of equal expression between the two protocols. Starting with the full set of 4331 genes, we removed all genes with p-values less than 0.1 from the t-test assuming unequal variances for the amplified and non-amplified material for each gene (1376 genes). In addition, we removed genes with p-values less than 0.1 when performing a two-sample t-test assuming equal variances for the amplified and non-amplified material for each gene (234 more genes subtracted from the set). This led to a "conservative set" of 2721 genes (2721/4331 = 63% of the genes investigated).
2) We defined a "rejected set" of genes for which we rejected the hypothesis of equal expression, and thus found the selected genes not to be consistently expressed between the amplified and the non-amplified material. We rejected hypotheses based on adjusted p-values from the two-sample t-test using the Benjamini-Hochberg multiple testing procedure [19], as implemented in the multtest package in the statistical language R. R packages for use with cDNA microarray data are described in Dudoit et al. [20]. Using a cut-off for these adjusted p-values of 0.01, led to a list of 154 genes. The expected false discovery rate (proportion of false positives) in this list was estimated to be 1%.
3) The remaining genes (1456 genes) were not classified as "conservative" nor "rejected" and we will refer to them as the "undetermined set".
The plot in Figure 2A shows the difference between the amplified and the non-amplified material for the log2-ratio of the two cell-lines vs. the p-value for each of the 4331 genes. Small p-values are mainly found for large differences, but large differences with high p-values and small differences with small p-values also exist. The plot in Figure 2B compares the ratio distribution of the 4331 genes obtained with amplified and non-amplified material. For both plots the genes in the "conservative set" are in green, the genes in the "rejected set" are in red, and the genes in the "undetermined set" are in black.
Figure 2.

(A) Plot of the difference in log2-ratio between the amplified and the non-amplified material for the log2-ratio of the two cell-lines (MT-1, U2OS) vs. the p-value for each of the 4331 genes, calculated for hypothesis testing. (B) Plot of the average log2-ratio of the two cell-lines (MT-1 and U2OS), for the amplified and the non-amplified material for each of the 4331 genes investigated during hypothesis testing. The genes in the "conservative set" are in green, the genes in the "rejected set" are in red and the genes in the "undetermined set" are in black.
The "conservative set" of genes was investigated further using our ANOVA procedure, and parameter estimates are presented in Table 3. The estimated variability for the "conservative set" had the same ranking as the total data set with one exception. The interaction between cell line, gene and protocol, CGP, was considerably lower in the "conservative set". The cell line, gene, and protocol effect was the only random effect in the log2-ratio that was dependent on the amplification protocol. This indicates that the noise due to amplification was low.
The SNR-criterion for the conservative data set is presented in Table 4. Inclusion of the cell line, gene, and protocol effect only reduced the SNR slightly from 5.38 to 5.30. We therefore conclude that the ratios for the genes in the "conservative set" seem to be preserved by the amplification protocol. This is in compliance with the results of the t-tests.
The SNR for the "conservative set" without σCGP was clearly lower than the SNR for the total set of genes. This result may have several explanations. One is that the total set of genes might have a greater variability in ratio and/or intensity than the ones in the conservative set. This is supported by the fact that both G and CG were lower in the "conservative set".
Assessing the percentage of genes where the ratio was preserved
For the data set under study (i.e. for the genes on our arrays, using the explained hybridization protocol, with our choice of scanning procedure, filtering and normalization), we found that the variability caused by amplification was substantial and the ratios seem not to be preserved. We found a "conservative set" of the genes where the variability caused by amplification was small, and the ratios seem to be preserved. Estimating the percentage of genes where the ratio was not preserved by the amplification protocol was difficult. Different strategies could be employed, but the validity of the assumptions underlying the different strategies is hard to check properly. If we believe that the assumptions underlying the two-sample t-test performed are true, based on the distribution of the p-values it is possible to estimate the number of true null-hypotheses (i.e. the number of genes where the ratio is preserved). Assuming that the test-statistics underlying the two-sample t-test were independent across genes and using the estimator of Schweder and Spjøtvoll [21], the number of genes with unchanged ratios was of the order 64%. Another strategy is to look at the percentage of genes not rejected at the level 5% of false discovery rate. This number was 90%. Turning this number around we could say that 10% of the genes showed significantly different expression when comparing amplified with non-amplified material. As an indication of the robustness of these findings we assessed the number of genes not rejected at the level 5% of false discovery rate when comparing two groups of arrays with amplified material. Comparing two groups of arrays with amplified material, we would expect the ratios between the two cell lines to be the same for both groups for all genes. In our data set there are eight arrays with amplified material, and there are 35 ways of dividing these arrays into two groups of four arrays each. Analysing these 35 data sets using multiple hypothesis testing the overall result was that 0.04% of the genes (compared to 10% for amplified vs. non-amplified) were found to show different expression ratios between the two cell lines for the two groups at a false discovery rate of 5%. This finding is in agreement with what was expected.
