

Abstract
Background
The retina, which is exposed to both sunlight and very high levels of oxygen, is exceptionally rich in polyunsaturated fatty acids, which makes it a favorable environment for the generation of reactive oxygen species. The cytotoxic effects of hydrogen peroxide (H2O2) induced oxidative stress on retinal pigment epithelium were characterized in this study.
Methods
The MTT cell viability assay, Texas-Red phalloidin staining, immunohistochemistry and Western blot analysis were used to assess the effects of oxidative stress on primary human retinal pigment epithelial cell cultures and the ARPE-19 cell line.
Results
The treatment of retinal pigment epithelial cells with H2O2 caused a dose-dependent decrease of cellular viability, which was preceded by a significant cytoskeletal rearrangement, activation of the Extracellular signal-Regulated Kinase, lipid peroxidation and nuclear condensation. This cell death was prevented partially by the prostaglandin derivative, 15d-PGJ2 and by the protein kinase inhibitor, AG126.
Conclusion
15d-PGJ2 and AG126 may be useful pharmacological tools in the future capable of preventing oxidative stress induced RPE cell death in human ocular diseases.
Background
The retina, which is exposed to both sunlight and very high levels of oxygen, is exceptionally rich in polyunsaturated fatty acids, which makes it a favorable environment for the generation of reactive oxygen species. For example, retinal pigment epithelium (RPE) generates a number of reactive oxygen species when illuminated with light [1]. Furthermore, phagocytosis of photoreceptor outer segments by RPE causes an increase in intracellular [2] and extracellular H2O2 generation [3]. Oxidative stress has been suggested as the cause of a number of retinal pathological conditions [4,5].
A number of in vitro studies have attempted to characterize the effect of oxidative stress on RPE. Depending on the experimental conditions, treatment of RPE with H2O2 can cause an alteration of cellular functions [6,7] or cell death [8,9]. Cytotoxic levels of H2O2 can cause significant mitochondrial [10] and genomic [11] DNA damage in RPE simulating the features of programmed cell death [8,12]. Studies from other cell types indicate that early cellular events following H2O2 treatment include morphological alterations and actin re-organization [13]. In addition, activation of the ERK (p42/44, Extracellular signal-Regulated Kinase) MAP kinases (Mitogen-Activated Protein kinases) can occur within minutes following cellular oxidative stress. Depending on the cell type examined, inhibition of the ERK activity can either prevent [14] or enhance H2O2-induced cell death [15]. The involvement of actin re-organization and ERK activation has not been examined in RPE under oxidative stress.
Lipid peroxidation products, such as 4-hydroxynonenal (4-HNE), have attracted considerable attention because of their potential involvement in aging and in a number of neuropathological conditions [16]. 4-HNE was implicated in the etiology of some pathological conditions in the eye. For example, levels of 4-HNE increased significantly in the vitreous of patients with proliferative vitreoretinopathy compared with controls [17]. Further, 4-HNE forms a stable adduct with rhodopsin in the photoreceptor, which may interfere with its functions [18]. 4-HNE even causes cataracts in cultured rat lens [19,20]. Whether oxidative stress on RPE can cause accumulation of cellular HNE-protein adducts remains unknown.
Peroxisome proliferator-activated receptors (PPARs) belong to a group of nuclear receptors that include steroid, retinoid, thyroid hormone receptors and others [21-23]. There are three types of PPARs: PPARα is expressed predominantly in the liver, heart, kidney, brown adipose and stomach mucosa, and is important for lipid catabolism. PPARγ is found in adipose tissues and is important for adipogenesis. PPARβ is found in most tissues, but its role is less well defined. RPE cells express all three forms of PPARs but PPARβ is the dominant isoform [24]. There is a 10-fold induction of PPARγ mRNA that reaches a peak at 4-hour after phagocytosis of photoreceptor outer segments. The levels of either PPARα mRNA or PPARβ mRNA are not altered under the same conditions. Increases in PPARγ in RPE cells may be important for handling the lipids generated during the phagocytosis of photoreceptor outer segments [24]. Since RPE cells generate large amounts of H2O2 during the phagocytosis of photoreceptor outer segments, there is a possibility that activation of PPARγ may assist in defending the oxidative stress associated with phagocytosis.
This study was designed to examine morphological alterations in RPE during oxidative stress, the involvement of ERK MAP kinase and 4-HNE and to test the hypothesis that PPARγ activation can prevent oxidative damage on RPE cells. The human RPE cell line, ARPE-19, and primary cultures of adult human RPE were used in this study. ARPE-19 is a spontaneous occurring, non-transformed RPE cell line which expresses RPE specific markers (such as CRALBP and RPE65) and exhibits morphological polarization and tight junctions similar to native RPE [25,26].
Methods
Materials
WY14643, 15d-PGJ2, azelaoyl PAF and ciglitazone were purchased from Cayman (Ann Arbor, MI). LY 171883 was purchased from Alexis Biochemicals (San Diego, CA). AG126 and PD98059 were purchased from Calbiochem (San Diego, CA). Hydrogen peroxide and other pharmacological and general biochemical reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated.
Cell cultures
ARPE-19 cells [25] were obtained from the American Type Culture Collection and grown in DMEM/F12 containing 10% heat-inactivated fetal bovine serum and 2.5 mM glutamine. Human primary RPE cultures were established from donor eyes obtained from the Arkansas Lions Eye Bank & Laboratory. The globe was sectioned at the ora serrata, and the lens and vitreous body were removed. The eye cup was then filled with 4% dispase (prepared in DMEM) containing 1× antibiotic-antimycotic reagents (final concentrations: 100 units/ml Penicillin G, 100 μg/ml Streptomycin and 0.25 μg/ml Amphotericin B) and incubated in a 37°C incubator with 5% CO2 for ~30 minutes. RPE cells were then removed under a dissecting microscope, dissociated by trituration using a Pasteur pipette, and then plated on poly-L-lysine (25 mg/liter in water) coated 35-cm2 culture dishes. The cell population was then expanded by sequentially subculturing into 25-cm2 tissue culture flasks, then to 75-cm2 tissue culture flasks. Cells of passages #3–#6 were used for this study. The culture medium consisted of 10% heat-inactivated fetal bovine serum, 2.5 mM glutamine and 1× antibiotic-antimycotic reagents in DMEM.
Cell viability
Cells were plated in 96-well plates (human primary RPE: 10,000 cells/well, ARPE-19: 15,000–20,000 cells/well, unless otherwise stated) overnight, fed with serum-free medium or treated with testing agents (prepared in serum-free medium) the next day followed by H2O2 treatment for a day, then the viability from each treatment was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [27]. Culture medium was removed after treatment and then 100 μl MTT solution (100 μg/ml prepared in DMEM) was added to each well. The cultures were incubated at 37°C for one hour in a tissue culture incubator. The MTT solution was then removed and the cells in each well were lysed by the addition of 100 μl dimethyl sulfoxide. The plate was placed on a shaker for one hour at room temperature to complete the lysing process, then the optical density of each well was measured by a 96-well plate reader with a filter setting at 570 nm (reference filter setting was 630 nm). When a pharmacological agent was used in the experiment, the MTT reading from cultures treated with that agent alone was used as 100% cellular viability.
