

Abstract
The binding between a PK and its target is highly specific, despite the fact that many different PKs exhibit significant sequence and structure homology. There must be, then, specificity-determining residues (SDRs) that enable different PKs to recognize their unique substrate. Here we use and further develop a computational procedure to discover putative SDRs (PSDRs) in protein families, whereby a family of homologous proteins is split into orthologous proteins, which are assumed to have the same specificity, and paralogous proteins, which have different specificities. We reason that PSDRs must be similar among orthologs, whereas they must necessarily be different among paralogs. Our statistical procedure and evolutionary model identifies such residues by discriminating a functional signal from a phylogenetic one. As case studies we investigate the prokaryotic two-component system and the eukaryotic AGC (i.e., cAMP-dependent PK, cGMP-dependent PK, and PKC) PKs. Without using experimental data, we predict PSDRs in prokaryotic and eukaryotic PKs, and suggest precise mutations that may convert the specificity of one PK to another. We compare our predictions with current experimental results and obtain considerable agreement with them. Our analysis unifies much of existing data on PK specificity. Finally, we find PSDRs that are outside the active site. Based on our results, as well as structural and biochemical characterizations of eukaryotic PKs, we propose the testable hypothesis of “specificity via differential activation” as a way for the cell to control kinase specificity.
Phosphorylation is central to signal transduction in living organisms, and PKs perform the vital task of phosphorylating a substrate. Approximately 2% of the eukaryotic genome codes for PKs (1), and the current estimate is that there are 518 different PKs in the human genome (2–4). In bacteria and archaea, >400 histidine PKs (HPKs) have been identified, and they serve a wide range of functions, including chemotaxis, osmoregulation, and nitrogen metabolism (5–8). HPK, with its cognate response regulator (RR) protein, constitutes the prototypical two-component system (9). The HPK consists of two modules: the highly variable sensing domain and the conserved kinase core (9). The kinase core has two parts (Fig. 1): the catalytic domain and the dimerization domain (DD). The catalytic domain hydrolyzes ATP and transfers the phosphate to a conserved His in the DD. The RR protein can also be separated into two moieties: the receiver and the effector domain. The receiver domain has an α–β Rossman fold that is highly conserved (blue and magenta in Fig. 1a), whereas the effector domain differs structurally from one system to the next (9). An Asp on the receiver domain accepts the phosphate from the His on the DD. The transfer of phosphate triggers the effector domain, which then binds on to DNA to activate or repress the appropriate genes (9). In summary, the phosphate is passed from the DD to the receiver domain of the RR. The only known exception is the chemotactic two-component system, where the phosphate is passed, not from the DD, but from a histidine-containing phosphotransfer domain, to the receiver domain of the RR (10). Therefore, in this work, we have eliminated the chemotactic proteins, and we will refer to the receiver domain of a RR protein simply as “RR.”
Figure 1.

The prokaryotic two-component system and its PSDRs. The x-ray structure (19) of the dimerized catalytic domains (red and cyan), their DDs (red and cyan helices in the center) and the receiver domain of the RR (blue and magenta). The PSDRs are shown as space-filling molecules and are colored the same as the chain in which they occur. A His-30–Asp-54 pair involved in phosphotransfer are shown in the yellow space-filling model. (a) Side view. Note the contacts between PSDRs of the DDs and PSDRs of corresponding RRs (red and magenta, blue and cyan). (b) Top view. Note the contacts between PSDRs of the DDs. Presumably, these interactions are responsible for correct dimerization. Numerous interacting PSDRs also surround the His-30–Asp-54 site.
In eukaryotes, PKs are classified into eight main groups (2, 11). Most of them are involved in signal transduction (12–14). In this paper, we focus on the cAMP-dependent PK, cGMP-dependent PK, and PKC (AGC) group of Ser/Thr PKs. The AGC group contains PKs such as cAMP-dependent PK (PKA), PKC, PKs related to PKA and PKC (RAC), G protein-coupled receptor kinase (GRK), ribosomal S6 PK, and the PVPK1 PK homologs in plants (4). The catalytic subunit of PKA is the best studied member of the AGC group (12). It has the typical “two-lobe” structure (Fig. 2a) that is conserved among Ser/Thr/Tyr PKs (15). The active cleft, where the substrate binds, lies between the two lobes (Fig. 2a). A general recognition motif exists among most PKA substrates (12, 16, 17). Interestingly, PKA must itself be phosphorylated on Thr-197 for the enzyme to be fully active (12).