Aspects of the results
In order to obtain reliable information from expression data from amplified material, it is important to be aware of how the amplification procedure may affect the data. By comparing expression data between amplified and non-amplified material, we have observed that the ratios were significantly different for a number of genes, in addition to a set of genes whose ratios were neither conserved nor rejected.
Although significant differences between ratios for a subset of genes was expected due to the nature of variability in microarray experiments, we examined whether there was a systematic skewing of the data by investigating molecular properties of the probes printed on the arrays.
We mapped the clone sequences of the genes to their reference mRNA sequence (RefSeq). Based on this mapping, we examined the GC content of the clones and of the remaining stretch between the 3' end of the clone to the poly (A) tail of corresponding mRNA. If the distribution of the GC content was different for the genes in the "conservative set" compared to the genes in the "rejected set" and "undetermined set", this could indicate that optimization in the form of increased temperatures during strand synthesis may be an improvement of the amplification protocol. Increased temperatures have been emphasized in certain publications [4,13]. We were able to match 40–50% of the clones under investigation with their corresponding mRNA sequence. For the 4331 genes for which ratio and p-values were calculated, the GC content displayed the same distribution in all subsets, "conservative ", " rejected", and "undetermined " (data not shown). This was also true for the set of genes with ratios obtained only from arrays with amplified material (4377 genes). Hence, there was no apparent bias towards sequences with low GC content during the amplification procedure.
As amplification of mRNA is a multistep procedure involving several polymerases synthesizing new strands, we were also interested in the position of the printed clones relative to the poly(A) tail. We knew that during amplification [10], the RNA transcripts were systematically reduced in length and were on the average 400–450 bp (data not shown), in addition to being 3' end biased due to the use of an oligo dT-primer. The question was whether the array probes that were positioned far from the poly (A) tail had an equal chance of being hybridized to labeled cDNA synthesized from both aRNA products and total RNA. We found that the average length of the printed clones was the same for all subgroups of genes mentioned above (data not shown), and the distance from the poly (A) sequence to the 3' end of the clones was also distributed evenly throughout the subgroups. The majority of the clones were 3' end biased. For a large fraction, the distance from the poly (A) tail to the 3' end was 0 bp. As there is a 3' end bias in the clone set, a drift away from the poly(A) tail caused by random priming in the second round of amplification could possibly affect the preservation of ratios. As the transcripts are reduced in length during the initial round of amplification, due to priming with anchored oligo dT, it is a reasonable assumption that a drift is more likely to affect ratio preservation of longer mRNA transcripts. However, this possibility seems to be less important, as the use of random priming does not appear to introduce variation between replicated amplifications. This is supported by the highly correlated array data when comparing aRNA samples generated from the same starting material.
One important feature of the amplified protocol was the increased total intensity on the arrays. The absolute intensity was doubled compared to standard total RNA experiments. One main difference is that 3 μg aRNA (used in labeling reaction) corresponds to approximately ten times more than 30 μg total RNA. Using equivalent amount of total RNA (300 μg) requires a substantial RNA source and is unrealistic in most experiments. Another difference was the high degree of purity of aRNA compared to total RNA as measured by optical density (2.4 versus 1.9). Greater purity implies reduced number of disturbing factors during the labeling reaction. In agreement with previous reports [13,14], we retained a considerably larger number of transcripts through the amplification protocol. After filtering procedures (see Table 1), 4377 genes were exclusively left in the amplified data set. We also observed that the average signal intensity for these 4377 gene transcripts was lower than for the amplified transcripts in the 4331 gene subset (with at least three observations per gene in each group of arrays (amplified and non-amplified). Hu et al. [14] suggested that this is due to improvement of signal intensities of genes with low transcript numbers.
