

Abstract
Insulin is thought to elicit its effects by crosslinking the two extracellular α-subunits of its receptor, thereby inducing a conformational change in the receptor, which activates the intracellular tyrosine kinase signaling cascade. Previously we identified a series of peptides binding to two discrete hotspots on the insulin receptor. Here we show that covalent linkage of such peptides into homodimers or heterodimers results in insulin agonists or antagonists, depending on how the peptides are linked. An optimized agonist has been shown, both in vitro and in vivo, to have a potency close to that of insulin itself. The ability to construct such peptide derivatives may offer a path for developing agonists or antagonists for treatment of a wide variety of diseases.
Insulin is one of the most studied peptide hormones because of its importance in maintaining glucose homeostasis. This 51-aa hormone is very well characterized with regard to its structure, both in crystal form and in solution. The insulin receptor (IR) is a transmembrane α2β2 glycoprotein whose intracellular tyrosine kinase domain is activated by binding of insulin, leading to a cascade of intracellular signaling events. The kinase domain of the IR (1) and an extracellular fragment of the related receptor for insulin-like growth factor I (IGF-IR; ref. 2) have been crystallized, but the structure of the insulin binding domain of the IR is not known, and the mechanism for the transmission of a signal through its transmembrane domain is not well understood. A model for the binding and activation has been proposed in which insulin uses two different sites on its surface to crosslink the two α-subunits of the IR, thus inducing a conformational change that activates the receptor (refs. 3 and 4; Fig. 1).
Figure 1.

Model for activation of the IR by binding of insulin. P denotes tyrosine phosphorylation.
In a previous report (5), we panned random, highly diverse peptide display libraries against the IR. By using this approach, we identified a large number of peptides binding to the IR and competing for insulin binding with micromolar or submicromolar affinity, although these peptides had no sequence homology with insulin. These peptides bound to two discrete hotspots on the receptor (designated site 1 and site 2), and these hotspots appeared to correspond to the two contact sites involved in insulin binding predicted by the crosslinking model (ref. 3 and J.B., unpublished results). At least two different sequence motifs were found for site 1 peptides, and some of these were full agonists but of low affinity. Other site 1 peptides were antagonists, whereas site 2 peptides were either antagonists or inactive. The mechanism behind the agonism of the site 1 peptides is not known, but it has been speculated that site 1 binding may be important for receptor activation, whereas the role of the site 2 interaction may be more related to affinity and selectivity. In addition to these two families of peptides, a third group was identified, but no further work has been done on this group. In the present work, we have used site 1 and site 2 peptides as building blocks to make synthetic and recombinant homodimers and heterodimers to create molecules that activate the IR with high potency and specificity.
Materials and Methods
Solid-Phase Peptide Synthesis.
Peptides were synthesized manually or on an Advanced ChemTech 396 synthesizer. Synthesis was performed on TentaGel S RAM resin (Rapp Polymere, Tübingen, Germany). Fluorenylmethoxycarbonyl amino acids purchased from Novabiochem were coupled by using a diisopropylcarbodiimide/1-hydroxy-7-azabenzotriazole coupling strategy. Peptides were cleaved as amides from the resin by using 90% trifluoroacetic acid, 5% triisopropylsilane, 3% thioanisol, and 2% phenol and precipitated by addition of diethyl ether followed by lyophilization. Disulfide formation was performed by air oxidation in 1% ammonium bicarbonate for 2 days, and the product was lyophilized and purified by reverse-phase HPLC. The identity of each peptide was confirmed by matrix-assisted laser desorption ionization MS.
Synthesis of Chemically Linked Dimers.
Peptide monomers were synthesized by standard solid-phase synthesis, and a serine residue was introduced either as the last amino acid in the synthesis or by coupling the fully protected serine to a selectively deprotected side-chain amino group of a C-terminal lysine. Peptides containing an aldehyde moiety were generated in solution by oxidation of the serine residue with 2 eq of NaIO4 in 20% DMSO/80% phosphate buffer (pH 7.5) for 5 min at room temperature. The peptides were purified immediately after the oxidation.
Oxyamino linkers of polyethylene glycol chains with two to six ethylene glycol units were prepared from commercially available homogeneous polyethylene glycol chains by tosylation followed by reaction with N-hydroxyphthalimide and treatment with hydrazine (6).