Figure 2.

The eukaryotic PK in the AGC group and its PSDRs. (a) The two-lobe structure of PKA (22) with PSDRs shown by space-fill. The two lobes are red and blue, respectively. The inhibitor substrate (yellow) lies between the lobes. Phospho-Thr-197 is green. (b) PSDRs are as follows: Lys-83, Gln-84, His-87 (blue, upper) Ser 53 (blue, lower) of the PKA and His-23 (yellow) of the inhibitor. Ala-21 (magenta) is the P site. His-23 is the P + 2 site. (c) Phe-129 and Arg-133 (blue) of the PKA, and Arg-18 and Arg-19 on the inhibitor (yellow). The P site is magenta. Arg-18 and Arg-19 correspond to P− 3 and P − 2, respectively. (d) PSDRs Val-191 (red, upper), Tyr-196 (red), Lys-83, and His-87 (blue). Phospho-Thr-197 is green. His-23 and Asp-24 (yellow) are the closest substrate residues to Tyr-196, but are clearly not in contact with Tyr-196.
One of the most remarkable properties of PKs is their specificity. For example, despite the highly conserved two-lobe fold and sequence, different Ser/Thr PKs recognize different consensus sequences in their substrates (16–18). In this work, the average sequence similarity is 63% among the Ser/Thr PKs. The two-component system in prokaryotes is another good example. An HPK phosphorylates only its cognate RR, and HPK-RR pairs for different functions usually do not crosstalk, even when all RRs share the same fold (19, 20).
How do PKs achieve such exquisite substrate specificity? There are several possible strategies. First, different PKs may be localized in distinct regions of a cell, or in different cell types. The GRKs are prime examples of this localization. Of the six families of GRKs, it seems that GRKs 1–3 are mostly cytosolic, but GRKs 4–6 are tightly bound to the cell membrane (14). Second, cofactor binding can selectively activate a PK toward its substrates. The best characterized case is cyclin-dependent kinase, whose activity is controlled by its cyclin partner (21). Finally, the active site of a PK may have evolved to accept only the appropriate substrates. Much work has been done by using peptide libraries (16–18) and x-ray crystallography (19, 22) to elucidate kinase–substrate interactions. Nevertheless, given the size of a typical PK (≈300 aa), it is difficult to know, a priori, the PK residues that are responsible for specificity. Locating these specificity-determining residues (SDRs) experimentally is labor-intensive and time-consuming (23).
To alleviate this problem, we have recently developed a bioinformatics approach to find putative SDRs (PSDRs) and applied it to bacterial transcription factors (24). Our method consists of two steps. First, computational analyses locate residues that exhibit specific conservation signatures. Second, statistical analyses distinguish such signatures from phylogenetic signals and position-specific conservation. In this work, we look for specific amino acids that are conserved within a family but vary across families in a multiple sequence alignment (MSA) of PKs. An important strength of our approach is its ability to predict a relatively small number of PSDRs, which can then be tested by point mutations (24).
In this paper, we predict the PSDRs in prokaryotic and eukaryotic PKs. By comparing our predictions with experiments, we are now able to understand existing data on kinase specificity in a unified framework. Furthermore, we pinpoint specific mutations that may convert the substrate specificity of a PK to another. Finally, we investigate the PSDRs outside the kinase active site. In particular, we focus on two residues, Val-191 and Trp-196 in PKA. We propose that different residues at positions 191 and 196 may influence kinase specificity by controlling Thr-197 phosphorylation. We call this concept “specificity via differential activation” and we suggest particular mutations that would test this concept.
Methods
Databases of Protein Sequences and MSA.
PK sequences have been downloaded from the Sentra (5) and SWISS-PROT databases (25), or found by a blast (26) search of the nonredundant database at the National Center for Biotechnology Information (Bethesda). clustalw (27) and HMMER2.2G (28) have been used to make the MSAs. We have used data from Pfam (29) to guide our alignments. For details, see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org.
Locating PSDRs with Mutual Information.