Compared to total RNA, hybridizations performed with aRNA generally provide superior Signal to Noise Ratio even for genes with low expression. In addition aRNA provides highly reproducible results and may in some situations give a better representation of the true transcript abundance than using total RNA. However, it is a plausible assumption, though not presently thoroughly tested, that a reduced Signal to Noise Ratio will result from increasingly lower sample start material for global mRNA amplification procedures. This will be a consequence of larger stochastic variation for transcripts of lower copy numbers. To distinguish which approach is the closest to the true standard, a detailed comparison of results obtained from experiments in a dilution series, or by other molecular techniques, including real time PCR or SAGE, may be warranted. These considerations also apply for similar methods, such as the recently introduced method of exponential amplification of transcripts by PCR, rather than amplification through in vitro transcription [17,22].
Conclusions
The present work has utilized a quantitative approach towards evaluating mRNA amplification procedures. Under the experimental conditions applied, we found that the impact of amplification was increased noise in the form of ratio distortions for some genes. We did not find that molecular properties of the clones could be used as a prediction of unfaithful ratio preservation. More than half of the genes in the undetermined group (27% of the 4331 gene subset), had a log2-ratio difference within the range of +/- 0.5. Distinguishing between genes that are truly expressed differently and genes that are simply affected by various sources of noise remains a challenge. Caution may therefore be necessary when interpreting results obtained from amplification procedures. In further studies, the same analysis set-up permits determination of the sensitivity level of the amplification procedure by assessing the effect of reducing the amount of total RNA in the first round of aRNA production. This will be useful in order to extract informative gene expression data from extremely limited cell samples.
Methods
cDNA arrays
The microarrays used in this paper were produced in house using a Micro Grid II robotic printer (Bio Robotics, Cambridge, UK). These 13 k human cDNA arrays were printed on amino silane coated slides (CMT GAPS, Corning Life Sciences, Corning, NY). For details on the arrays, we refer to: http://www.med.uio.no/dnr/microarray/index.html.
RNA purification
RNA from two cell lines, MT-1 (human breast cell line, kindly provided by Dr. I Fitchner, (Naundorf et al. [23]) and U2OS (human osteosarcoma line, ATCC, Rockville, MD), were used in all experiments. The cell lines were maintained in RPMI media (Bio Whittaker Europe) supplemented with 10% fetal calf serum (PAA Laboratories, Linz, Austria). The cell cultures were grown without antibiotics and tested negative for mycoplasma infection. Total RNA was isolated using GenElute Mammalian Total RNA kit (Sigma, St. Louis, MO) according to the manufacturer's protocol. RNA concentrations were determined by OD260 reading in 50 mM NaOH (GeneQuant, Amersham Pharmacia Biotech AB, Uppsala, Sweden). Total RNA from the respective cell lines were aliquoted to 30 μg total RNA per sample. In addition, an aliquot of RNA from each cell line was diluted to 1 μg/ul total RNA concentration and further down to 0.1 μg/ul.
RNA amplification
RNA amplification was carried out as published by Baugh et al. [5] though slightly modified. The modifications mainly involved a different choice of enzyme in respect to the original protocol. A detailed version of the protocol can be found at the following website: http://www.med.uio.no/dnr/microarray/Mat_and_met_amp.pdf. Briefly, the initial amount of total RNA used for all amplifications was 0.2 μg and all samples went through two rounds of amplification. In a 10 μl reverse transcription reaction, 0.2 μg total RNA was transcribed by priming with a dT/T7-primer. Second strand synthesis was initiated by RNase digestion. The purified double stranded cDNA served as the template for the first round of in vitro aRNA transcription. For the subsequent round of amplification, the first strand cDNA was primed with random hexamers. The synthesis of the second strand was initiated by annealing a dT/T7-primer to the aRNA:cDNA heteroduplex with partially digested aRNA. After purification, a second round of in vitro transcription followed.