Actin staining
This was performed as described by Carter et al. [28]. Cells grown on coverslips were treated with H2O2 for various period of time as indicated in figure legends, washed three times with phosphate-buffered saline (PBS), then fixed for 10 minutes with 3.7% formaldehyde (prepared in PBS). The cultures were then washed three times with PBS, extracted with -20°C acetone for 5 minutes followed by three washes with PBS. Each culture was then stained with Texas Red-phalloidin (Molecular Probes, Eugene, OR) prepared in PBS containing 1% bovine serum albumin for 20 minutes at room temperature in a lightproof box. The cultures were then washed three times with water, mounted with GEL/MOUNT (Biomedia, Foster City, CA). The slides were examined under a Nikon microscope Eclipse E600 (Nikon Instruments Inc., Melville, NY). Cells were photographed digitally at a fixed exposure time by using a Photometrics CoolSNAP fx camera (Roper Scientific, Inc., Tucson, AZ) and the software MetaVue, Meta Imaging Series 4.5 (Universal Imaging Corporation, Downingtown, PA).
Immunohistochemical and immunofluorescent staining
Cultures grown on coverslips were used in this set of experiments. Phospho-ERK was detected by mouse anti-phospho-ERK antibody (Santa Cruz Biotechnology, Santa Cruz, CA, used at 1:1500) and the Vectastain ABC kit (Vector Lab., Burlingame, CA) with diaminobenzidine as the chromogen. Cellular 4-HNE protein adducts were detected by rabbit anti-4-HNE antibodies (Calbiochem, San Diego, CA, used at 1:250) followed by anti-rabbit secondary antibody labeled with FITC (Dako Corporation, Carpinteria, CA). The cultures were then mounted with GEL/MOUNT. The slides were examined under Nikon microscope Eclipse E600 (Nikon Instruments Inc., Melville, NY). Cells were photographed at fixed exposure time with Nikon digital still camera DXM 1200 (Nikon Instruments Inc., Melville, NY) and the software Nikon ACT-1 version 2.11 (Nikon Corporation, Tokyo, Japan).
Nuclear staining
Cells grown on coverslips were treated with H2O2 for various periods of time as indicated in figure legends, fixed for 10 minutes with 3.7% formaldehyde (prepared in PBS), stained with 1 μg/ml bisbenzimide solution (Hoechst 33258, Sigma, St. Louis, MO) for 10 minutes at room temperature, mounted with GEL/MOUNT, then observed under a fluorescent microscope as described in the previous section.
Western blot analysis
RPE cells were plated in T-150 flasks overnight, fed with serum-free medium for a day, then treated with H2O2 as indicated in figure legends. For sample preparation, cultures were detached with a scraper in PBS then centrifuged. The cell pellets were suspended in 150–300 μl ice-cold RIPA lysis buffer (1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS in PBS, 0.5 mM Phenylmethylsulfonyl fluoride (PMSF), 0.02 mg/ml Aprotinin, 1 mM Na3VO4) and incubated for 30 min on ice. Each sample was further homogenized by passage through a 21-gauge needle followed by an addition of 10 μl PMSF (stock solution: 50 mM). The samples were then incubated for 30 min on ice and subsequently centrifuged at 14,000 RPM for 20 min at 4°C. The supernatant from each sample was collected and an aliquot was taken for protein concentration determination by Micro BCA Protein Assay Reagent Kit (Pierce, Rockford, IL). Protein samples were stored at -70°C until ready for electrophoretic analysis. Equal amounts of protein samples (20 μg/well) were heated at 95°C for 6 min then loaded onto 1% SDS, 10% polyacrylamide gels. BenchMark pre-stained protein ladder (Invitrogen life technologies, Carlsbad, California) was used as molecular weight standards. Following electrophoretic separation, the proteins were transferred to nitrocellulose membranes. Equal loading and appropriate transfer of each lane was confirmed by staining the membrane with the Ponceau S solution (Sigma, St. Louis, MO). Membranes were blocked in 5% nonfat dried milk for one hour, washed in 10 mM Tris-buffered saline (pH 7.5) containing 0.1% Tween-20 (TBST) and probed with primary antibody overnight at 4°C. Membranes were washed with TBST and then incubated for 1 hour at room temperature with a secondary antibody followed by washing three times with TBST, immersed in ECL Plus western blotting detection system (Amersham Pharmacia Biotech, Buckinghamshire, England) for 1 min and then exposed to Kodak X-ray film (Rochester, NY). The films were analyzed by densitometry using a ChemiImager 5500 imaging system with AlphaEaseFC software (Alpha Innotech Corporation, San Leandro, CA). Results, expressed as percentage of control, from replicate experiments were pooled and averaged. The primary antibodies used in this set of experiments were from Santa Cruz (anti-ERK 1/2, used at 1:5000; anti-phospho-ERK, used at 1:1000; anti-PPARγ, used at 1:2000) or from Calbiochem (anti-4-HNE, used at 1:2,000).
Statistical analysis
Unless otherwise stated, results of cell viability experiments were pooled from 12 replicate samples derived from 3 independent experiments, and expressed as mean ± SEM. Statistical analyses were performed by analysis of variance (one-way ANOVA) followed by the Bonferroni test to determine the significance of difference.
Results
Cytotoxicity of H2O2 toward primary human RPE and ARPE-19 cells
Initial experiments analyzing the cytotoxic effect of oxidative stress on primary cultures of human RPE cells indicated that H2O2 at 1 mM or less did not affect cellular viability. Higher concentrations caused a sharp decrease of viability in human RPE such that a 24-hour treatment of cells with 1.2 mM H2O2 lowered the viability to 13% of controls (Fig. 1A, squares). This concentration lowered the viability of human RPE to 62% after a 4-hour treatment (Fig. 1A, circles).
Figure 1.
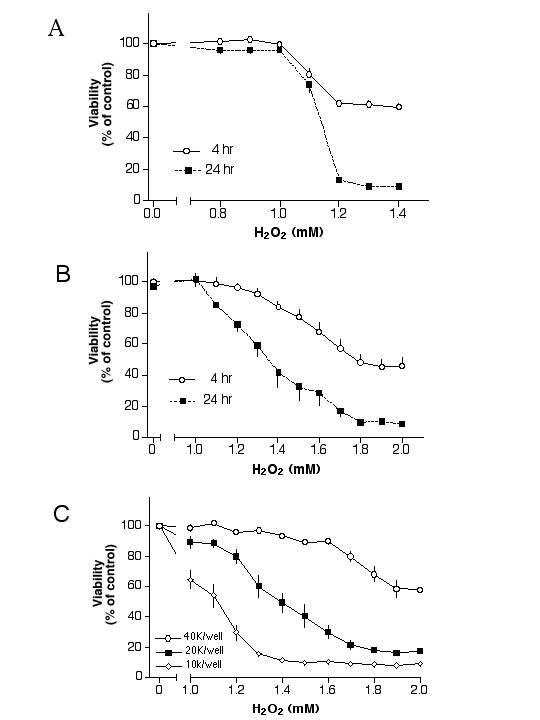
Cytotoxicity of H2O2 on human RPE cells. Fig. 1A: Primary human RPE cells were plated in 96-well plate, fed with serum-free medium the next day, then treated with H2O2 for 4 hours (circles) or 24 hours (squares). Fig. 1B: ARPE-19 cells were plated in 96-well plate, fed with serum-free medium the next day, then treated with H2O2 for 4 hours (circles) or 24 hours (squares). Fig. 1C: ARPE-19 cells were plated with a density of 10,000 cells/well, 20,000 cells/well or 40,000 cells/well in 96-well plates, fed with serum-free medium the next day, then treated with H2O2 for 24 hours. The viability of each treatment was determined by the MTT assay. Cultures at a density of 40,000 cells/well were 100% confluent.
In a set of analogous experiments, ARPE-19 cells treated with various concentrations of H2O2 for 4 hours or 24 hours showed that a 4-hour treatment with 1 mM H2O2 did not affect viability. An increase of H2O2 concentrations beyond 1 mM decreased viability such as 2 mM H2O2 decreased the viability to 46% of controls in 4 hours (Fig. 1B, circles). Treatment of cells with H2O2 for 24 hours caused a dose-dependent decrease of viability with a LD50 of ~1.35 mM (Fig. 1B, squares).