For the preparation of homodimers, the unprotected and oxidized peptide (4 eq) was dimerized on the dioxyamino-ethylene glycol linker (1 eq) in 90% DMSO at 38°C. Heterodimers were synthesized in two steps. First, an aldehyde containing peptide (1 eq) in 90% DMSO was allowed to react with dioxyamino-ethylene glycol (100 eq) at room temperature followed by reverse-phase HPLC purification. Subsequently, the peptide-ethylene glycol linker (1 eq) was chemically ligated to another oxidized peptide (1 eq) at 38°C. The peptide dimer was purified and its identity was confirmed by matrix-assisted laser desorption ionization MS.
Recombinant Peptide Dimers.
Site 1- and site 2-specific monomer peptides were linked to create RB537 and RB539 and expressed as protein fusions to a chitin binding domain affinity tag (New England Biolabs). The peptide dimers were linked via a 6-aa sequence (GGSGGS) chosen for inherent flexibility and solubility. An inducible self-cleavage (by addition of DTT) activity of a protein splicing element (termed intein) is present in the construct to separate the target protein from the affinity tag in the purification. In the DNA construct, the C terminus of the peptide dimer sequence is fused to the N terminus of the intein/chitin binding domain sequence. Two peptide-flanking epitope tags were included, FLAG (DYKDDDDK) at the N terminus and E-Tag (GAPVPYPDPLEPR) at the C terminus. Escherichia coli ER2566 (New England Biolabs) harboring the plasmids encoding the fusion proteins was grown in 2× yeast extract/tryptone/ampicillin/glucose (YT-AG) media for 8 h (250 rpm, 37°C). The cultures were subcultured to 2-liter volumes in 2× yeast extract/tryptone/ampicillin (YT-A) to achieve an OD600 of 0.1. When the cultures reached an OD600 of 0.5, isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.3 mM, and the cultures were grown at 30°C overnight. The next day, cells were removed from the medium by centrifugation (3,000 × g). Samples of the cell pellet were analyzed by SDS/PAGE followed by Western blot analysis by using mouse mAb anti-E-Tag-horseradish peroxidase conjugate (Amersham Biosciences) to visualize the expressed product. The cell pellets were disrupted mechanically by sonication. After removal of cell debris by centrifugation, the soluble proteins (clarified lysate) were prepared for chromatographic purification by dilution or dialysis into the appropriate starting buffer. The chitin binding domain fusions were purified by chitin affinity chromatography. The lysate was loaded onto a chitin affinity column, and the column was washed with 10 volumes of column buffer (20 mM Tris/500 mM NaCl/1 mM EDTA, pH 9.0). Three bed volumes of the cleavage buffer (20 mM Tris/1 mM EDTA/50 mM DTT/0.2% PMSF, pH 9.0) were loaded onto the column, and the column was incubated overnight. The next day, the target protein was eluted by continuing the flow of the cleavage buffer without DTT. The purified proteins were analyzed for purity and integrity by SDS/PAGE and Western blot analysis according to standard protocols.
Receptor Binding Assays.
Human IR (the isoform without exon 11) or human IGF-IR was partially purified by wheat germ agglutinin chromatography (7) from transfected baby hamster kidney cells after solubilization with Triton X-100 and was stored at −80°C. Receptors were incubated with 125I-labeled insulin or IGF-I at various concentrations of peptide. The assay buffer contained 100 mM Hepes (pH 7.8), 100 mM NaCl, 10 mM MgCl2, 0.5% human serum albumin, 0.2% gamma globulin, and 0.025% Triton X-100. The receptor concentration was chosen to give 30–60% binding of 2,000 cpm (3 pM) of its 125I-labeled ligand [TyrA14-125I-HI (HI, human insulin) or Tyr-31-125I-IGF-I], and a dilution series of the substance to be tested was added. After equilibration for 2 days at 4°C, each sample (200 μl) was precipitated by addition of 400 μl of 25% polyethylene glycol 6000, centrifuged, washed with 1 ml of 15% polyethylene glycol 6000, and counted in a gamma counter.
Kinase Assay.