RNA labeling
Three μg of aRNA and 30 μg of total RNA was labeled with Cy3- or Cy5-labeled dCTP (Amersham Pharmacia Biotech AB) during reverse transcription. The reaction mixture consisted of random hexamers (16 μg) or anchored oligo-dT 20-mer primers (4 μg) depending on whether aRNA or total RNA was labeled. Further, 40 U RNAsin (Promega, Madison, WI), 1st strand buffer, 0.1 M DTT, 0.5 mM of dATP, dCTP, dGTP and 0.2 mM dTTP was added and the mix was incubated for 5 min at 65°C in a water bath. The tube was then transferred to a heating block (42°C), and 4 μl of either fluorophore was added to the respective tubes followed by 400 U SuperScript II (Invitrogen, Groningen, The Netherlands). After 60 min the reaction was stopped by adding 5 μl 0.5 M EDTA (pH = 8.0). Residual RNA was hydrolyzed with 10 μl 1 M NaOH and incubated at 65°C for either 60 or 15 min depending on whether total RNA or aRNA was reverse transcribed. Twenty-five μl 1 M Tris-HCl (pH 7.5) was added to neutralize the mixture. Micro Bio-Spin columns, P-6 (Bio-Rad Laboratories, Hercules, CA) were used to remove unincorporated dye. Labeled Cy3- and Cy5-cDNA was respectively diluted with TE-buffer (pH 7.5) before Microcon YM-30 columns (Amicon, Millipore Corporation, Bedford, MA) were applied to concentrate the samples to 17–20 μl.
Hybridization and scanning
The hybridization volume of 40 μl consisted of 17 μl of each of the labeled probes, 3.5 × SSC (pH = 7.5), 0.3% SDS, 1.25 × Denhardt's solution, 4 μg yeast tRNA, and 4 μg BSA. Additionally, 16 μg poly A (Amersham Pharmacia Biotech AB) was used if the probe was based on total RNA. The final mix was heated for 2 min at 100°C and spun down for 10 min at 13 K before it was applied on a microarray slide under the LifterSlip (Erie Scientific Company, Portsmouth, NH). The slide was then placed in an ArrayIT hybridization chamber (Telechem, Sunnyvale, CA) and submersed into a water bath for over night hybridization at 65°C. Prior to scanning, the coverslip was removed in a solution of 2 X SSC and 0.1% SDS, and the slide was washed in the following solutions for 5 min at room temperature; 1 X SSC, 0.2 X SSC and 0.05 X SSC. The slide was dried by centrifugation. Scanning was performed with a ScanARRAY 4000 (Packard Biosciences, Biochip Technologies LLC, Meriden, CT) scanner, and data was acquired from the images using GenePix Pro 3.0 software (Axon Instruments Inc., Union City, CA). A background-corrected intensity for each spot was formed by subtracting the mean of the pixels in the local background from the mean of the pixels in the spot.
Description of experiments
To avoid variability due to total RNA preparation, we pooled purified total RNA into a single large sample for each of the two RNA sources, the MT-1 and U2OS cell lines. The RNAs used in the experiments were based on these two initial samples. Aliquots from the initial sample represented a batch and there were six batches per cell line. Two of these batches were directly labeled in duplicated experiments using a dye swap strategy, while the remaining 4 diluted batches were used as starting material for independent, parallel amplifications. Corresponding pairs of aRNA, aRNA (MT-1) and aRNA (U2OS), were labeled and co-hybridized in duplicated experiments using (again) a dye swap strategy. Thus, in total there were 4 data sets based on non-amplified RNA and 8 based on amplified RNA. The microarray slides were all from the same print batch. Since it is impossible to hybridize non-amplified and amplified material on the same array, we chose to use two different cell lines on each microarray.
Data preparation
From the data for each microarray we first manually removed technically flawed spots. Then spots automatically flagged by the GenePix software were removed. Finally, all spots where the spot intensity (uncorrected foreground intensity) was lower than twice the background intensity in any of the two channels were removed (i.e. ((spot-background)/background) < 1). An overview of the number of filtered genes is given in Tables 1 and 2.
Systematic errors were corrected by normalizing the data using the locally weighted scatterplot smoother, lowess, as described in Yang et al. [24] and implemented in the sma package in the statistical language R. This routine normalized the ratios for each spot on each array by subtracting a value s, depending on the value of the log2-transformed total intensity of the spot on the array. In addition we normalized log2-transformed intensities by adding the value s/2 to the log2-transformed intensity of the Cy3 (green) channel and subtracting the value s/2 from the log2-transformed intensity of the Cy5 (red) channel. This normalization of intensities was done because the ANOVA model is based on log2-transformed intensities.
Analysis of variance modeling
The first step in the ANOVA modeling is to evaluate the sources of variability present, and their interactions. The next step is to construct the model and experimental design accordingly. We identified the following 6 main effects or factors for the design:
-We used 12 different microarrays (A).
-There were 2 cell lines (C).
-Each cell line was divided into 6 batches, giving a total of 12 batches (B).
-The material was labeled red (Cy5) or green (Cy3), so the dye (D) effect had 2 levels.