H2O2 induced cytotoxicity in RPE cells appears to be affected by the density of cells in cultures [29]. To study this phenomenon, ARPE-19 cells plated in 96-well plates with densities of 10,000, 20,000 or 40,000 cells/well were subjected to H2O2 treatment. Cells grown in a high-density culture were indeed more resistant to H2O2 as compared to those grown in a low-density culture (Fig. 1C). For example, H2O2 at 1.4 mM reduced cellular viability to 11%, 49% or 94% of control in cultures with 10,000, 20,000 or 40,000 cells/well, respectively.
H2O2 induced morphological alterations and formation of 4-HNE-adducts
To analyze morphological changes of RPE cells treated with a cytotoxic concentration of H2O2, ARPE-19 cells were treated with 1.5 mM H2O2 for various periods of time and then processed for actin staining. Without H2O2 treatment, stress fibers appeared thin and diffuse in control cells (Fig. 2A, arrow). At 15-min after treatment, these cells appeared to round up and significant ruffling occurred at the edge of their plasma membrane (Fig. 2B, arrows). Overall thickening of stress fibers occurred at two hours after treatment in addition to the membrane ruffling (Fig. 2C, arrows). Additional morphological changes after 4 hours of H2O2 treatment (Fig. 2D,2E,2F) included distinctive dense bands at the periphery of some cells (Fig. 2D, arrows), peri-nuclear staining of actin fibers in other cells (which made the nuclei prominent, Fig. 2E, arrows) and microspikes (Fig. 2F, arrow) on the cell surface of some cells.
Figure 2.
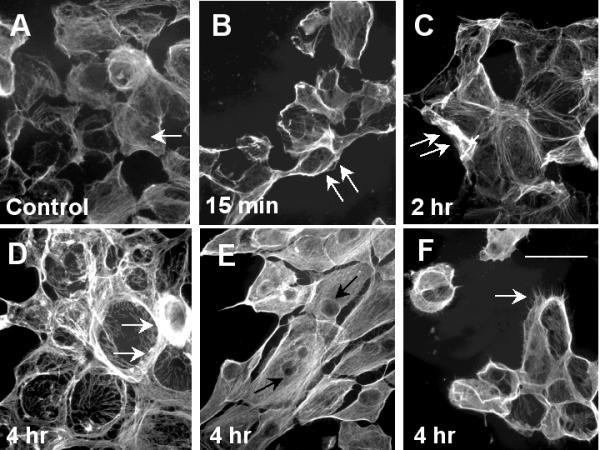
Reorganization of actin fibers caused by oxidative stress. ARPE-19 cells were treated with 1.5 mM H2O2 then processed for actin stain as described in the Methods. Fig. 2A: untreated; Fig. 2B: 15-min; Fig. 2C: 2-hour; Figs. 2D,2E and 2F: 4-hour after treatment. See text for detailed discussion of the results. Scale bar in Fig. 2F: 50 μm.
Subsequent experiments with immunofluorescent staining indicated that 1.5 mM H2O2 treatment of ARPE-19 cells for 4 hours led to the formation of 4-HNE protein adducts both in the cytoplasm and nucleus, presumably as a result of lipid-peroxidation (Fig. 3A,3B). Various protein bands on Western blots were positive for anti-HNE antibody reactivity, further intensified as a result of H2O2 treatment (Fig 3C). Densitometry measurements were performed to estimate the collective increase of band intensity in each lane between 39 Kd and 87 Kd. Results derived from a total of six independent experiments indicated that treatment of cells with 0.5, 1, 1.5 or 2 mM H2O2 caused an increase of intensity to 2.5-, 3.1-, 3.7- or 7.1-fold of control, respectively.
Figure 3.
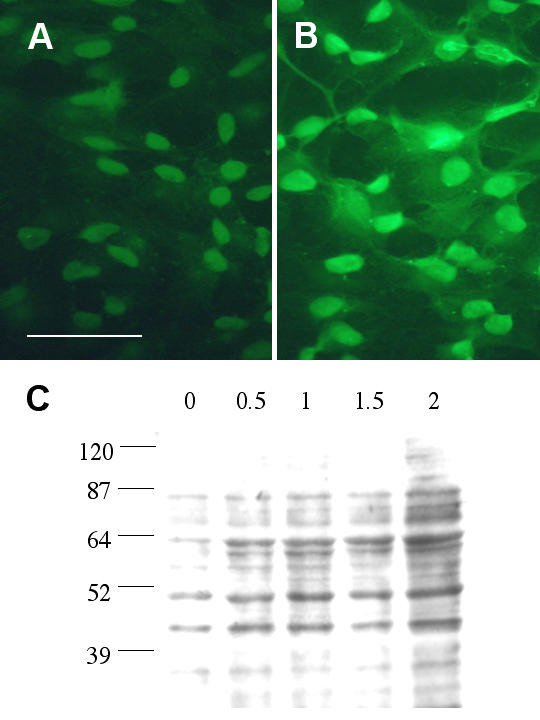
H2O2 induced lipid peroxidation in RPE. ARPE-19 cells were treated with 1.5 mM H2O2 for 4 hours then processed for immunofluorescent staining using antibodies against 4-HNE. Fig. 3A: untreated cells. Scale bar: 50 μm. Fig. 3B: H2O2 treated cells. Note the overall enhanced staining in nucleus and cytoplasm. Fig. 3C: ARPE-19 cells were treated with various concentrations (0.5, 1, 1.5, 2 mM) of H2O2 for 24 hours then processed for Western blot analysis using anti-4-HNE antibodies. These polyclonal antibodies recognize cysteine-, histidine- and lysine-4-HNE Michael adducts. This is a representative result from 6 similar experiments.
Though there was significant cytoskeletal alterations and lipid peroxidation at 4 hours after H2O2 treatment, nuclear staining using bisbenzimide (Hoechst 33258) indicated that most cells maintained normal nuclear morphology at this time (Fig. 4A, untreated cells; Fig. 4B, 4 hours after treatment). Nuclear condensation increased with length of H2O2 exposure, which was clearly evident at 12 hours after H2O2 treatment (Fig 4C, arrows), and even more prominent at 16 hours after treatment (Fig. 4D). These results suggest that H2O2treatment caused apoptosis in RPE. Nuclear condensation appeared to start at the edge of the culture (relatively lower density area, Fig. 4C, arrows) and spread to the center part of the culture (higher density, Fig. 4D) over time. This phenomenon is consistent with the viability assay in which higher density cultures were more resistant to oxidative stress (Fig. 1C).
Figure 4.
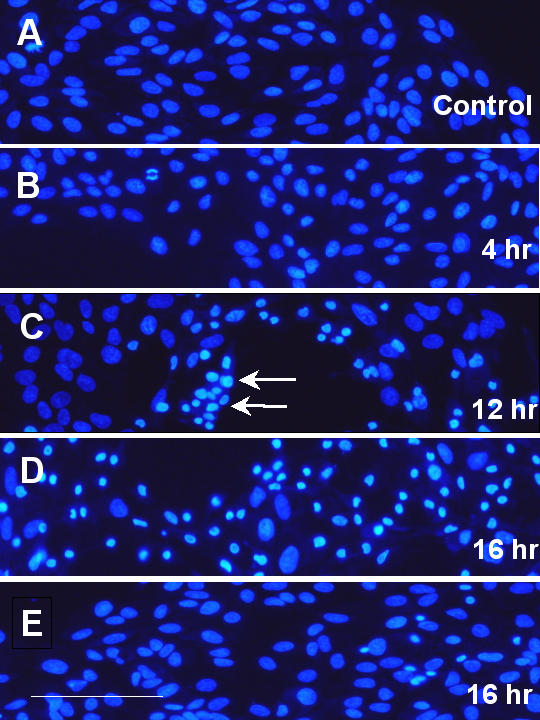
H2O2 induced nuclear condensation in ARPE-19 cells. ARPE-19 cells were treated with 1.5 mM H2O2 for various periods of time, and then processed for nuclear staining. Fig. 4A: untreated cells; Fig. 4B: 4-hour; Fig. 4C: 12-hour; Fig. 4D: 16-hour after treatment. Fig. 4E: Cells were pretreated with 1 μM 15d-PGJ2 overnight, followed by 1.5 mM H2O2 for 16 hours (without 15d-PGJ2). See text for detailed description of the results. Scale bar in Fig. 4E: 100 μm.