Wheat germ agglutinin-purified recombinant human IR was mixed with either insulin or peptide in varying concentrations in substrate phosphorylation buffer (50 mM Hepes, pH 8.0/3 mM MnCl2/10 mM MgCl2/0.05% Triton X-100/0.1% BSA/12.5 μM ATP). Synthetic biotinylated substrate peptide (biotin-KSRGDYMTMQIG) was added to a final concentration of 2 μg/ml. After a 1-h incubation at room temperature, the reactions were stopped by addition of 50 mM EDTA. The reactions were transferred to streptavidin-coated 96-well microtiter plates (Nunc, catalog no. 236001) and incubated for 1 h at room temperature. The plates were washed three times with TBS (10 mM Tris, pH 8.0/150 mM NaCl), and a 2,000-fold dilution of horseradish peroxidase-conjugated phosphotyrosine Ab (Transduction Laboratories, Lexington, KY; catalog no. E120H) in TBS was added. The plates were incubated for 30 min and washed three times with TBS. TMB ONE substrate from Kem-En-Tec (Copenhagen) was added, and the reaction was stopped with 1% H2SO4 after 15 min. The absorbance, representing the extent of substrate phosphorylation, was measured in a spectrophotometer at 450 nM.
Mouse Adipocyte Lipogenesis Assay.
Glucose incorporation into the lipid phase was determined in primary mouse adipocytes. Epididymal fat pads were dissected from B6D2F1 mice (Charles River Breeding Laboratories) and minced, and adipocytes were prepared by collagenase degradation during shaking for 1.5 h at 36°C in 110 mM NaCl/4.8 mM KCl/1.2 mM KH2PO4/1.2 mM MgSO4/2.5 mM CaCl2/25 mM Hepes, pH 7.9/4% human serum albumin/1.1 mM glucose/0.4 mg/ml collagenase type 1 (Worthington). Cells were then filtered through two layers of gauze and centrifuged briefly at 500 × g. Cells were then washed two to four times (110 mM NaCl/5 mM KCl/1.2 mM KH2PO4/1.2 mM MgSO4/2.5 mM CaCl2/25 mM Hepes, pH 7.9/1% human serum albumin), and 100-μl aliquots were pipetted into 96-well Picoplates (Packard). Ten microliters of insulin and 10 μl of compound or control solvent were added, and the assay was initiated by the addition of 10 μl of d-[3-3H]glucose and glucose to a final concentration of 0.5 mM glucose. The plates were shaken for 2 h, and the assay was stopped by the addition of MicroScint-E (Packard). Plates were counted in a TopCount (Packard). Full insulin dose responses were run on all plates. Triplicate samples and a full dose–response curve for insulin was run for every experiment. Data were normalized to the full insulin response and fitted to a sigmoidal dose response with variable slope (constrained to 100% maximal response for full agonists). Data are presented ± SD (insulin, n = 10; S371, n = 4; S374, n = 3; S519, n = 7; others, n = 1; SD calculated in triplicate).
In Vivo Effect of Peptides in Wistar Rats.
Twenty-one male Wistar rats, weighing 200–225 g and fasted for 18 h, were anesthetized by using 2 ml/kg Hypnorm-Dormicum (1.25 mg/ml Dormicum/2.5 mg/ml fluanisone/0.079 mg/ml fentanyl citrate) as a priming dose 30 min before test-substance dosing and an additional 1 ml/kg every 20 min (at time points −10 min, 10 min, and 30 min relative to test-substance dosing).
The rats were allocated into three groups. The animals were dosed with a 2-ml/kg i.v. injection (tail vein) of either vehicle (n = 8) or 20 nmol/kg S519 peptide (n = 8) or 2.5 nmol/kg human insulin (n = 5). Blood samples for the determination of whole-blood glucose concentration were collected in heparinized 10-μl glass tubes by puncture of the capillary vessels in the tail tip at −20 min and 0 min (before dosing) and at 10, 20, 30, 40, 60, 80, 120, and 180 min after dosing. Blood glucose concentrations were measured after dilution in analysis buffer by the immobilized glucose oxidase method by using an EBIO Plus autoanalyzer (Eppendorf).
Results and Discussion
Assembly of Peptide Building Blocks.
An affinity-optimized site 1 peptide (S371, GSLDESFYDWFERQLGKK) and a site 2 peptide (S446, EWLDQEWAWVQCEVYGRGCPSEE) were chosen as building blocks to make the homodimers and heterodimers. Two methods were used to investigate the importance of the relative orientation of the peptides. In one approach, recombinant homodimers and heterodimers were prepared with glycine/serine linkers of different lengths. A second method used chemoselective ligation of peptides without side-chain protecting groups (8). In the latter method, a serine was attached, either to the N-terminal amino group or to the side-chain amino group of a C-terminal lysine, and subsequently oxidized to an aldehyde function. Triethylene or tetraethylene glycol was functionalized with an oxyamino function at each end (6) and used to chemoselectively ligate two aldehydes by formation of stable oxime bonds. By using these methods, all of the four possible end-to-end orientations (N-N, C-C, C-N, N-C) could be synthesized (Fig. 2).