-The material was amplified on 8 of the microarrays and not amplified on the other 4, i.e. two levels for the protocol (P) effect.
-A total of 10759 genes (G) remained after filtering (these genes were present on at least one microarray).
Figure 1 presents an overview of the experimental design. For a particular cell line, the material was divided into 6 batches. These batches were split in two and labeled Cy5 (red) or Cy3 (green). The design is not balanced, since we chose to use twice as many microarrays with amplified material, but we wanted to be able to handle the possibility that each amplification event induced a larger variability between the arrays with amplified material than between the arrays with the non-amplified material.
Statistical model for the ANOVA analyses
To investigate the different sources of variability, we set up an ANOVA-based statistical model. Related models are found in Kerr et al. [25], Wolfinger et al. [26] and Jin et al. [27]. Let log2 yijklmn denote the log2-transform of the normalized measured intensity on array i, for cell line j, for batch k, using dye l and protocol m for gene n. We explain the log2-transformed intensity by the following model:
log2 yijklmn = μ + Ai + Cj + Bk + Dl + Pm + Gn
+ADil + CPjm + GPnm + CGjn + AGin + DGln + CGPjnm + εijklmn,
with i = 1,...,12, j = 1,2, k = 1,...,12, l = 1,2, m = 1,2, n = 1,...,10759, where μ is the overall mean, Ai is the overall array effect on array i, Cj is the overall cell line effect of cell-line j, Bk is the overall batch effect of batch k, Dl is the overall dye effect of dye l, Pm is the overall effect of the amplification protocol and Gn is the overall gene effect of gene n. Furthermore, ADil is the interaction between array and dye, so if this effect is significant, the dye has different effect for different arrays. CPjm models different effect of amplification on different cell lines. GPnm is of significant size if the amplification protocol has a different effect on different genes and CGjn represents the gene-specific cell line effect. This is usually an effect of interest e.g. if we want to measure different expression for the two cell lines for each gene. AGin is the spot effect, it models variation in the amount of cDNA deposited in each spot during the printing process. DGln represents the gene-specific dye effect. CGPjmn is the cell line specific GPnm effect, and is our interaction of primary interest.
Only the cell line effect (Cj), the dye effect (Dl), the protocol effect (Pm) and the cell line protocol interaction (CPjm) were modeled to be fixed effects (the first three have only two levels and the last is a combination of two fixed effects). CPjm is confounded with Cj and Pm, so we only estimated CP22. All other effects were modeled as independent normally distributed random effects with zero mean and constant standard deviation. This means that Ai ~ N(0, 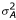 ), Bk ~ N(0,
), Bk ~ N(0, 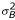 ), and similarly for all the other random effects. εijklmn is the modeling and measurement error, and it was also assumed to be normally distributed, εijklmn ~ N(0,
), and similarly for all the other random effects. εijklmn is the modeling and measurement error, and it was also assumed to be normally distributed, εijklmn ~ N(0, 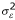 ). This is a mixed-effects model, since some of the effects are fixed and others are random.
). This is a mixed-effects model, since some of the effects are fixed and others are random.
We were interested in the preservation of the ratios after amplification and used the model for the log2-transformed intensity, log2 yijklmn, to set up an expression for the ratio of cell line 1 and cell line 2 for each spot on each array. Considering a randomly chosen spot, we wrote the observation of cell line 1 (j = 1) as log2 yijklmn and cell line 2 (j = 2) as log2 yijk'l'mn (the same array, i, was used, the same protocol, m, was used, it was the same gene, n, but batch, k and k', and dye, l and l', were different). The effects that were equal for each spot cancelled when looking at the ratio:
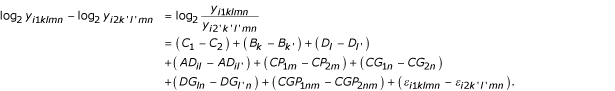
We wanted to see how the overall signal was affected by an amplification protocol that possibly did not fully preserve the ratios. The signal we are usually interested in is the gene expression using a specific cell type for a specific gene, i.e. CGjn. The Signal to Noise Ratio (SNR) is a valuable measure for answering this question. The SNR is defined as signal divided by noise. CGjn is the signal and the other effects are noise. Using the expression for the ratios, we see that the variance of the ratio of interest is
![]()
(the variance for the fixed effects are VAR(Cj) = VAR(Dl) = VAR(CPjm) = 0) so the Signal to Noise Ratio is
![]()
By comparing the Signal to Noise Ratio with and without the CGPjmn interaction present in the model, we were able to see how large impact amplification had on the signal. Hence, we estimated the parameters in the ANOVA model, and used the parameters estimated for the random effects to form the SNR to be investigated.