Effect of oxidative stress on PPARγ protein expression
During rod outer segment ingestion, there is a generation of H2O2 [2,3] and an up-regulation of PPARγ mRNA expression [24] in RPE, experiments were performed to determine if H2O2 could induce PPARγ protein expression over the control levels. ARPE-19 cells were treated with various concentrations (0.5, 1, 1.5, 2 mM) of H2O2 for 24 hours and processed for Western blot analysis. Results indicated a slight induction of PPARγ protein levels in some experiments. This induction, however, was not apparent in other experiments (Results not shown). Densitometry analyses from a total of six independent experiments indicated that treatment of these cells with 0.5, 1, 1.5 or 2 mM H2O2 for 24 hours caused an alteration of band intensity to 76%, 108%, 113% or 104% of controls, respectively. There was no significant change of PPARγ protein level after 30-min or 12-hour H2O2 treatment, either (results not shown). A separate set of experiments also indicated that treatment of primary human RPE cells with 1 mM H2O2 for 24 hours did not induce PPARγ protein expression over the control levels (results not shown).
Prevention of oxidative stress induced cytotoxicity by PPARγ agonists
Even though H2O2 did not increase PPARγ protein expression significantly as indicated in the previous study, experiments were performed to determine whether PPARγ agonists could activate the existing PPARγ thus preventing H2O2 induced cytotoxicity in RPE cells. Cells were pretreated with the PPARγ agonist 15d-PGJ2 overnight followed by H2O2 for one day (without 15d-PGJ2) and processed for the MTT viability assay. Results indicated that 15d-PGJ2 prevented H2O2 induced cytotoxicity in primary human RPE cultures in a dose-dependent manner (Fig. 5A). Subsequent experiments with ARPE-19 cells also indicated this saving effect. While H2O2 at 1.3 mM, 1.4 mM or 1.5 mM lowered the ARPE-19 viability to 64%, 46% or 28% of control, respectively; pretreatment of these cells with 1 μM 15d-PGJ2 raised the viabilities to 95%, 84% or 68% of control, respectively (Fig. 5B). Cells stained with Hoechst 33258 indicated that 15d-PGJ2 prevented H2O2 induced nuclear condensation (Fig. 4E; compare this with Fig. 4D). The other PPARγ agonists, ciglitazone [30], azelaoyl PAF [31] and LY171883 [32], however, were not effective (tested between 1–10 μM, 3 independent experiments for each agent, results not shown). The PPARα agonist WY14643 had some saving effect at a high concentration (40 μM), however, the difference was not statistically significant (results not shown).
Figure 5.
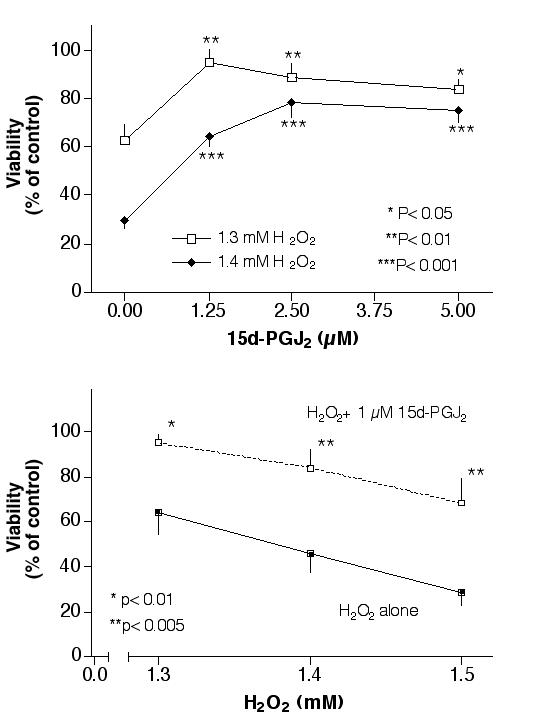
Prevention of H2O2 induced cytotoxicity by 15d-PGJ2 in RPE cells. Fig. 5A: Primary RPE cells were pretreated with various concentrations of 15d-PGJ2 overnight, followed by 1.3 mM or 1.4 mM H2O2 for 1 day (without 15d-PGJ2), then processed for the MTT viability assay. Fig. 5B: ARPE-19 cells were pretreated with 1 μM 15d-PGJ2 overnight, followed by various concentrations of H2O2 for 1 day (without 15d-PGJ2), then processed for the MTT viability assay. Statistical analyses were performed comparing cells treated with H2O2 alone versus H2O2 plus 15d-PGJ2.
Activation of ERK by H2O2
A set of experiments was performed to determine the effects of H2O2 on ERK activation. ARPE-19 cells were treated with 1.5 mM H2O2 for 1 hour and processed for immunohistochemical staining using antibodies against phospho-ERK. Untreated cells showed a faint phospho-ERK staining while there was an apparent increase of phospho-ERK staining in the cytoplasm and nuclei in H2O2 treated cells (Fig. 6A,6B). A separate set of cultures was treated with H2O2 for 30 min and then processed for Western blot analysis. This H2O2 treatment of the cells caused only a slight alteration in ERK levels but led to a dose-dependent increase of phospho-ERK, with a maximal stimulation observed at 1.5 mM (Fig. 6C). Densitometry analyses from 3 independent experiments indicated that 0.5, 1, 1.5 or 2 mM H2O2 caused an increase of p42ERK (ERK2) phosphorylation to 2.1-, 12.7-, 18.8- or 2.0- fold of control, respectively. Similar treatment caused an increase of p44 ERK (ERK1) phosphorylation to 1.5-, 15.7-, 21.4- or 3.4-fold of control, respectively. The phospho-ERK in H2O2 treated samples remained higher than control at 24-hour after treatment (results not shown).
Figure 6.
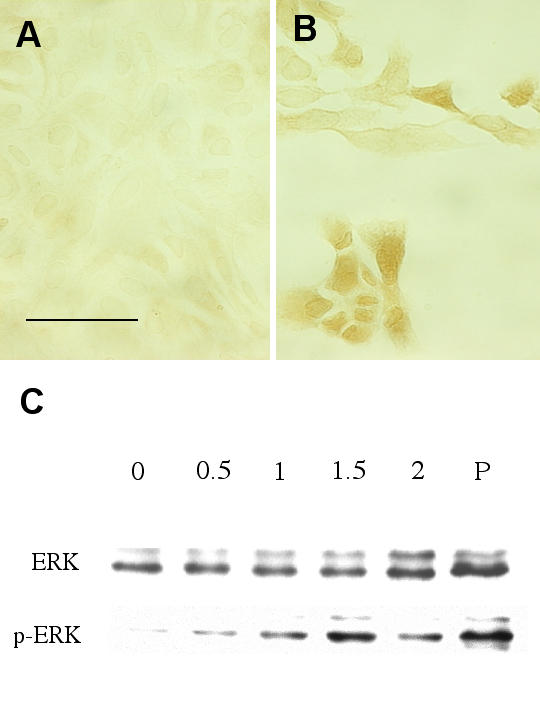
Activation of ERK by H2O2 treatment. ARPE-19 were treated with 1.5 mM H2O2 for one hour, then processed for immunohistochemical staining using anti-phospho-ERK antibody. Fig. 6A: untreated cells. Scale bar: 50 μm. Fig. 6B: H2O2 treated cells. Fig. 6C: ARPE-19 were treated with various concentrations (0.5, 1, 1.5, 2 mM) of H2O2 for 30 minutes, and then processed for Western blot analysis. Antibodies against ERK and phospho-ERK were used in this study. P: positive controls. This is a representative result from 3 similar experiments.
