

Abstract
In signal-recognition particle (SRP)-dependent protein targeting to the bacterial plasma membrane, two GTPases, Ffh (a subunit of the bacterial SRP) and FtsY (the bacterial SRP receptor), act as GTPase activating proteins for one another. The molecular mechanism of this reciprocal GTPase activation is poorly understood. In this work, we show that, unlike other GTPases, free FtsY exhibits only low preference for GTP over other nucleotides. On formation of the SRP⋅FtsY complex, however, the nucleotide specificity of FtsY is enhanced 103-fold. Thus, interactions with SRP must induce conformational changes that directly affect the FtsY GTP-binding site: in response to SRP binding, FtsY switches from a nonspecific “open” state to a “closed” state that provides discrimination between cognate and noncognate nucleotides. We propose that this conformational change leads to more accurate positioning of the nucleotide and thus could contribute to activation of FtsY's GTPase activity by a novel mechanism.
GTPases control a variety of biological processes, including signal transduction, translation, vesicle transport, and cotranslational protein targeting. The function of GTPases is regulated by a GTPase “switch” mechanism, in which the GTPases undergo conformational changes between an active GTP-bound form and an inactive GDP-bound form that are driven by cycles of GTP binding and hydrolysis. For many GTPases, the rate of interconversion between the active and inactive forms is modulated by external factors, such as guanine nucleotide exchange factors, guanine nucleotide dissociation inhibitors, and GTPase activating proteins (1, 2).
Two interacting GTPases, Ffh and FtsY, mediate cotranslational protein targeting to the bacterial plasma membrane (3). Ffh and FtsY are homologs of the core components of the eukaryotic signal-recognition particle (SRP) and SRP receptor, respectively. Ffh, in complex with a 4.5S RNA, forms a minimal bacterial SRP that recognizes a nascent polypeptide chain bearing a signal sequence as it emerges from the ribosome. The complex of ribosome, nascent chain, and SRP is then targeted to the plasma membrane via interaction of Ffh with FtsY. On this interaction, the ribosome⋅nascent chain complex is transferred to the translocation machinery or “translocon,” which either integrates the protein into or translocates it across the membrane. Like most GTPases, Ffh and FtsY (and their eukaryotic homologs) engage in functional cycles that are tightly coupled to their GTPase cycles (3). During each targeting reaction, GTP binding is required for complex formation between SRP and FtsY, allowing the nascent polypeptide chain to be delivered to the plasma (or endoplasmic reticulum) membrane; subsequent GTP hydrolysis then drives disassembly of the SRP⋅FtsY complex, allowing the GTPases to be recycled.
The structure of Ffh and FtsY defines them as a unique subgroup in the GTPase superfamily (4–7). Both proteins contain a central GTPase domain that shares homology with other members of the GTPase superfamily, such as Ras and EF-Tu. On the other hand, all SRP family GTPases also contain an N-terminal four-helix bundle not present in other GTPases. The four-helix bundle and the GTPase domain form a structural and functional unit, called collectively the NG domain (4, 6). In addition, Ffh and FtsY each possesses a specialized domain that enables it to play its unique role in protein targeting. FtsY has an acidic preN domain, or “A domain,” which enables it to interact with the membrane (8), and Ffh has a C-terminal methionine-rich domain, or M domain, which contains binding sites for signal sequences (5, 9–11) and for the SRP RNA (5, 11–13).
The biochemical properties also distinguish Ffh and FtsY from other classical GTPases. Unlike canonical GTPases that bind guanine nucleotides tightly and require nucleotide exchange factors to convert from the GDP- to GTP-bound forms, Ffh and FtsY have weak nucleotide affinities, with GDP binding and dissociating quickly (14–16). Therefore, there is no requirement for nucleotide exchange factors. Further, conversion from their GTP- to GDP-bound forms is catalyzed by a direct interaction between Ffh and FtsY, with both proteins acting as GTPase activating proteins for one another (17). The molecular mechanism of this reciprocal GTPase activation remains poorly understood.