Similarly to our previous study (24), we assume that PSDRs would be conserved among enzymes of the same substrate, but would vary among enzymes of different substrates. For example, the red column in Fig. 3 may be a PSDR, but the blue and magenta columns are not. To quantify the difference between the red and the blue (or magenta) column, we use “mutual information,” defined as
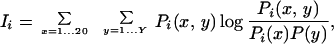 |
1 |
where i, x, and y denote the position in the alignment, the amino acid type, and the family number, respectively (30). Y is the total number of families in an alignment (i.e., Y = 12 in Tables 1 and 2; Y = 6 in Table 3). Pi(x, y) is the probability of finding amino acid type x at position i and in family y. Pi(x) is the probability of finding amino acid type x at position i regardless of family and P(y) is simply the fraction of proteins belonging to family y. Importantly, Ii measures the correlation between x and y, and Ii = 0 if and only if x and y are statistically independent (30). Therefore, the red column in Fig. 3 has a high Ii, whereas the blue and magenta columns have low Iis. However, Ii alone cannot define a PSDR because it strongly depends on the amino acid composition of the column (24). Instead, we must estimate the statistical significance of Ii. A similar method has been used recently to find potentially important sites in proteins (31). Our method also assumes similar binding geometries in most PK–substrate pairs. This assumption is supported by available crystal structures (22, 32), experiments with peptide libraries (16–18), and recent computational studies (33).
Figure 3.

An illustrative example of how to locate PSDRs from an alignment. The red column has high mutual information and may be a PSDR. The blue and magenta columns have low mutual information and are not PSDRs.
Table 1.
PSDRs and their identities among prokaryotic two-component systems: DD of histidine kinase
| Sequence position in Sp0B*
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 24 | 25 | 33 | 34 | 38 | 42 | 43 | 45 | 74 | 78 | |
| Ii† | 1.38 | 1.22 | 1.43 | 1.36 | 1.33 | 1.55 | 1.25 | 1.45 | 1.81 | 1.42 |
| Family‡ | ||||||||||
| PhoR | F | F/L | R/K | T | V | Y/T | L | M/T | Q/T | L |
| BaeS | F/S | M/N | R | T | V/I | E/R | L | A/G | D/E | L |
| CreC | Y | V | K/I | S | A | A | A | I/L/S | T/R/N | Q/L |
| KdpD | L | L | R | T/S | A/V/G | A | A/V | T | N | M |
| PhoQ | T | L | K | T | V | T | L | S | Y | R |
| NarX | L | V/L | T/G | A/V | L/G | Q/E | E | Q/K | C/M | Q/A |
| AutS | L | A/G | E | L | E | K | L | K | K | L |
| CitA | A | L | M | N | V/T | L | V/L | L | S/N | A |
| EnvZ | L | M | R | T | R | A | T | M | Y | G |
| FixL | M | A | N | Q | A | Y | I/M/S | G | I | L |
| NtrB | L/M | A/V | K | N | G | A | A | L | R | F/L |
| Sp0B | L | L | M | N | L | N | L | L | P | F |
Table 2.
PSDRs and their identities among prokaryotic two-component systems: RR
| Sequence position in Sp0F*
|
||||||
|---|---|---|---|---|---|---|
| 18 | 56 | 84 | 90 | 103 | 107 | |
| Ii† | 1.78 | 1.55 | 1.58 | 1.26 | 1.49 | 1.41 |
| Family‡ | ||||||
| PhoB | M/L | M | R/K | R/K | T | S |
| BaeR | L/I | M/K | K/L | R/K | C/V | S/N |
| CreB | T | G | R | R | A/V | S |
| KdpE | F | G | R | K | T | G |
| PhoP | H | G | R | K | T | H |
| NarL | G | N/D | S | V/L | L | E/N |
| AutR | E | D/H | Y | A | T/L | D/E |
| CitB | I/L | Y/H | A | I | I | T |
| OmpR | L | M | K | R | P | N |
| FixJ | S | R | H | A | E | E/D |
| NtrC | V | V/R | Q/H | A | P | D |
| Sp0F | L | K | Y | I | A | D |
| VanR | T | — | L | Q | — | R |
Table 3.