Due to the large dimensions in the mixed-effects model, we estimated the parameters of interest (the fixed effects and the variances of the random effects) using Gibbs-sampling. Gibbs-sampling is an iterative simulation method, which was run on the computer to provide our parameter estimates. We refer to the web page http://www.nr.no/documents/samba/research_areas/SMBI/NAR/ for details.
Hypothesis testing
Preservation of ratios can be addressed by performing hypothesis testing by comparing ratios between the two cell lines for the amplified material with ratios between the two cell lines for the non-amplified material for each gene. We have used this strategy as a complement to our preferred ANOVA-based procedure. Let νn(1) be the expected log2-ratio between the two cell lines for the non-amplified material for gene n, and let νn(2) be the expected log2-ratio between the two cell-lines for the amplified material for gene n. We wanted to test the following hypothesis for each gene n.
H0n: νn(1) = νn(2) vs H1n: νn(1) ≠ νn(2)
Many methods for performing hypothesis testing exist, and we chose to use a two-sample t-test for each gene separately. Thus, we estimated the variability for each gene in each group of samples separately, amplified and non-amplified (allowing the variability for the amplified samples to be different from the variability for the non-amplified sample). The Smith-Welch-Satterthwaite approximation to the degrees of freedom in the two-sample t-test was used, see any introductory statistical text e.g. Walpole, Myers and Myers [28], chapter 10.8.
Authors' contribution
VN participated in the experiment design, cultivated cells, isolated RNA, performed the amplifications and hybridizations, participated in the clone sequence analysis and drafted the manuscript. AL, MH, and ML designed the statistical models, performed the statistical analyses and wrote the statistics part of the manuscript. HR wrote the Gibbs-sampling software that the ANOVA-analyses are based on. FL participated with the clone sequence analysis. OM was in charge of the microarray production. ØF supervised the editing. EH participated in the experiment design, statistical approach and editing of manuscript. BSS guided the practical work, data processing, analyses and editing of manuscript.
Acknowledgments
Acknowledgements
We would like to thank Dr. Magne Aldrin and Professor Arnoldo Frigessi at the Norwegian Computing Center, for their contributions and valuable comments concerning the statistical parts of this paper. Vigdis Nygaard is a Research Fellow of the Norwegian Cancer Society.
Contributor Information
Vigdis Nygaard, Email: vigdisny@radium.uio.no.
Anders Løland, Email: Anders.Loland@nr.no.
Marit Holden, Email: Marit.Holden@nr.no.
Mette Langaas, Email: mettela@math.ntnu.no.
Håvard Rue, Email: hrue@stat.ntnu.no.
Fang Liu, Email: fangli@radium.uio.no.
Ola Myklebost, Email: olam@radium.uio.no.
Øystein Fodstad, Email: fodstad@radium.uio.no.
Eivind Hovig, Email: ehovig@radium.uio.no.
Birgitte Smith-Sørensen, Email: bss@radium.uio.no.