In an attempt to address how the targeting cycle carried out by Ffh and FtsY is coupled to their GTPase cycles, we probed their GTPase sites using an FtsY mutant that harbors an Asp → Asn substitution, FtsY(D449N) (17). This Asp residue, located in the GTP-binding consensus motif (N/TKxD), is conserved throughout the GTPase superfamily and engages in a hydrogen bond network with the N2 and N3 amino protons of the guanine ring (Fig. 1A; refs. 18–21). In many GTPases, mutation of this Asp residue to Asn weakens their affinity for GTP and strengthens their affinity for xanthosine 5′-triphosphate (XTP), resulting in a switch in nucleotide specificity by up to 105-fold (Fig. 1B; see, e.g., refs. 22–25). Accordingly, we previously showed that, in the presence of SRP, GTP is hydrolyzed preferentially by wild-type FtsY, whereas XTP is hydrolyzed preferentially by mutant FtsY(D449N) (17). Surprisingly, we show here that FtsY acquires nucleotide specificity only on formation of the SRP⋅FtsY complex. These results provide evidence for conformational changes in FtsY during interaction with SRP and suggest a novel mechanism for the activation of FtsY's GTPase reaction by SRP.
Figure 1.

Hydrogen bonding interactions between the base moiety of GTP (G) and the conserved aspartate residue (A), and between the base moiety of XTP (X) and a corresponding asparagine residue (B). The Asp-449 residue of FtsY was mislabeled as Asp-441 in a previous publication (17).
Materials and Methods
Materials.
XTP, GTP, and 5′-guanylylimidodiphosphate (GppNHp) were from Sigma and were purified over a MonoQ column (Amersham Biosciences). Xanthosine 5′-diphosphate and GDP were from Sigma (98%). [γ-32P]GTP and [γ-32P]XTP were from ICN and Amersham Biosciences. A truncated version of Escherichia coli FtsY, FtsY (47–497), was used for this study because of its higher solubility and expression level (26). The expression and purification of Ffh, FtsY, and 4.5S RNA have been described (26, 27). For arguments against concerns about GTPase contaminants in FtsY preparations, see Supporting Text and Fig. 7, which are published as supporting information on the PNAS web site, www.pnas.org.
Kinetics.
All reactions were carried out at 25°C in assay buffer [50 mM KHepes/150 mM KOAc/2 mM Mg(OAc)2/2 mM DTT/0.01% Nikkol (Anatrace, Maumee, OH), pH 7.5] with cold GTP or XTP doped with trace amounts of [γ-32P]GTP or [γ-32P]XTP. Reactions were initiated by addition of GTP or XTP and were followed and analyzed as described (26). Single turnover reactions were followed for ≥3 half-lives except for very slow reactions. Good first-order fits to the data, with end points of ≥90%, were obtained in all cases. For multiple turnover reactions, the initial linear portion of the reaction (≤20% reaction) was followed, and an end point of 90% was assumed to obtain observed rate constants from the initial rates.
Determination of the Basal GTPase and XTPase Activity.
The basal GTPase and XTPase activities of FtsY were measured in single turnover reactions with FtsY in excess over GTP and XTP, respectively. The [FtsY] dependence of observed rate constants of the reaction (kobs) was fit to Eq. 1,
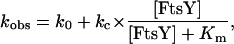 |
1 |
in which kc is the maximal rate constant with saturating FtsY, Km is the FtsY concentration that gives half the maximal rate constant, and k0 is a fudge factor that represents the reaction rate in the absence of FtsY.
Determination of Nucleotide Affinity.
The GTP affinity of wild-type FtsY and mutant FtsY(D449N) was determined from the FtsY concentration dependence of the basal GTPase reactions according to Eq. 1. Because the chemical step is rate limiting for the basal GTPase reaction, Km is equal to Kd, the dissociation constant of GTP (26). The affinity of other nucleotides was determined by inhibition methods as described (26).
Determination of Stimulated GTPase and XTPase Reactions.
The SRP-stimulated XTPase reaction of FtsY (XTP* + FtsY + GTP·SRP → products) was determined in single turnover experiments with a small fixed amount of FtsY (0.1 μM, wild type or D449N) and varying concentrations of SRP. The SRP-stimulated GTPase reaction was determined in multiple turnover experiments in the presence of a small fixed amount of SRP (0.05 μM) and varying concentrations of wild-type FtsY or mutant FtsY(D449N). The FtsY concentration dependence was fit to single binding curves analogous to Eq. 1.
Fluorescence Measurements.
Fluorescence emission spectra were acquired as described (26). The association reactions of SRP and FtsY (wild type and mutant) were carried out in assay buffer in the presence of 1 μM FtsY and 2 μM SRP. The spectra for uncomplexed FtsY were first determined in the presence of 2 mM EDTA, and Mg2+ was added to a final concentration of 10 mM to initiate complex formation.
Results
Free FtsY Has Low Nucleotide Specificity.