Effect of ERK inhibition on H2O2 induced cytotoxicity in ARPE-19
Since ERK activation appeared to be an early event associated with H2O2 induced cytotoxicity, experiments were performed to determine if inhibitors of ERK activation could affect the cytotoxicity caused by H2O2. ARPE-19 cells were pretreated with 10 μM PD98059 (a MEK inhibitor which functions upstream of ERK [33]) followed by H2O2 challenge. Results from 3 independent experiments indicated that this agent could prevent low-dose (1–1.2 mM) H2O2 induced cell death slightly. In contrast, it enhanced high-dose (1.3–1.6 mM) H2O2 induced cell death. These effects in either case were not remarkable (results not shown). This agent at concentrations higher than 10 μM was toxic to ARPE-19 cells by itself and could enhance H2O2 toxicity (results not shown).
In a separate set of experiments, ARPE-19 cells were pretreated with 10 μM AG126 (a protein tyrosine kinase inhibitor [34]) followed by H2O2 challenge. The treatment of cells with 1.3 mM, 1.4 mM or 1.5 mM H2O2 decreased the cellular viability to 66%, 40% or 22% of control, respectively. Pretreatment of cells with AG126 raised the viability to 72% (p < 0.05 compared to H2O2 treatment only), 63% (p < 0.005) or 35% (p < 0.01) of control, respectively. AG126 at concentrations higher than 10 μM was toxic to ARPE-19 cells by itself and could enhance H2O2 toxicity (results not shown).
Both AG126 (10 μM) and 15d-PGJ2 (1 μM) could prevent H2O2 induced cytotoxicity in ARPE-19, a set of experiments was also performed to determine whether these two agents had additive saving effects. While 1.4 mM H2O2 treatment reduced the viability to ~40% of control, AG126 raised this viability to ~55% of control and 15d-PGJ2 raised the viability to 75% of control. A combination of these two agents did not raise the viability above 15d-PGJ2 alone (Fig. 7).
Figure 7.
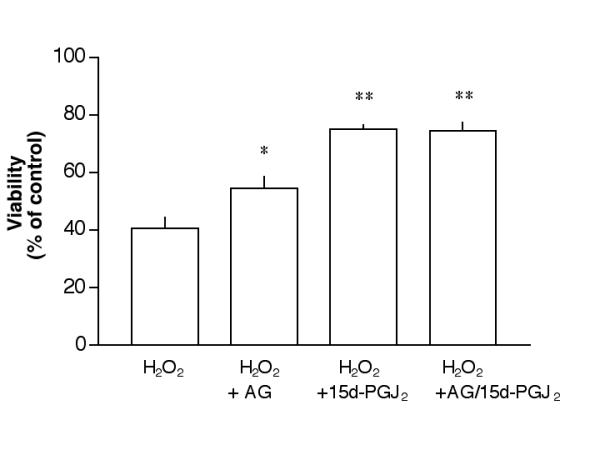
Saving of RPE cells by AG126 and 15d-PGJ2. ARPE-19 cells were treated with 10 μM AG126, 1 μM 15d-PGJ2 or a combination of these two agents overnight, followed by 1.4 mM H2O2 for 1 day (without testing agents), then processed for the MTT viability assay. *P < 0.05 as compared to H2O2 alone; **p < 0.001 as compared to H2O2 alone. There was no statistical difference between H2O2 +15d-PGJ2 and H2O2 +AG/15d-PGJ2.
Discussion
Cytotoxicity of oxidative stress on RPE
Results from this study indicated that both primary human RPE cultures and ARPE-19 cells were susceptible to H2O2 treatment. It is important to point out that H2O2 showed a very steep dose-response curve in primary RPE cultures under our experimental conditions such that an increase of H2O2 concentration from 1 mM to 1.2 mM for one-day treatment caused a decrease in viability from ~100% to ~10% of control (Fig. 1A). Though the H2O2 titration curve in ARPE-19 was not as steep as observed in primary RPE, one-day treatment of these cells with 1.8 mM H2O2 could decrease the cellular viability to ~10% of control while 1 mM H2O2 did not affect cellular viability (Fig. 1B). The sharp dose-response curves demonstrated in this study suggests that it is important to have an effective cellular anti-oxidation mechanism to prevent the build-up of oxidative stress over a critical level. An increase of oxidative stress in RPE is associated with an increase of cellular catalase, metallothionein [3] and glutathione S-transferase [35], which should serve as a protective mechanism to decrease the cytotoxicity caused by H2O2 and other reactive oxygen species. This protective mechanism declines with age because a study analyzing metallothionein levels in RPE of macular region showed a dramatic decrease in aged donors (mean age = 80-yr) as compared to those from younger donors (mean age = 58-yr) [36]. A separate report concluded that there was also an age-dependent decrease of catalase activity in RPE [37]. These studies suggest RPE in the elderly are more susceptible to oxidative stress induced damage, which may contribute to age-related RPE dysfunction and vision loss. The oxidative stress induced RPE cytotoxicity can be aggravated by zinc deficiency, as previously reported by Tate et al. [9].
Significant morphological changes were observed in RPE cells treated with a cytotoxic concentration of H2O2. Actin fiber re-organization was noticed as early as 15 minutes after treatment (Fig. 2). Those images presented in Fig. 2D,2E and 2F are three representatives of RPE cells at 4 hr after H2O2 treatment. The cells shown in 2D and 2F are commonly seen at different locations on the same slide. In Fig. 2D, the dense peripheral bands of actin fibers were very prominent in some cells and the high intensity fluorescence caused an impression that the cells were relatively empty. The thickening of dense peripheral bands in this study was in contrast to previous observations in endothelial cells. For example, Zhao and Davis reported that cultured pulmonary endothelial cells treated with H2O2 underwent a morphological change that was accompanied by an accumulation/increase in stress fibers running across the cells while the dense peripheral bands of most cells were disrupted or eliminated [38]. Similar observations on endothelial cells were made by Liu and Sundqvist [39]. The dense peripheral bands are important in cell-cell adhesion, which maintains the structural integrity. While it is apparent that endothelial cells used in those studies and epithelial cells used in this study respond differently to H2O2, the underlying reason for this difference is currently unknown. One may speculate that RPE cells use this build-in mechanism to defend the oxidative stress that they are constantly exposed to. We currently have no data to support or dismiss this possibility. In Fig. 2E, some cultures responded to H2O2 treatment by a perinuclear distribution of actin fibers. In Fig. 2F, we observed microspikes at the periphery of the cells. A detailed study of microspikes by Adams indicated that these structures consist of actin, the actin binding protein, 55 kd/fascin and myosin [40]. The perinuclear staining and microspikes of cells under oxidative stress in this study are very similar to those observed by Huot et al. in endothelial cells treated with H2O2 [41]. The functional significance of these structural changes in RPE after H2O2 treatment is not clear. There is a possibility that the appearance of microspikes around the cells under stressed conditions is an adapted behavior of the cells in order to attach to the substratum firmly. Prolonged H2O2 treatment led to cell death with the appearance of condensed nuclei is an indication of apoptosis (Fig. 4C,4D). This general finding is consistent with that observed by others [8,12].