References
- Stears RL, Getts RC, Gullans SR. A novel, sensitive detection system for high-density microarrays using dendrimer technology. Physiol Genomics. 2000;3:93–99. doi: 10.1152/physiolgenomics.2000.3.2.93. [DOI] [PubMed] [Google Scholar]
- Karsten SL, Van Deerlin VM, Sabatti C, Gill LH, Geschwind DH. An evaluation of tyramide signal amplification and archived fixed and frozen tissue in microarray gene expression analysis. Nucleic Acids Res. 2002;30:E4. doi: 10.1093/nar/30.2.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci U S A. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM. High-fidelity mRNA amplification for gene profiling. Nat Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- Baugh LR, Hill AA, Brown EL, Hunter CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res. 2001;29:E29. doi: 10.1093/nar/29.5.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Mimori K, Yoshikawa Y, Shibuta K, Utsunomiya T, Sadanaga N, Tanaka F, Matsuyama A, Inoue H, Sugimachi K. Analysis of the gene-expression profile regarding the progression of human gastric carcinoma. Surgery. 2002;131:S39–47. doi: 10.1067/msy.2002.119292. [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Khanna C, Jazaeri AA, Petersen D, Liu ET. Core biopsies can be used to distinguish differences in expression profiling by cDNA microarrays. J Mol Diagn. 2002;4:30–36. doi: 10.1016/S1525-1578(10)60677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidl SJ, Nilsson S, Kalen M, Hellstrom M, Takemoto M, Hakansson J, Lindahl P. mRNA expression profiling of laser microbeam microdissected cells from slender embryonic structures. Am J Pathol. 2002;160:801–813. doi: 10.1016/S0002-9440(10)64903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Bowman ED, Simon R, Peng AC, Robles AI, Jones RT, Katagiri T, He P, Mizukami H, Charboneau L, et al. Laser capture microdissection and microarray expression analysis of lung adenocarcinoma reveals tobacco smoking- and prognosis-related molecular profiles. Cancer Res. 2002;62:3244–3250. [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier GM, Pyati J, Wan JS, Erlander MG. Screening differentially expressed cDNA clones obtained by differential display using amplified RNA. Nucleic Acids Res. 1997;25:913–914. doi: 10.1093/nar/25.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison RD, Robinson GA. lambda RNA internal standards quantify sensitivity and amplification efficiency of mammalian gene expression profiling. Biotechniques. 1998;25:504–508. doi: 10.2144/98253rr06. [DOI] [PubMed] [Google Scholar]
- Puskas LG, Zvara A, Hackler L, Jr, Van Hummelen P. RNA amplification results in reproducible microarray data with slight ratio bias. Biotechniques. 2002;32:1330–1334. doi: 10.2144/02326mt04. [DOI] [PubMed] [Google Scholar]
- Hu L, Wang J, Baggerly K, Wang H, Fuller GN, Hamilton SR, Coombes KR, Zhang W. Obtaining reliable information from minute amounts of RNA using cDNA microarrays. BMC Genomics. 2002;3:16. doi: 10.1186/1471-2164-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Hastie T, Whitfield ML, Borresen-Dale AL, Jeffrey SS. Optimization and evaluation of T7 based RNA linear amplification protocols for cDNA microarray analysis. BMC Genomics. 2002;3:31. doi: 10.1186/1471-2164-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman AL, Costouros NG, Wang E, Qian M, Marincola FM, Alexander HR, Libutti SK. Advantages of mRNA amplification for microarray analysis. Biotechniques. 2002;33:906–912. doi: 10.2144/02334mt04. [DOI] [PubMed] [Google Scholar]
- Smith L, Underhill P, Pritchard C, Tymowska-Lalanne Z, Abdul-Hussein S, Hilton H, Winchester L, Williams D, Freeman T, Webb S, et al. Single primer amplification (SPA) of cDNA for microarray expression analysis. Nucleic Acids Res. 2003;31:E9–9. doi: 10.1093/nar/gng009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Salunga RC, Guo H, Bittner A, Joy KC, Galindo JE, Xiao H, Rogers KE, Wan JS, Jackson MR, et al. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat Med. 1999;5:117–122. doi: 10.1038/4806. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JRSS. 1995;57:289–300. [Google Scholar]
- Dudoit S, Yang YH, Bolstad B. Using R for the analysis of DNA microarray data. R news. 2002;2:24–32. [Google Scholar]
- Schweder T, Spjøtvoll E. Plots of p-values to evaluate many tests simultaneously. Biometrika. 1982;69:493–502. [Google Scholar]
- Aoyagi K, Tatsuta T, Nishigaki M, Akimoto S, Tanabe C, Omoto Y, Hayashi S, Sakamoto H, Sakamoto M, Yoshida T, et al. A faithful method for PCR-mediated global mRNA amplification and its integration into microarray analysis on laser-captured cells. Biochem Biophys Res Commun. 2003;300:915–920. doi: 10.1016/S0006-291X(02)02967-4. [DOI] [PubMed] [Google Scholar]
- Naundorf H, Rewasowa EC, Fichtner I, Buttner B, Becker M, Gorlich M. Characterization of two human mammary carcinomas, MT-1 and MT-3, suitable for in vivo testing of ether lipids and their derivatives. Breast Cancer Res Treat. 1992;23:87–95. doi: 10.1007/BF01831480. [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet. 2001;29:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- Walpole RE, Myers SL, Myers RH. Prentice-Hall International, Upper Saddle River, NJ. 6 1998. Probability and statistics for engineers and scientists. [Google Scholar]