GTPases are generally characterized by a high degree of specificity toward their nucleotide substrates. The magnitude of discrimination between cognate and noncognate bases is typically 102- to 103-fold for well studied GTPases (see, e.g., refs. 22–25 and 28 and references therein).
We were therefore surprised to find that FtsY shows little discrimination between nucleotides. As shown in Fig. 2 and summarized in Table 1, FtsY hydrolyzes GTP with a rate constant (kc/Km) of 230 M−1 min−1, no more than 13-fold faster than the hydrolysis of noncognate nucleotides such as XTP and ATP. Similarly, when the binding affinity of cognate nucleotides is compared with those of noncognate nucleotides (Kds in Table 2), GTP and GDP bind no more than 30-fold stronger than noncognate nucleotides, including ATP and CTP. This is remarkable, because it demonstrates that FtsY does not discriminate strongly even between purine and pyrimidine containing nucleotides.
Figure 2.

Basal GTPase (A) and XTPase (B) reactions of wild-type FtsY (□ and ○) and mutant FtsY(D449N) (■ and ●), determined as described in Materials and Methods. The solid lines are fits of the FtsY concentration dependences to Eq. 1. For the GTPase reactions (A), the fit gave kc values of 0.0039 and 0.0010 min−1 and Km values of 15 and 19 μM for FtsY(wt) and FtsY(D449N), respectively.
Table 1.
Basal nucleotide hydrolysis rate constants for wild-type FtsY and mutant FtsY (D449N)
| FtsY construct |
kc/Km (M−1 min−1)*
|
GTP specificity†
|
|||
|---|---|---|---|---|---|
| GTP | XTP | ATP | GTP/XTP | GTP/ATP | |
| Wild type | 230 | 17 | 120 | 13 | 1.9 |
| D449N | 53 | 55 | 58 | 0.96 | 0.91 |
| Rel (kwt/kD449N) | 4.3 | 0.31 | 2.1 | na | na |
na, Not applicable.
The second-order rate constant for the reaction: FtsY + NTP → products (kc/Km; N = G, X, or A) was determined from the [FtsY] dependence in Fig. 2. For GTP, this is obtained from the kc and Km values from the fit of data to Eq. 1. For XTP and ATP, this is obtained from the slope of the initial linear range of the FtsY concentration dependence (Fig. 2 and data not shown).
GTP specificity is defined as the preference of the FtsY constructs to hydrolyze GTP relative to other nucleotides such as XTP and ATP, and was obtained from the ratio of kc/Km values for GTP over XTP or ATP.
Table 2.
Nucleotide affinities of wild-type FtsY and mutant FtsY (D449N)
| FtsY construct |
Kd, μM
|
||||||
|---|---|---|---|---|---|---|---|
| GTP | GDP | GppNHp | XTP | ITP | ATP | CTP | |
| Wild type | 15 | 24 | 38 | 460 | 150 | 250 | 490 |
| D449N | 19 | 26 | 24 | 260 | 510 | 670 | |
The nucleotide specificity of GTPases typically arises from a network of interactions with the guanine base, the most conserved interaction is the hydrogen bonding network between a conserved Asp residue in the GTP-binding pocket and the exocyclic amine of guanine (Fig. 1). In many GTPases, mutation of this Asp residue to Asn results in a change in the nucleotide preference from GTP to XTP by up to 105-fold (see, e.g., refs. 22–25 and 28 and references therein). To probe the contribution of the conserved Asp residue to the nucleotide specificity of FtsY, we next explored the properties of FtsY bearing the Asp → Asn mutation, FtsY(D449N). As shown in Fig. 2 and quantitated in Table 1, mutant FtsY(D449N) hydrolyzes XTP only 3-fold faster and GTP only 4-fold slower than wild-type FtsY, resulting in an indistinguishable rate constant for GTP and XTP hydrolysis by mutant FtsY(D449N), which is nowhere near the discrimination seen in other GTPases. There are also <2-fold changes in the nucleotide-binding affinity between wild-type FtsY and mutant FtsY(D449N) (Table 2). Thus, we conclude from the data presented so far that free FtsY has a very low specificity in recognizing different nucleotides and that Asp-449 contributes little, if anything, to the selective recognition of the cognate nucleotide.
The Presence of SRP Confers Nucleotide Specificity to FtsY.