Figure 2.

Synthesis of chemically linked peptide dimers.
Properties of Peptide Homodimers.
To establish whether IR activation could be achieved by site 1–site 1 crosslinking, homodimers of the site 1 peptide S371 (S374 and S378) were prepared. These dimers had a strongly improved potency in a mouse adipocyte lipogenesis assay (ref. 9; Fig. 3A) but showed no increase in either binding affinity to solubilized human IR in a competition assay vs. 125I-insulin (Tables 1 and 2) or potency in a soluble IR kinase activation assay relative to the monomer (Fig. 3D). These results suggest that the mechanism of receptor, activation differs from that of insulin. One possible explanation for the activity of the homodimer could be that the two ends of the peptide bind to two adjacent receptor molecules, resulting in intermolecular activation of the receptors. In this case, the potency of the peptide would depend on the concentration of the receptors, i.e., it would be more potent when the receptors were concentrated on a cell surface rather than in solution. A similar transactivation mechanism has been described for activation of the insulin receptor by molecules such as lectins, polylysine, and receptor Abs (10–12). On the other hand, homodimers of site 2 peptides showed no increase in binding affinity (regardless of orientation) and, like the site 2 monomers, had no effect in the lipogenesis assay (data not shown).
Figure 3.

In vitro properties of peptides. (A) Stimulation of glucose incorporation into lipids in mouse adipocytes by peptides, human insulin (*), S371 (■), S446 (○), S374 (▾), and S378 (▵). (B) Stimulation of glucose incorporation into lipids in mouse adipocytes by peptides, human insulin (*), RB539 (□), S454 (■), S455 (▵), S456 (▴), and S519 (●). (C) Inhibition of insulin-stimulated glucose incorporation by RB537 (○). (D) Activation of the IR kinase by peptides, human insulin (*), S371 (■), S374 (▾), RB537 (○), RB539 (□), S455 (▵), S456 (▴), and S519 (●).
Table 1.
Sequences of peptide monomers and dimers
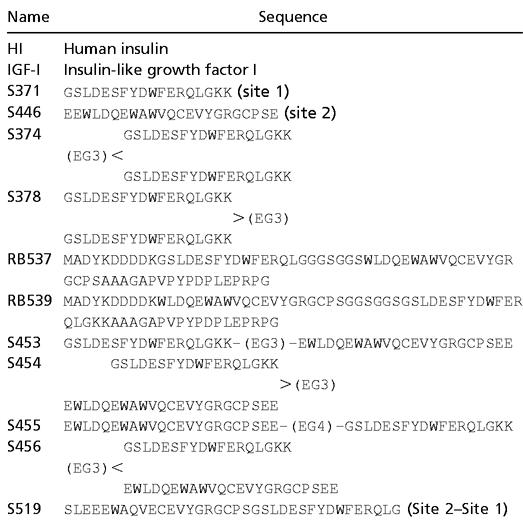 |
(EG3) and (EG4) are tri- and tetraethylene glycol-based linkers. All S-numbered peptides are C-terminal amides. The two cysteines in the site 2 sequence are oxidized to form a disulfide bridge in all peptides.
Table 2.
Properties of peptides in IR binding and mouse adipocyte lipogenesis assays
| Name | IRKd (mol/liter) | IGF-IRKd (mol/liter) | Lipogenesis EC50 (mol/liter) |
|---|---|---|---|
| HI | 8.0 × 10−12 | 2.0 × 10−8 | 4.7 × 10−11 |
| IGF-I | 1.0 × 10−9 | 1.5 × 10−11 | 8.0 × 10−8 |
| S371 | 1.6 × 10−7 | 7.3 × 10−7 | >1 × 10−5 |
| S446 | 4.9 × 10−7 | >2 × 10−5 | No effect |
| S374 | 1.8 × 10−7 | ND | 3.2 × 10−7 |
| S378 | 6.5 × 10−8 | ND | 7.6 × 10−8 |
| RB537 | 6.0 × 10−11 | 9.8 × 10−7 | 4.4 × 10−8 (antagonist) |
| RB539 | 7.0 × 10−10 | 1.5 × 10−6 | 1.9 × 10−8 |
| S453 | 5.7 × 10−10 | ND | ND |
| S454 | 3.8 × 10−10 | ND | Partial agonist |
| S455 | 1.1 × 10−9 | ND | 1.9 × 10−7 |
| S456 | 2.4 × 10−9 | ND | >1 × 10−6 |
| S519 | 2.0 × 10−11 | 2.5 × 10−6 | 4.2 × 10−9 |
IR is the human IR (the isoform without exon 11); IGF-IR is the human receptor for IGF-I. ND, Not determined.