It is of interest to note that cultures with a reduced density are more vulnerable to H2O2 treatment (Fig. 1C). One straightforward explanation for the vulnerability of low-density cultures treated with H2O2 is that more molecules of H2O2 per cell were present in these low-density cultures, which caused a lower viability. If this were the case, a mathematic analysis of the H2O2 molecules per cell should be able to estimate the cytotoxicity in this experimental system. For example, 1.4 mM H2O2 in a culture plated at a density of 40,000 cells/well should result in a viability close to twice of that generated in 20,000 cells/well and four times of that in 10,000 cells/well. Results from Fig. 1C indicated that the viability of each condition above was 94%, 49% or 11%, respectively. It is apparent that H2O2 killed more cells in cultures of lower density (10,000 cells/well) than expected. Because the result could not always be predicted by a mathematic estimation, there is a possibility that cell-cell contacts and some autocrine/paracrine production in a culture may alter the microenvironment thus determine the cytotoxicity caused by H2O2. Since cells with lower density are more susceptible to oxidative stress, there is a possibility that a vicious cycle exists such that when an area of RPE loses some of the constituent cells by oxidative stress, the remaining cells will become even more vulnerable to this insult.
PPARs in RPE
This study indicated that the PPARγ agonist 15d-PGJ2 could prevent H2O2 induced RPE cell death (Figs. 5A,5B and Fig. 4E). This suggests that this agent can prevent oxidative stress induced RPE damage. This finding is significant given prior reports that oxidative stress on RPE is an important contributing factor of age-related macular degeneration [5,42]. An earlier report also indicated that PPARγ agonists could prevent laser photocoagulation induced choroidal neovascularization in rat eyes and monkey eyes, which suggests the possible application of these agents on the exudative form of age-related macular degeneration [43]. It is important to note that the other PPARγ agonists tested in this study (ciglitazone, azelaoyl PAF and LY171883) were ineffective in saving cells under the same experimental conditions. Since three out of four PPARγ agonists tested were ineffective, this raises the possibility that the saving effect of 15d-PGJ2 is not through PPARγ activation. In this respect, it should be noted that this agent has been shown to exert some biological effects that are independent of PPARγ activation [44-46].
We have previously shown that 15d-PGJ2 can prevent the cytotoxicity of cholesterol oxides, end products of cholesterol under oxidative stress [27]. Together with results from the current study, this agent may have a general protective effect against oxidative stress itself or against cytotoxic agents generated by oxidative stress. It is important to note that 15d-PGJ2 functions as a general inhibitor of immune activities. For example, this agent is a potent inhibitor of microglia, the macrophage-like cells in the central nervous system [47-49] and shown to be effective in reducing the symptoms of experimental autoimmune encephalomyelitis (EAE), an animal model for the human disease multiple sclerosis [50]. Together, these results suggest that this agent may be useful in preventing ocular diseases that result from oxidative stress or inflammation.
ERK activation and 4-HNE protein adducts formation as a result of oxidative stress
Oxidative stress is a potent stimulator of the MAP kinase activities [14,15,51]. Phosphorylation of ERK could be observed within 30–60 minutes after H2O2 treatment (Fig. 6). There are conflicting results in literature regarding whether an increase of ERK phosphorylation is beneficial or detrimental to cell survival. Bhat et al. demonstrated that 5–25 μM PD98059 could prevent H2O2 induced cell death in an oligodendrocyte cell line [14]. In contrast, 20 μM PD98059 was shown to enhance cell death caused by H2O2 treatment in HeLa cells [15]. This reagent had no significant effect on H2O2 treated ARPE-19 cells under our experimental conditions. AG126, on the other hand, appeared to reduce the cytotoxicity caused by oxidative stress (Fig. 7). While this suggests that inhibition of tyrosine phosphorylation or ERK activation after H2O2 treatment might be beneficial for the cells, results from PD98059 could not support this statement.
There is a possibility that the saving effect of AG126 is not related to ERK inhibition. Under the current experimental conditions, AG126 at 10 μM was protective but it enhanced H2O2 toxicity at 20 μM. These results were consistent with those reported by Sagara et al. [52]. AG126 belongs to a group of tyrosine kinase inhibitors named tyrphostins. Many of these agents can prevent cellular oxidative stress including that generated by glutamate. There is no correlation between the kinase inhibition and the observed protective effect. Overnight treatment of cells with AG126 can increase cellular glutathione levels. Furthermore, this agent has some anti-oxidant effect based on the Trolox Equivalent Activity Concentration [52]. Collectively, this may explain the observation that AG126 at 10 μM could partially prevent H2O2 induced cytotoxicity. This agent can also function as a mitochondria uncoupler, thus it actually enhances H2O2 cytotoxicity at higher concentrations [52].
An accumulation of 4-HNE-conjugated protein has been detected in lesions of aging-related diseases, including Parkinson's disease [53] and Alzheimer's disease [54]. It also accumulates in the affected neuronal tissues of experimental animals experiencing brain ischemia [55,56] and traumatic spinal cord injury [57]. 4-HNE can impair synaptosomal membrane proteins, glutamate transport and importantly, mitochondrial functions [58,59]. Evidence indicates that 4-HNE increases neuronal susceptibility to oxidative stress [60] and serves as a mediator of oxidative stress that leads to neuronal apoptosis [61]. As indicated in the Introduction, 4-HNE has been implicated in some ocular pathological conditions [19,20].
Several laboratories have attempted to document the formation of 4-HNE protein adducts during oxidative stress by Western blot analysis. Uchida et al. first reported oxidative stress caused by t-butylhydroperoxide or iron/ascorbate could lead to 4-HNE protein adducts in hepatocytes [62]. Further studies by these researchers attempted to identify the common epitope recognized by the 4-HNE antibodies. However, no specific protein was positively identified [63]. More recently, the formation of 4-HNE protein adducts was also demonstrated by Western blot analysis from the heart [64], the liver [65] and the kidney [66] under oxidative stress. It is important to point out that the patterns of 4-HNE protein adducts, as shown by these Western blot analyses, were different among these studies, probably as a result of tissue specificity. No particular protein was identified as a specific target for 4-HNE adduct formation in these studies. Results from the current study indicate that H2O2 treatment of RPE could lead to the accumulation of 4-HNE protein adducts in the cytoplasm and the nuclei of these cells (Fig. 3). There was a dose-dependent increase of 4-HNE protein adduct after H2O2 treatment with a maximal intensity of ~7X control at 2 mM H2O2 (Fig. 3C). This may partially contribute to oxidative stress induced ocular disease. Even though ocular tissues, including the retina, contain glutathione S-transferases that can actively detoxify 4-HNE [35,67], adverse events may occur if aging or pathological conditions hinder this normal defense from functioning properly. It warrants further studies to identify those proteins species that are modified by 4-HNE.
Conclusions
This study demonstrates that oxidative stress induced by H2O2 could lead to RPE cell death, which is preceded by distinct morphological changes, ERK activation and 4-HNE protein adduct formation. The cell death can be prevented partially by AG126, and the prostaglandin derivative, 15d-PGJ2. These agents may be useful pharmacological tools in the future capable of preventing oxidative stress induced RPE cell death in human ocular diseases.
Competing Interests
None declared.
Authors' Contributions
TKG and JYC were both involved in the experimental design and data collection. Both authors read and approved the final manuscript.