The low nucleotide specificity of FtsY described above was puzzling in light of a previous study from this lab, which showed that FtsY and FtsY(D449N) preferentially hydrolyze their cognate nucleotides (17). One possible resolution of this paradox was suggested by the fact that the previous results were obtained in the presence of bacterial SRP (Ffh bound to 4.5S RNA) and thus measured the SRP-stimulated reaction, whereas the properties of free FtsY were examined above. We therefore asked whether SRP modulates the nucleotide specificity of FtsY.
To test this possibility, we determined the rate constant for XTP hydrolysis by FtsY and FtsY(D449N) in the presence of increasing amounts of SRP (Fig. 3). The rate constant for the reaction XTP + FtsY + SRP → products [(kc/Km)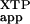 in Table 3], which includes both binding of SRP to FtsY and SRP-dependent stimulation of FtsY's XTPase activity, was compared between wild-type FtsY and mutant FtsY(D449N). Because SRP also contains a GTPase domain that needs to be nucleotide triphosphate-bound to interact with FtsY, we also added GTP to occupy the GTPase site in SRP. The concentration of GTP (10 μM) was chosen so that the GTP-binding site in SRP was saturated, whereas that of FtsY was not. This was possible because GTP binds tighter to SRP than to FtsY (Kd = 0.39 and 15 μM for SRP and FtsY, respectively; ref. 26 and Table 2).
in Table 3], which includes both binding of SRP to FtsY and SRP-dependent stimulation of FtsY's XTPase activity, was compared between wild-type FtsY and mutant FtsY(D449N). Because SRP also contains a GTPase domain that needs to be nucleotide triphosphate-bound to interact with FtsY, we also added GTP to occupy the GTPase site in SRP. The concentration of GTP (10 μM) was chosen so that the GTP-binding site in SRP was saturated, whereas that of FtsY was not. This was possible because GTP binds tighter to SRP than to FtsY (Kd = 0.39 and 15 μM for SRP and FtsY, respectively; ref. 26 and Table 2).
Figure 3.

SRP preferentially stimulates the XTPase activity of mutant FtsY(D449N). Observed rate constants for XTP hydrolysis were determined as described in Materials and Methods with 0.1 μM mutant FtsY(D449N) (●) or wild-type FtsY (○), or with no FtsY added (×).
Table 3.
Rate constant for SRP-stimulated XTP hydrolysis by wild-type FtsY and mutant FtsY (D449N)
| FtsY construct | (kc/Km)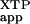 *, M−1 min−1 *, M−1 min−1
|
(kc/Km)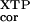 †, M−1 min−1 †, M−1 min−1
|
(kc/Km)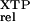 ‡ ‡
|
|---|---|---|---|
| D449N | 1.5 × 105 | 1.5 × 105 | 1.6 × 103 |
| Wild type | 270 | 94 | (1) |
| — | 180 | (0) |
The apparent second-order rate constant (kc/Km)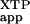 was determined from the slope of the initial linear portion of the SRP concentration dependence (Fig. 3). The rate constant observed in the absence of FtsY (—) reflects the slow rate of XTP hydrolysis by SRP.
was determined from the slope of the initial linear portion of the SRP concentration dependence (Fig. 3). The rate constant observed in the absence of FtsY (—) reflects the slow rate of XTP hydrolysis by SRP.
The corrected rate constant (kc/Km)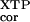 is the apparent rate constant [(kc/Km)
is the apparent rate constant [(kc/Km)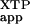 ] with wild-type FtsY and mutant FtsY (D449N) subtracted by that without added FtsY. This corrects for the background XTP hydrolysis reaction from SRP.
] with wild-type FtsY and mutant FtsY (D449N) subtracted by that without added FtsY. This corrects for the background XTP hydrolysis reaction from SRP.
The relative rate constant (kc/Km)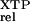 is the rate constant for XTP hydrolysis [(kc/Km)
is the rate constant for XTP hydrolysis [(kc/Km)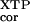 ] by mutant FtsY (D449N) relative to wild-type FtsY.
] by mutant FtsY (D449N) relative to wild-type FtsY.