Properties of Peptide Heterodimers.
To make molecules that might activate the receptor by an insulin-like intramolecular mechanism, we prepared heterodimers of site 1 and site 2 peptides in different orientations. Fusion peptides were prepared recombinantly in E. coli (RB537 and RB539), and both of these peptides had improved IR affinity compared with the monomer peptides, suggesting that both sites contributed to the binding. RB537 (site 1–site 2) had no effect in either the kinase assay or the lipogenesis assay but was found to be an antagonist when tested in the presence of a submaximally stimulating concentration of insulin (Fig. 3C). In contrast, RB539 (site 2–site 1) was an agonist and gave rise to the same maximal response as insulin (Fig. 3B).
To further elucidate the effects of coupling orientation on the properties of dimer peptides, we prepared chemically linked heterodimeric site 1–site 2 peptides in all four possible end-to-end orientations (C-N, N-C, C-C, N-N). All showed improved binding relative to the monomers, and their properties in the kinase and lipogenesis assays strongly depended on the orientation. Dimers coupled at both N termini or both C termini (S454 and S456) were weak or partial agonists. A dimer coupled C terminal to N terminal in the order site 1–site 2 (S453) was an antagonist (data not shown), and a dimer coupled C terminal to N terminal in the order site 2–site 1 (S455) was a full agonist (Fig. 3 B and D). Somewhat surprisingly, the length of the linker was not critical because linkers containing two to six ethylene glycol units gave similar results (data not shown). Because all four linkage isomers show a similarly increased affinity for the receptor relative to the monomers, it must be assumed that the binding involves contacts with both binding sites, but the antagonist somehow locks the receptor in its inactive state rather than eliciting the conformational change required for activation. The results from studies with the chemically linked dimers were consistent with the results obtained with the recombinant dimers, and, because the optimal orientation for agonism (site 2–site 1) could be obtained by simple peptide synthesis, further optimization of the agonist sequences was performed on linear two-site peptides synthesized by standard solid-phase peptide synthesis. A number of amino acid changes (suggested by positional analysis of changes observed in secondary phage display libraries of the monomers) were incorporated, and the peptide was reduced in size by deleting some biologically unimportant residues. These changes resulted in an improved peptide designated S519, which is a single chain peptide of 36 aa with one internal disulfide bridge.
Properties of the Optimized IR Agonist, S519.
In the IR competition binding assay, S519 was found to have a receptor affinity of 20 pM compared with 8 pM for insulin. This is an improvement of 3–4 orders of magnitude over the affinities of the original peptide building blocks. The potency of S519 in the IR kinase activation assay was ≈10% relative to human insulin. In the lipogenesis assay, S519 was an insulin agonist with an EC50 of 4 nM, which corresponds to ≈1% potency relative to human insulin. When 20 nmol/kg S519 was administered i.v. to anesthetized Wistar rats, the fall in plasma glucose was comparable in magnitude and duration with that induced by human insulin at 2.5 nmol/kg (Fig. 4). The potency of S519 in the mouse adipocyte assay and the rat in vivo model was lower than would be expected from its affinity to the human IR, but preliminary results indicate that species differences may be at least partly responsible for these differential effects (data not shown). The peptides were selected and optimized with respect to affinity for the human IR, and, because the receptor interactions are not likely to be exactly the same as those of insulin, the peptides may exhibit a species selectivity not seen for insulin.
Figure 4.

Blood glucose after i.v. administration of vehicle (n = 8), human insulin (n = 5), or S519 (n = 8) to anesthetized Wistar rats.