Abbreviations
15d-PGJ2, 15-deoxy-delta 12, 14-Prostaglandin J2; ERK, Extracellular signal-Regulated Kinase; 4-HNE, 4-hydroxynonenal, MAP kinases, Mitogen-Activated Protein kinases; MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide); PBS, phosphate-buffered saline; PPARs, peroxisome proliferator-activated receptors; RPE, retinal pigment epithelium.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgments
This work was supported by funds from Fight For Sight and Research to Prevent Blindness. Primary human RPE cell cultures were prepared from specimens provided by the Arkansas Lions Eye Bank & Laboratory. Support by the Pat & Williard Walker Eye Research Center is greatly appreciated.
Contributor Information
Tarun K Garg, Email: gargtarunk@uams.edu.
Jason Y Chang, Email: changjasony@uams.edu.
References
- Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem. 1995;270:18825–18830. doi: 10.1074/jbc.270.32.18825. [DOI] [PubMed] [Google Scholar]
- Miceli MV, Liles MR, Newsome DA. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp Cell Res. 1994;214:242–249. doi: 10.1006/excr.1994.1254. [DOI] [PubMed] [Google Scholar]
- Tate D.J., Jr., Miceli MV, Newsome DA. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995;36:1271–1279. [PubMed] [Google Scholar]
- Cai J, Nelson KC, Wu M, Sternberg P., Jr., Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/S1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32–42. [PMC free article] [PubMed] [Google Scholar]
- Wills NK, Weng T, Mo L, Hellmich HL, Yu A, Wang T, Buchheit S, Godley BF. Chloride channel expression in cultured human fetal RPE cells: response to oxidative stress. Invest Ophthalmol Vis Sci. 2000;41:4247–4255. [PubMed] [Google Scholar]
- Jahngen-Hodge J, Obin MS, Gong X, Shang F, Nowell T.R., Jr., Gong J, Abasi H, Blumberg J, Taylor A. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. J Biol Chem. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- Barak A, Morse LS, Goldkorn T. Ceramide: a potential mediator of apoptosis in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42:247–254. [PubMed] [Google Scholar]
- Tate DJ, Miceli MV, Newsome DA. Zinc protects against oxidative damage in cultured human retinal pigment epithelial cells. Free Radic Biol Med. 1999;26:704–713. doi: 10.1016/S0891-5849(98)00253-6. [DOI] [PubMed] [Google Scholar]
- Ballinger SW, Van Houten B, Jin GF, Conklin CA, Godley BF. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp Eye Res. 1999;68:765–772. doi: 10.1006/exer.1998.0661. [DOI] [PubMed] [Google Scholar]
- Verna LK, Holman SA, Lee VC, Hoh J. UVA-induced oxidative damage in retinal pigment epithelial cells after H2O2 or sparfloxacin exposure. Cell Biol Toxicol. 2000;16:303–312. doi: 10.1023/A:1026798314217. [DOI] [PubMed] [Google Scholar]
- Jin GF, Hurst JS, Godley BF. Hydrogen peroxide stimulates apoptosis in cultured human retinal pigment epithelial cells. Curr Eye Res. 2001;22:165–173. doi: 10.1076/ceyr.22.3.165.5517. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med. 2001;31:1624–1632. doi: 10.1016/S0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal- regulated kinase in hydrogen peroxide-induced cell death. J Neurochem. 1999;72:112–119. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidative stress: influences of mitogen- activated protein kinase signalling pathways on cell survival. Biochem J. 1998;333:291–300. doi: 10.1042/bj3330291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Mattson MP. Roles of lipid peroxidation in modulation of cellular signaling pathways, cell dysfunction, and death in the nervous system. Rev Neurosci. 1998;9:105–116. doi: 10.1515/revneuro.1998.9.2.105. [DOI] [PubMed] [Google Scholar]
- Verdejo C, Marco P, Renau-Piqueras J, Pinazo-Duran MD. Lipid peroxidation in proliferative vitreoretinopathies. Eye. 1999;13:183–188. doi: 10.1038/eye.1999.48. [DOI] [PubMed] [Google Scholar]
- van Kuijk FJ. 4-Hydroxynonenal interaction with rhodopsin. Biochem Biophys Res Commun. 1997;230:275–279. doi: 10.1006/bbrc.1996.5942. [DOI] [PubMed] [Google Scholar]
- Ansari NH, Wang L, Srivastava SK. Role of lipid aldehydes in cataractogenesis: 4-hydroxynonenal-induced cataract. Biochem Mol Med. 1996;58:25–30. doi: 10.1006/bmme.1996.0028. [DOI] [PubMed] [Google Scholar]
- Srivastata SK, Awasthi S, Wang L, Bhatnagar A, Awasthi YC, Ansari NH. Attenuation of 4-hydroxynonenal-induced cataractogenesis in rat lens by butylated hydroxytoluene. Curr Eye Res. 1996;15:749–754. doi: 10.3109/02713689609003458. [DOI] [PubMed] [Google Scholar]
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Corton JC, Anderson SP, Stauber A. Central role of peroxisome proliferator-activated receptors in the actions of peroxisome proliferators. Annu Rev Pharmacol Toxicol. 2000;40:491–518. doi: 10.1146/annurev.pharmtox.40.1.491. [DOI] [PubMed] [Google Scholar]
- Bishop-Bailey D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br J Pharmacol. 2000;129:823–834. doi: 10.1038/sj.bjp.0703149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ershov AV, Bazan NG. Photoreceptor phagocytosis selectively activates PPARgamma expression in retinal pigment epithelial cells. J Neurosci Res. 2000;60:328–337. doi: 10.1002/(SICI)1097-4547(20000501)60:3<328::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Marmorstein AD, Bonilha VL, Rodriguez-Boulan E, Giordano F, Hjelmeland LM. Use of the ARPE-19 cell line as a model of RPE polarity: basolateral secretion of FGF5. Invest Ophthalmol Vis Sci. 1998;39:2744–2749. [PubMed] [Google Scholar]
- Chang JY, Liu L. Peroxisome proliferator-activated receptor agoinsts prevent 25-OH-cholesterol induced c-jun activation and cell death. BMC Pharmacol. 2001;1:10. doi: 10.1186/1471-2210-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CA, Parham GP, Chambers T. Cytoskeletal reorganization induced by retinoic acid treatment of human endometrial adenocarcinoma (RL95-2) cells is correlated with alterations in protein kinase C-alpha. Pathobiology. 1998;66:284–292. doi: 10.1159/000028035. [DOI] [PubMed] [Google Scholar]
- Wada M, Gelfman CM, Matsunaga H, Alizadeh M, Morse L, Handa JT, Hjelmeland LM. Density-dependent expression of FGF-2 in response to oxidative stress in RPE cells in vitro. Curr Eye Res. 2001;23:226–231. doi: 10.1076/ceyr.23.3.226.5467. [DOI] [PubMed] [Google Scholar]
- Willson TM, Cobb JE, Cowan DJ, Wiethe RW, Correa ID, Prakash SR, Beck KD, Moore LB, Kliewer SA, Lehmann JM. The structure-activity relationship between peroxisome proliferator-activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones. J Med Chem. 1996;39:665–668. doi: 10.1021/jm950395a. [DOI] [PubMed] [Google Scholar]
- Davies SS, Pontsler AV, Marathe GK, Harrison KA, Murphy RC, Hinshaw JC, Prestwich GD, Hilaire AS, Prescott SM, Zimmerman GA, McIntyre TM. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor gamma ligands and agonists. J Biol Chem. 2001;276:16015–16023. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.36.21040. [DOI] [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Singhal SS, Godley BF, Chandra A, Pandya U, Jin GF, Saini MK, Awasthi S, Awasthi YC. Induction of glutathione S-transferase hGST 5.8 is an early response to oxidative stress in RPE cells. Invest Ophthalmol Vis Sci. 1999;40:2652–2659. [PubMed] [Google Scholar]