PSDRs and their identities among eukaryotic PKs in the AGC group
| PSDRs | Sequence position in structure of PKA*
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 48 | 53 | 56 | 58 | 83 | 84 | 87 | 115 | 129 | 133 | 191 | 196 | 243 | 247 | 249 | 253 | |
| Ii | 0.99 | 0.95 | 1.10 | 1.01 | 0.98 | 1.25 | 1.18 | 1.01 | 1.10 | 0.98 | 1.11 | 1.03 | 1.25 | 1.02 | 1.11 | 1.12 |
| Family† | ||||||||||||||||
| PKA | T | S/G | R | M/H/E | K | Q | H | N/Y | F/W | R | V/I | W | P | Y | K/N | G |
| PKC | V | S/N | K | M/L | D/E | D/E | C/S | R | M | Q | N/G | S/R/K | E | F/Y | S/A | D/H |
| RAC | L | T | K | I | D | E | H | R | F | S | G | K | H | F | L | E |
| GRK | V/I | G | E | C/Y | K/Q | G | M/L | A/K | K/H | Y/S | I/F/V | R/H/K | K/D | V/I | R | E/T/N |
| S6 PK | V | G/S/A | K | F | A/N | D | H/R | K | F | S/E/Y | S/A/F | Y/H | R | M/I | K/L | A |
| PVPK1 | R/P | D | S | Y/H/F | N/K | K | R | F/H | H/F | Q/D | C/T | M/N | R | F/L | N/V | P/K |
Sequence positions according to the x-ray structure of cAMP-dependent PK (22). See Table 1 for the meanings of Ii and X/Y(/Z).
See text for definitions of abbreviations. The number of sequences for each family is 69 (PKA), 139 (PKC), 23 (RAC), 58 (GRK), 41 (S6 PK), and 50 (PVPK1). The PKA family includes all cyclic nucleotide-dependent PKs (4).
Evaluating Statistical Significance and Choosing PSDRs.
To evaluate the statistical significance of an Ii, we need “control” MSAs to estimate the P value (= the probability of observing this Ii in the control). We base our choice on the following reasoning. There are two major mechanisms of conservation for an amino acid position. The first mechanism is independent of amino acid position in a protein. Examples of the first mechanism are chance and phylogeny (34). The second mechanism is position-specific, and conserves residues that have important structural, functional, or kinetic roles (35). In this work, we are searching for specificity-determining residues, which are important for protein function. PSDRs, then, are probably under position-dependent conservation. Therefore, the ideal control MSA should take into account the position-independent conservation, but should ignore any position-specific conservation that may be in the MSA of real proteins. We use the linear transformation method (24) to generate control MSAs that satisfy this requirement. The I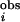 s and I
s and I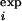 s for the RR are plotted in Fig. 4a. In addition, for the eukaryotic PKs, we find the method of Wollenberg and Atchley (34) to be useful in locating the cutoff for I
s for the RR are plotted in Fig. 4a. In addition, for the eukaryotic PKs, we find the method of Wollenberg and Atchley (34) to be useful in locating the cutoff for I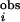 (Fig. 4b). For the prokaryotic PKs, we define a PSDR as any residue with I
(Fig. 4b). For the prokaryotic PKs, we define a PSDR as any residue with I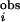 ≥ 1.2 and P ≤ 0.0013. For the eukaryotic PKs, we choose any residues with I
≥ 1.2 and P ≤ 0.0013. For the eukaryotic PKs, we choose any residues with I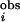 ≥ 0.95 and P ≤ 0.0003 as PSDRs. Details of the methods are given in Supporting Text, which is published as supporting information on the PNAS web site.
≥ 0.95 and P ≤ 0.0003 as PSDRs. Details of the methods are given in Supporting Text, which is published as supporting information on the PNAS web site.
Figure 4.

Mutual information (Ii) and its statistical significance. (a) The Ii values for the RR in the prokaryotic two-component system. The abscissa and ordinate represent the sequence position and mutual information, respectively. The blue points are the observed values and the red points are the expected values. The error bars are drawn at three standard deviations from the mean of the expected value and correspond to P = 0.0013. (b) The distribution of Iis for the eukaryotic PKs in the AGC group. The abscissa and ordinate represent, respectively, the mutual information and the probability of observing a particular mutual information. The blue line is the observed result and the red line is the control from the method cited in ref. 34. The vertical line means that there is a probability of <0.001 of observing an Ii ≥ 1.0 in the control MSAs from the method cited in ref. 34. Note that “p” in this figure does not correspond to the P value described in the text, which has been determined by the linear transformation method from ref. 24. See Methods for details.
Results and Discussion
Prokaroytic HPK Two-Component System.
Our analysis predicts 10 and 6 PSDRs in the DD and RR, respectively (Tables 1 and 2 and Fig. 1). There are no PSDRs on the helix that does not make contact with the RR (Fig. 1b). All six PSDRs on the RR are near the active site of the Rossman fold, even though they are wide apart in the amino acid sequence (Fig. 1 and Table 2). In the DD, the 10 PSDRs segregate into two groups: one near the turn of the antiparallel helical bundle, and the other close to the termini of the helices (Fig. 1).