The results of this experiment show that XTP hydrolysis by mutant FtsY(D449N) is stimulated by increasing amounts of SRP (Fig. 3, filled circles), consistent with the role of SRP as the GTPase activating protein for FtsY (17, 26). In contrast, wild-type FtsY shows little enhancement of XTP hydrolysis (open circles) over a control reaction in which FtsY was omitted (crosses). The rate constant for XTP hydrolysis with the mutant FtsY(D449N) is 1.6 × 103-fold faster than with wild-type FtsY [Table 3, (kc/Km)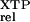 ]. This represents a 103-fold discrimination between the cognate and noncognate nucleotides, comparable to the level of discrimination typically observed in other GTPases. A similar result was obtained when the nonhydrolyzable GTP analog, GppNHp, was used instead of GTP, giving an XTPase rate constant that is 1.1 × 103-fold faster with mutant FtsY(D449N) than with wild-type FtsY (not shown). Thus, the presence of SRP substantially increases the nucleotide specificity of FtsY, and the amino acid at position 449 (Asp or Asn) plays a major role in the SRP-induced specificity change.
]. This represents a 103-fold discrimination between the cognate and noncognate nucleotides, comparable to the level of discrimination typically observed in other GTPases. A similar result was obtained when the nonhydrolyzable GTP analog, GppNHp, was used instead of GTP, giving an XTPase rate constant that is 1.1 × 103-fold faster with mutant FtsY(D449N) than with wild-type FtsY (not shown). Thus, the presence of SRP substantially increases the nucleotide specificity of FtsY, and the amino acid at position 449 (Asp or Asn) plays a major role in the SRP-induced specificity change.
FtsY Acquires Nucleotide Specificity in the SRP⋅FtsY Complex Formation Step.
The experiment described above showed that FtsY can discriminate between the cognate and noncognate nucleotides in a reaction that includes two steps: (i) formation of the SRP⋅FtsY complex and (ii) stimulation of XTPase activity once the complex is formed. To determine during which of these two steps FtsY gains nucleotide specificity, we compared the protein concentration dependence for the SRP-stimulated GTPase reaction of FtsY when it is bound to the cognate and noncognate nucleotides. If only complex formation is impaired when a noncognate nucleotide is bound to FtsY, then SRP should still be able to substantially stimulate FtsY's GTPase activity if complex formation is forced at high concentrations of FtsY. In contrast, if only the stimulation of nucleotide hydrolysis by SRP is impaired when a noncognate nucleotide is bound to FtsY, then no significant stimulation of FtsY's GTPase activity would be observed even at high concentrations of FtsY.
We therefore determined the rate of GTP hydrolysis in the presence of a small fixed amount of SRP and varying concentrations of FtsY or FtsY(D449N) (Fig. 4). The concentration of FtsY, instead of SRP, was varied because FtsY is much more soluble than SRP, allowing us to vary its concentration over a wider range. The concentration of GTP (100 μM) in the reaction ensures that the majority of SRP and FtsY (wild type or mutant) molecules are bound with GTP. The data show that at low concentration, the mutant FtsY(D449N) exhibits a lower rate of GTP hydrolysis than wild-type FtsY (Fig. 4, filled vs. open circles). At high FtsY concentrations, however, both the mutant and wild-type FtsY approach similar levels of maximal hydrolysis rate. These results are thus consistent with the model that efficient complex formation requires FtsY to be bound to its cognate nucleotide, but once the complex is formed, the GTPase activation step is insensitive to the identity of the bound nucleotide.
Figure 4.

The SRP-stimulated GTPase reaction of wild-type FtsY (○) and mutant FtsY(D449N) (●), determined as described in Materials and Methods. The lines are fits of the FtsY concentration dependences to a single binding curve analogous to Eq. 1 and gave Km values of 0.77 and 8.2 μM and maximal GTPase rate constants (kcat) of 54 and 35 min−1 for wild-type FtsY and mutant FtsY(D449N), respectively.
In Fig. 3, we showed that XTP preferentially interacts with mutant FtsY(D449N) over wild-type FtsY. The data in Fig. 4 complement these results by demonstrating that, reciprocally, GTP preferentially interacts with wild-type FtsY over mutant FtsY(D449N).
To provide independent evidence for the notion that complex formation requires FtsY to be bound to its cognate nucleotide, we directly monitored complex formation using a fluorescence assay described previously (15, 29). The assay is based on the observation that FtsY contains a tryptophan residue whose fluorescence properties change on binding SRP. In the course of the studies described here, we observed that formation of this complex requires the presence of Mg2+ ions (Fig. 5A), which provided a convenient way to initiate the reaction while keeping all other constituents in the solution constant. In agreement with previous results, the fluorescence spectrum of wild-type FtsY exhibits a blue shift and an almost 2-fold increase in intensity on complex formation in the presence of saturating GppNHp and Mg2+ (Fig. 5A). In contrast, mutant FtsY(D449N) showed no fluorescence change on addition of Mg2+ when GppNHp was present as the sole nucleotide (Fig. 5B). When the reaction is supplemented with XTP in addition to GppNHp, however, the fluorescence increase is partially restored for mutant FtsY(D449N) (Fig. 5D).