Another interesting result pointed to differences in S519 binding to the IR vs. the closely related receptor for IGF-IR. Previously, it has been shown that most site 1 peptides show only a slight preference for the IR vs. the IGF-IR, whereas the site 2 peptides have no measurable affinity for the IGF-IR. This was also reflected in the properties of S519, which had an affinity for the IGF-IR comparable with that of the site 1 component. Thus, the relative improvement in IR affinity for S519 vs. S371 was accompanied by a similar increase in selectivity for the IR, and the resulting selectivity of S519 for the IR vs. the IGF-IR was higher than that of insulin itself. Because the main function of the IR is to mediate the metabolic effects of insulin, whereas the IGF-IR is involved in mitogenic signaling, it remains to be seen whether the increased selectivity of the insulin mimetic peptides will be reflected in a different profile with regard to cellular signaling and mitogenic activity.
Conclusions
In summary, this work shows that de novo construction of an artificial high-affinity ligand to a receptor can be accomplished if the appropriate building blocks are available, here in the form of peptides identified by panning of phage display libraries. Also, knowledge about the receptor and a model for the binding of the native hormone can aid in constructing artificial ligands and in interpreting the results. It should be stressed that, although the results are consistent with the IR activation model, they do not prove its validity. There are a number of issues to be addressed, most notably a more detailed understanding of the alternative ways of activating the receptor and the relative importance of the two binding sites, as well as a molecular explanation of the mechanism of action of the antagonists.
Because of their small size and simple structure, the insulin mimetic peptides identified in this work could find a use in the treatment of diabetes, either as a replacement for insulin (e.g., for alternative routes of administration) or as a tool for gene therapy in which the peptides might be more efficiently expressed than insulin, which requires proteolytic processing. In addition, the peptides could serve as pharmacophores for the development of nonpeptide structures with similar properties and eventually lead to orally available insulin mimetics. Finally, the methods described could lead to construction of artificial ligands with a full spectrum of agonist or antagonist properties for a number of other receptors, including those for which the natural ligand is unknown or nonexisting.
Abbreviations
- IR
insulin receptor
- IGF-IR
insulin-like growth factor I receptor
Note Added in Proof.
Additional experiments indicate that the affinity of S519 for the rat insulin receptor is about five times lower than for the human insulin receptor (unpublished data).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hubbard S R, Wei L, Ellis L, Hendrickson W A. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 2.Garrett T J, McKern N M, Lou M Z, Frenkel M J, Bentley J D, Lovrecz G O, Elleman T C, Cosgrove L J, Ward C W. Nature. 1998;394:395–399. doi: 10.1038/28668. [DOI] [PubMed] [Google Scholar]
- 3.Schäffer L. Eur J Biochem. 1994;221:1127–1132. doi: 10.1111/j.1432-1033.1994.tb18833.x. [DOI] [PubMed] [Google Scholar]
- 4.De Meyts P. Diabetologia. 1994;37:S135–S148. doi: 10.1007/BF00400837. [DOI] [PubMed] [Google Scholar]
- 5.Pillutla R C, Hsiao K C, Beasley J R, Brandt J, Østergaard S, Hansen P H, Spetzler J C, Danielsen G M, Andersen A S, Brissette R E, et al. J Biol Chem. 2002;277:22590–22594. doi: 10.1074/jbc.M202119200. [DOI] [PubMed] [Google Scholar]
- 6.Shtamburg V G, Dmitrenko A A, Pleshkova A P, Pritykin L M. Zh Org Khim. 1993;29:1762–1771. [Google Scholar]
- 7.Yamada K, Goncalves E, Kahn C R, Shoelson S E. J Biol Chem. 1992;267:12452–12461. [PubMed] [Google Scholar]
- 8.Rose K. J Am Chem Soc. 1994;116:30–33. [Google Scholar]
- 9.Moody A J, Stan M A, Stan M, Gliemann J. Horm Metab Res. 1973;6:12–16. doi: 10.1055/s-0028-1093895. [DOI] [PubMed] [Google Scholar]
- 10.Shiba T, Tobe K, Koshio O, Yamamoto R, Shibasaki Y, Matsumoto N, Toyoshima S, Osawa T, Akanuma Y, Takaku F, et al. Biochem J. 1990;267:787–794. doi: 10.1042/bj2670787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison B D, Feltz S M, Pessin J E. J Biol Chem. 1989;264:9994–10001. [PubMed] [Google Scholar]
- 12.Soos M A, O'Brien R M, Brindle N P J, Stigter J M, Okamoto A K, Whittaker J, Siddle K. Proc Natl Acad Sci USA. 1989;86:5217–5221. doi: 10.1073/pnas.86.14.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]