A fascinating picture emerges when we view the PSDRs on the DD and RR together (Fig. 1). Most of PSDRs are involved in contacts between DD and RR, or between DDs of the two monomers. The first group may be responsible for specific DD–RR recognition, whereas the second group may play a role in correct dimerization.
Leu-18 of the RR forms a tight hydrophobic minicore with Leu-38, Asn-42, and Leu-45 on the DD (Fig. 1). The side chain of Leu-18 is <5.5 Å away from the side chain of Leu-38, Asn-42, or Leu-45. For three reasons, we believe that these four residues play a primary role in determining specificity of DD–RR recognition. First, the residues have statistically significant Ii values (Tables 1 and 2). Second, the residues are in close contact with each other. It should be noted that the DD and RR sequences have been analyzed separately, without using structural information, and Leu-18, Leu-38, Asn-42, and Leu-45 are found to be in contact only after the mutual information analysis. Third, among the families in Tables 1 and 2, there are correlated mutations between position 18 of the RR and positions 38, 42, and 45 of the DD. The most dramatic example is between the sporulation (Sp0F–Sp0B in Tables 1 and 2) and the autolysin system (AutS-AutR in Tables 1 and 2). When Leu-18 in Sp0F changes to Glu in AutR, Leu-38, Asn-42, and Leu-45 in Sp0B turn to Glu, Lys, and Lys, respectively, in AutS. The hydrophobic contacts among Leu-18, Leu-38, Asn-42, and Leu-45 in Sp0F–Sp0, then, seem to have been replaced by a mixture of hydrophobic and electrostatic interactions in AutS-AutR. This hydrophobic-to-electrostatic switch may prevent Sp0F from binding to AutS in Bacillus subtilis. A simple experiment will confirm or refute this hypothesis: changing Leu-18 to Glu in Sp0F should weaken the binding between Sp0F and Sp0B, but further mutating Leu-38, Asn-42, and Leu-45 in Sp0B to Glu, Lys, and Lys should restore, at least partially, the affinity between Sp0F and Sp0B.
Haldimann et al. (23) have done a similar experiment on the PhoR–PhoB system, trying to make PhoB recognize the noncognate HPK VanS. Comparing their results with our predictions, it is very encouraging that Met-17 in PhoB (= Leu-18 in Sp0F), is involved in altering the specificity of PhoB (23). However, our analysis misses some of the specificity-altering mutations in PhoB, such as T97A (23). Interestingly, Haldimann et al. (23) find no PhoB mutant that recognizes only VanS, and not PhoR. To make such a mutant, Table 2 suggests the following method: Arg-85 (= 84 in Table 2), Arg-91 (= 90 in Table 2), and Ser-108 (= 107 in Table 2) in PhoB should be mutated to Leu, Gln, and Arg, respectively. Furthermore, Met-17 could be replaced by Thr, rather than by Val (23).
Four other observations are noteworthy in Fig. 1 and Tables 1 and 2. First, in the Sp0F–Sp0B system, the phosphate is transferred from His-30 on the DD to Asp-54 on the RR (20). These two residues (yellow in Fig. 1) are not PSDRs as they are conserved in all families, but are surrounded by several PSDRs. Enzyme-substrate specificity, then, may be determined by amino acids outside the active site. This phenomenon appears again in our analysis of Ser/Thr PKs (see next section). Second, two PSDRs on Sp0F, Asp-107 and Lys-56, are in contact with Sp0B. However, the Sp0B contact partners of Asp-107 and Lys-56 are not PSDRs, because they have mutual information values that are within expectation (data not shown). Asp 107 and Lys 56, then, may play a secondary role in determining specificity. Third, some residues in Table 1 (e.g., Phe-78 in Sp0B) makes no DD–RR contacts in the Sp0F–Sp0B complex, but they have been determined by our method to be PSDRs. Such residues form contacts between DDs of the two chains, and therefore may be responsible for specific dimerization. Other PSDR residues may affect specificity in a way that is not revealed by the x-ray structure, may be false positives, or may be responsible for specificity of some other interactions formed by these proteins. Finally, our previous work has demonstrated that certain amino acids may have been conserved for protein folding kinetics (35, 36), and the kinetically important residues (nucleating residues) in the Rossman fold have been identified (35). The six PSDRs in the RR are aligned with Ile-20, Asn-59, Asp-89, Ile-96, Val-108, and Thr-112 in the CheY structure of ref. 35. Not surprisingly, there is no overlap between the PSDRs and the nucleating residues in the Rossman fold. Although both PSDRs and nucleating residues tend to be conserved, they exhibit different evolutionary signatures. PSDRs are conserved in each family, but need to be different across various families to avoid crosstalk among paralogous proteins. In contrast, nucleating residues do not necessarily change from one family to the other, but are conserved for rapid folding.