- Tate DJ, Newsome DA, Oliver PD. Metallothionein shows an age-related decrease in human macular retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1993;34:2348–2351. [PubMed] [Google Scholar]
- Liles MR, Newsome DA, Oliver PD. Antioxidant enzymes in the aging human retinal pigment epithelium. Arch Ophthalmol. 1991;109:1285–1288. doi: 10.1001/archopht.1991.01080090111033. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Davis HW. Hydrogen peroxide-induced cytoskeletal rearrangement in cultured pulmonary endothelial cells. J Cell Physiol. 1998;174:370–379. doi: 10.1002/(SICI)1097-4652(199803)174:3<370::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Liu SM, Sundqvist T. Nitric oxide and cGMP regulate endothelial permeability and F-actin distribution in hydrogen peroxide-treated endothelial cells. Exp Cell Res. 1997;235:238–244. doi: 10.1006/excr.1997.3675. [DOI] [PubMed] [Google Scholar]
- Adams JC. Formation of stable microspikes containing actin and the 55 kDa actin bundling protein, fascin, is a consequence of cell adhesion to thrombospondin-1: implications for the anti-adhesive activities of thrombospondin-1. J Cell Sci. 1995;108 ( Pt 5):1977–1990. doi: 10.1242/jcs.108.5.1977. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Rousseau S, Deschesnes RG, Shah GM, Landry J. SAPK2/p38-dependent F-actin reorganization regulates early membrane blebbing during stress-induced apoptosis. J Cell Biol. 1998;143:1361–1373. doi: 10.1083/jcb.143.5.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell SR, Bressler NM. Age-related macular degeneration. In: Regillo CD, Brown GC and Flynn HW Jr, editor. Vitreoretinal disease: The essentials. New York, Thieme Medical Publishers, Inc.; 1999. pp. 213–240. [Google Scholar]
- Murata T, He S, Hangai M, Ishibashi T, Xi XP, Kim S, Hsueh WA, Ryan SJ, Law RE, Hinton DR. Peroxisome proliferator-activated receptor-gamma ligands inhibit choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41:2309–2317. [PubMed] [Google Scholar]
- Vaidya S, Somers EP, Wright SD, Detmers PA, Bansal VS. 15-Deoxy-Delta12,1412,14-prostaglandin J2 inhibits the beta2 integrin-dependent oxidative burst: involvement of a mechanism distinct from peroxisome proliferator-activated receptor gamma ligation. J Immunol. 1999;163:6187–6192. [PubMed] [Google Scholar]
- Ward C, Dransfield I, Murray J, Farrow SN, Haslett C, Rossi AG. Prostaglandin D2 and its metabolites induce caspase-dependent granulocyte apoptosis that is mediated via inhibition of I kappa B alpha degradation using a peroxisome proliferator-activated receptor-gamma-independent mechanism. J Immunol. 2002;168:6232–6243. doi: 10.4049/jimmunol.168.12.6232. [DOI] [PubMed] [Google Scholar]
- Jozkowicz A, Dulak J, Prager M, Nanobashvili J, Nigisch A, Winter B, Weigel G, Huk I. Prostaglandin-J2 induces synthesis of interleukin-8 by endothelial cells in a PPAR-gamma-independent manner. Prostaglandins Other Lipid Mediat. 2001;66:165–177. doi: 10.1016/S0090-6980(01)00155-1. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR- gamma) and its natural ligand 15-deoxy-Delta12, 14-prostaglandin J2 in the regulation of microglial functions. Eur J Neurosci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down- regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14- prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PD, Chavis JA. The cyclopentone prostaglandin 15-deoxy-Delta(12,14) prostaglandin J2 represses nitric oxide, TNF-alpha, and IL-12 production by microglial cells. J Neuroimmunol. 2001;115:28–35. doi: 10.1016/S0165-5728(01)00267-3. [DOI] [PubMed] [Google Scholar]
- Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- Yoshizumi M, Abe J, Haendeler J, Huang Q, Berk BC. Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. J Biol Chem. 2000;275:11706–11712. doi: 10.1074/jbc.275.16.11706. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Ishige K, Tsai C, Maher P. Tyrphostins protect neuronal cells from oxidative stress. J Biol Chem. 2002;277:36204–36215. doi: 10.1074/jbc.M203895200. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Smith MA, Avila J, Nunomura A, Siedlak SL, Zhu X, Perry G, Sayre LM. In Alzheimer's disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J Neurochem. 2000;75:1234–1241. doi: 10.1046/j.1471-4159.2000.0751234.x. [DOI] [PubMed] [Google Scholar]
- Urabe T, Yamasaki Y, Hattori N, Yoshikawa M, Uchida K, Mizuno Y. Accumulation of 4-hydroxynonenal-modified proteins in hippocampal CA1 pyramidal neurons precedes delayed neuronal damage in the gerbil brain. Neuroscience. 2000;100:241–250. doi: 10.1016/S0306-4522(00)00264-5. [DOI] [PubMed] [Google Scholar]
- Yoshino H, Hattori N, Urabe T, Uchida K, Tanaka M, Mizuno Y. Postischemic accumulation of lipid peroxidation products in the rat brain: immunohistochemical detection of 4-hydroxy-2-nonenal modified proteins. Brain Res. 1997;767:81–86. doi: 10.1016/S0006-8993(97)00616-1. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Mark RJ, Begley JG, Waeg G, Mattson MP. 4-hydroxynonenal, a lipid peroxidation product, rapidly accumulates following traumatic spinal cord injury and inhibits glutamate uptake. J Neurochem. 1997;68:2469–2476. doi: 10.1046/j.1471-4159.1997.68062469.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;80:685–696. doi: 10.1016/S0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69:1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. 4-hydroxynonenal increases neuronal susceptibility to oxidative stress. J Neurosci Res. 1999;58:823–830. doi: 10.1002/(SICI)1097-4547(19991215)58:6<823::AID-JNR9>3.3.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Szweda LI, Chae HZ, Stadtman ER. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proc Natl Acad Sci USA. 1993;90:8742–8746. doi: 10.1073/pnas.90.18.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Itakura K, Kawakishi S, Hiai H, Toyokuni S, Stadtman ER. Characterization of epitopes recognized by 4-hydroxy-2-nonenal specific antibodies. Arch Biochem Biophys. 1995;324:241–248. doi: 10.1006/abbi.1995.0036. [DOI] [PubMed] [Google Scholar]
- Eaton P, Li JM, Hearse DJ, Shattock MJ. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am J Physiol. 1999;276:H935–NaN. doi: 10.1152/ajpheart.1999.276.3.H935. [DOI] [PubMed] [Google Scholar]
- Zhang C, Walker LM, Hinson JA, Mayeux PR. Oxidant stress in rat liver after lipopolysaccharide administration: effect of inducible nitric-oxide synthase inhibition. J Pharmacol Exp Ther. 2000;293:968–972. [PubMed] [Google Scholar]
- Walker LM, York JL, Imam SZ, Ali SF, Muldrew KL, Mayeux PR. Oxidative stress and reactive nitrogen species generation during renal ischemia. Toxicol Sci. 2001;63:143–148. doi: 10.1093/toxsci/63.1.143. [DOI] [PubMed] [Google Scholar]
- Singhal SS, Awasthi S, Srivastava SK, Zimniak P, Ansari NH, Awasthi YC. Novel human ocular glutathione S-transferases with high activity toward 4-hydroxynonenal. Invest Ophthalmol Vis Sci. 1995;36:142–150. [PubMed] [Google Scholar]