AGC Group of Eukaryotic PKs.
Our analysis reveals 16 PSDRs in this group (Table 3). Interestingly, seven of them are near, or in contact with, the inhibitor substrate: Ser-53, Lys-83, Gln-84, His-87 (all shown in Fig. 2b), Phe-129 (Fig. 2c), Arg-133 (Fig. 2c), and Pro-243. Two PSDRs (Val-191 and Trp-196) are in close proximity to Thr-197 (Fig. 2d). Arg-56 is next to Phe-54 and Gly-55, the two ATP-binding residues in PKA (12).
We first analyze the PSDRs near or in contact with the substrate, using the well known PKA–inhibitor-complex structure from ref. 21 (Fig. 2a). In the peptide inhibitor (yellow in Fig. 2a), the P (phosphorylation) site is Ala-21 (22). Ser-53, Gln-84, Phe-129, and Pro-243 are within 4.8 Å of their substrate partner, whereas His-87 is 6.3 Å from its partner on the inhibitor. Ser-53, Gln-84, and His-87 interact with His-23 at the P + 2 site on the substrate (Fig. 2b). The side chains of Phe-129 and Arg-133 are tightly packed against the Arg side chains at P − 2 and P − 3 (Fig. 2c). Pro-243 makes hydrophobic contacts with the side chain of Arg-15, which corresponds to P − 6.
Importantly, our results elucidate a considerable part of existing data on AGC specificity, and they predict mutations that could switch specificity from one family to another in Table 3. For example, considering the tight interactions among positions 129, P − 2 and P − 3 (Fig. 2c), the residue at position 129 is expected to determine the variation at P − 2 and P − 3. This expectation is fulfilled in Table 3. In PKA, PKC, RAC, and S6 PK, position 129 is occupied mostly by residues that can make hydrophobic interactions (Phe, Trp, and Met), whereas in GRK, a basic residue (Lys) is commonly found at position 129. There is a corresponding change in the substrate residues at P − 2 and P − 3. Both PKA and PKC select amino acids with a long hydrocarbon chain (Arg or Lys) at P − 2 and P − 3 on the substrate (16). Arg is also strongly favored at the P − 3 site for RAC and S6 PKs (16). In contrast, for GRK2, the substrate consensus sequence has acidic residues (Asp or Glu) at P − 2 and P − 3 (16). Electrostatics seems, then, to govern the specificity between position 129 and the P − 2 or P − 3 site in GRK, whereas hydrophobic packing appears to dominate in PKA, PKC, RAC, and S6 PK. This hypothesis could be tested by mutating Phe-129 in PKA to Lys. The substrate consensus sequence should then prefer Asp or Glu at P − 2 and P − 3.
Position 84 is another clear example of how Table 3 illuminates and unifies experimental data on specificity. For the P + 2 site, there seems to be no preference in PKA (16). On the other hand, PKC (16) and RAC (16) show a significant preference for the basic Arg at P + 2. In PKA, Gln-84 holds on to His 23 at P + 2 through hydrophobic and electrostatic interactions (Fig. 2b). In PKC and RAC, the counterpart of Gln-84 is either Asp or Glu (Table 3), which explains why Arg is preferred at P + 2. PKC and RAC, then, seem to use a salt bridge to achieve specificity for P + 2 recognition, whereas PKA may rely on a combination of hydrophobic and electrostatic interactions. We can readily test this hypothesis by mutating Gln-84 to an acidic residue. An enhanced preference for Arg at P + 2 would then confirm the hypothesis.
We now turn our attention to positions 191 and 196. The residues are not in contact with the inhibitor in PKA (Fig. 2d), so it seems unlikely that Val-191 or Trp-196 plays a direct role in recognizing the substrate. Why, then, do these two positions show intrafamily conservation but interfamily diversity (Table 3)? The answer may lie in the proximity among Val-191, Trp-196, and Thr-197 (Fig. 2d). As stated before, Thr-197 in PKA, and its counterpart in several other PKs, must be phosphorylated for the enzyme to be fully active (12). When PKA is activated by phosphorylation, it could be detrimental for the cell to simultaneously activate other PKs such as PKC and RAC. Therefore, the different residues at positions 191 and 196 may be a method for the cell to regulate the phosphorylation rate and thereby to achieve enzyme-substrate specificity. In particular, the side chain of Trp-196 is largely solvent-exposed (Fig. 2d) and may be involved in controlling access and guiding any PKA kinase to Thr-197. This example illustrates the hypothesis of specificity via differential activation in the Introduction.
Experiments show that phosphoinositide-dependent PK1 (PDK1) phosphorylates Thr-197 in PKA, RAC, and S6 PK at different rates (37–39). At 30°C, PDK1 takes ≈25 min to achieve 50% phosphorylation at Thr-197 (37), whereas the same degree of phosphorylation requires almost 50 min in RAC (38). The wild-type S6 PK is minimally phosphorylated by PDK1, even after 45 min of incubation at 30°C (39). Interestingly, there appears to be a correlation between the size of the side chain at position 196 and the phosphorylation rate at Thr-197: PKA (Trp-196) > RAC (Lys-196) > S6 PK (His196). However, the experimental conditions for the three experiments were not identical (37–39). Therefore, to rigorously test the hypothesis of specificity via differential activation, we propose the mutation of Trp-196 in PKA to Lys (as in RAC) and His (as in S6 PK). If specificity via differential activation is true, we expect the rate of Thr-197 phosphorylation to differ considerably among Trp-196, Lys-196, and His-196 under the same experimental conditions.
Conclusions
We have achieved three important goals in this work. First, without using experimental data, we predict PSDRs in prokaryotic and eukaryotic PKs, and we refine our statistical procedure for the discovery of PSDRs by using paralogous and orthologous proteins. Second, we compare our predictions with current experimental results and we obtain considerable agreement. More importantly, our analysis has enabled us to understand, within a unified framework, how different PKs distinguish their substrates from nonsubstrates. In particular, the hydrophobic/electrostatic balance between a PK and its substrate appears to be a major determinant of enzyme-substrate specificity. Finally, we find PSDRs that are outside the active site in prokaryotic and eukaryotic PKs. Based on our results, as well as structural and biochemical characterizations of eukaryotic PKs, we propose the hypothesis of specificity via differential activation as a way for the cell to control kinase specificity. Very importantly, for every prediction or hypothesis we make, we outline specific mutations that could confirm or refute the prediction or hypothesis.
Supplementary Material
Acknowledgments
This work has been supported by National Institutes of Health Grant 52126 (to E.I.S.), an NEC fund, and a John F. and Virginia T. Taplin Award (to L.A.M.).
Abbreviations
- SDR
specificity-determining residues
- PSDR
putative SDR
- HPK
histidine PK
- DD
dimerization domain
- RR
response regulator
- AGC
cAMP-dependent PK, cGMP-dependent PK, and PKC
- GRK
G protein-coupled receptor kinase
- MSA
multiple sequence alignment
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Rubin G M, Yandell M D, Wortman J R, Gabor Miklos G L, Nelson C R, Hariharan I K, Fortini M E, Li P W, Apweiler R, Fleischmann W, et al. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning G, Whyte D N, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Hunter T. Cell. 1987;50:823–829. doi: 10.1016/0092-8674(87)90509-5. [DOI] [PubMed] [Google Scholar]
- 4.Hanks S K, Hunter T. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 5.Maltsev N, Marland E, Yu G-X, Bhatnagar S, Lusk R. Nucleic Acids Res. 2002;30:349–350. doi: 10.1093/nar/30.1.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grebe T W, Stock J B. Adv Microb Physiol. 1999;41:139–227. doi: 10.1016/s0065-2911(08)60167-8. [DOI] [PubMed] [Google Scholar]
- 7.Koretke K K, Lupas A N, Warren P V, Rosenberg M, Brown J R. Mol Biol Evol. 2000;17:1956–1970. doi: 10.1093/oxfordjournals.molbev.a026297. [DOI] [PubMed] [Google Scholar]
- 8.Kim D-J, Forst S. Microbiology. 2001;147:1197–1212. doi: 10.1099/00221287-147-5-1197. [DOI] [PubMed] [Google Scholar]
- 9.Stock A M, Robinson V L, Goudreau P N. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 10.Dutta R, Qin L, Inouye M. Mol Microbiol. 1999;34:633–640. doi: 10.1046/j.1365-2958.1999.01646.x. [DOI] [PubMed] [Google Scholar]
- 11.Hanks S K, Quinn A M. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- 12.Smith C M, Radzio-Andzelm E, Madhusudan, Akamine P, Taylor S S. Prog Biophys Mol Biol. 1999;71:313–341. doi: 10.1016/s0079-6107(98)00059-5. [DOI] [PubMed] [Google Scholar]
- 13.Mellor H, Parker P J. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pitcher J A, Freedman N J, Lefkowitz R J. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 15.Huse M, Kuriyan J. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 16.Pinna L A, Ruzzene M. Biochim Biophys Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 17.Songyang Z, Lu K P, Kwon Y T, Tsai L-H, Filhol O, Cochet C, Brickey D A, Soderling T R, Bartleson C, Graves D J, et al. Mol Cell Biol. 1996;16:6486–6493. doi: 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yaffe M B, Leparc G G, Lai J, Obata T, Volinia S, Cantley L C. Nat Biotechnol. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- 19.Zapf J, Sen U, Madhusudan, Hoch J A, Varughese K I. Structure (London) 2000;8:851–862. doi: 10.1016/s0969-2126(00)00174-x. [DOI] [PubMed] [Google Scholar]
- 20.Varughese K I. Curr Opin Microbiol. 2002;5:142–148. doi: 10.1016/s1369-5274(02)00305-3. [DOI] [PubMed] [Google Scholar]
- 21.Peeper D, Parker L L, Ewen M E, Toebes M, Hall F L, Xu M, Zantema A, van der Eb A J, Piwnica-Worms H. EMBO J. 1993;12:1947–1954. doi: 10.1002/j.1460-2075.1993.tb05844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knighton D R, Zheng J, Ten Eyck L F, Xuong N-H, Taylor S S, Sowadski J M. Nature. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 23.Haldimann A, Prahalad M K, Fisher S L, Kim S-K, Walsh C T, Wanner B L. Proc Natl Acad Sci USA. 1996;93:14361–14366. doi: 10.1073/pnas.93.25.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirny L A, Gelfand M S. J Mol Biol. 2002;321:7–20. doi: 10.1016/s0022-2836(02)00587-9. [DOI] [PubMed] [Google Scholar]
- 25.Bairoch A, Apweiler R. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eddy S R, Mitchison G, Durbin R. J Comput Biol. 1995;2:9–23. doi: 10.1089/cmb.1995.2.9. [DOI] [PubMed] [Google Scholar]
- 29.Batesman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cover T, Thomas J. Elements of Information Theory. New York: Wiley; 1991. [Google Scholar]
- 31.Bickel P J, Kechris K J, Spector P C, Wedemayer G J, Glazer A N. Proc Natl Acad Sci USA. 2002;99:14764–14771. doi: 10.1073/pnas.222508899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook A, Lowe E D, Chrysina E D, Skamnaki V T, Oikonomakos N G, Johnson L N. Biochemistry. 2002;41:7301–7311. doi: 10.1021/bi0201724. [DOI] [PubMed] [Google Scholar]
- 33.Brinkworth R I, Breinl R A, Kobe B. Proc Natl Acad Sci USA. 2003;100:74–79. doi: 10.1073/pnas.0134224100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wollenberg K R, Atchley W R. Proc Natl Acad Sci USA. 2000;97:3288–3291. doi: 10.1073/pnas.070154797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirny L A, Shakhnovich E I. J Mol Biol. 1999;291:177–196. doi: 10.1006/jmbi.1999.2911. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Mirny L A, Shakhnovich E I. Nat Struct Biol. 2000;7:336–341. doi: 10.1038/74111. [DOI] [PubMed] [Google Scholar]
- 37.Cheng X, Ma Y, Moore M, Hemmings B A, Taylor S S. Proc Natl Acad Sci USA. 1998;95:9849–9854. doi: 10.1073/pnas.95.17.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alessi D R, James S R, Downes C P, Holmes A B, Gaffney P R J, Reese C B, Cohen P. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 39.Pullen N, Dennis P B, Andjelkovic M, Dufner A, Kozma S C, Hemmings B A, Thomas G. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.