Figure 5.

Formation of the SRP⋅FtsY complex requires FtsY to be bound with the cognate nucleotide. The fluorescence emission spectrum of SRP mixed with wild-type or mutant FtsY was determined in the presence of various nucleotides with or without 10 mM added Mg2+ (filled and open symbols, respectively), as described in Materials and Methods. (A) Wild-type FtsY in the presence of 100 μM GppNHp. (B) FtsY(D449N) in the presence of 100 μM GppNHp. (C) Wild-type FtsY in the presence of 100 μM GppNHp and 500 μM XTP. (D) FtsY(D449N) in the presence of 100 μM GppNHp and 500 μM XTP. In C and D, the dashed and dotted lines are the spectra of FtsY (wild type or mutant) without added XTP in the presence and absence of Mg2+, respectively (data from A and B).
The smaller increase in fluorescence with mutant FtsY(D449N) relative to that with wild-type FtsY is expected because the mutant FtsY(D449N) binds XTP weakly, especially in the presence of GppNHp. In addition, XTP can also bind to Ffh, and this binding would inhibit complex formation by forcing a noncognate nucleotide into SRP's GTP-binding site [SRP binds XTP and GppNHp with dissociation constants of 22 and 3.3 μM, respectively (S.S. and P.W., unpublished results)]. Therefore, at any given concentration of GppNHp and XTP, only a fraction of proteins is occupied with a cognate nucleotide, Ffh with GppNHp and FtsY(D449N) with XTP. Indeed, addition of XTP into a reaction containing wild-type FtsY and SRP reduces the fluorescence change on Mg2+ addition (Fig. 5C), demonstrating that the restoration of fluorescence increase by XTP is specific for mutant FtsY(D449N) (Fig. 5D). Taken together, the results of the fluorescence experiments provide independent evidence that FtsY acquires the ability to discriminate between cognate and noncognate nucleotides during formation of the SRP⋅FtsY complex.
Discussion
We have characterized here the nucleotide recognition properties of FtsY, one of the two directly interacting GTPases involved in protein targeting. Our results further support the notion that FtsY show many distinct features compared with the classical “GTPase switch” paradigm that has been described for other GTPases (2). Thus, elucidation and comparison of the mechanism of SRP subfamily of GTPases to those of other members of the GTPase superfamily significantly broaden our view of the basic principles of molecular recognition and regulation of nucleotide-dependent molecular switches.
An Open, Nondiscriminating Nucleotide-Binding Site in Free FtsY.
GTPases are typically characterized by a high specificity for their cognate guanine-containing nucleotides over noncognate nucleotides. Unlike other GTPases, however, we show here that FtsY displays surprisingly low nucleotide specificity in its free, uncomplexed form.
The small difference in the nucleotide affinity and hydrolysis rate between wild-type FtsY and mutant FtsY(D449N) shows that in free FtsY the conserved Asp-449 does not form specific interactions with the exocyclic amine of the guanine ring. Indeed, the weak GTP affinity of FtsY and its ability to bind and hydrolyze a variety of nucleotides suggest an open, floppy GTPase site that is prone to rearrangements [Fig. 6, (FtsY)o]. Consistent with this model, the crystal structure of FtsY in complex with GDP or GppNHp revealed an elongated nucleotide-binding cleft in which the distance between Asp-449 and the guanine ring appears to be too long to make a strong hydrogen bond [D = 4.6 and 3.4 Å] between the carboxylic oxygen of Asp-449 and N2 of guanine in two different crystal structures of FtsY bound with GppNHp from Thermus aquaticus (C. Reyes, E. Rutenber, P.W., and R. Stroud, unpublished results); in contrast, the corresponding distance is 2.9 Å in the Ras structure (21) and 3.0 Å in the EF-Tu structure (18)]. Consistent with this notion, the GTP-binding site also appeared wide open in the crystal structure of apo-FtsY from E. coli (6).
Figure 6.

Model for SRP-induced conformational change of FtsY. (FtsY)o and (FtsY)c denote the open and closed states of FtsY, respectively. KΔ and KΔ′ are the equilibrium constants for the open → closed conformational change with free FtsY and FtsY bound to SRP, respectively, and Kb,open and Kb,closed denote the equilibrium for SRP binding in the open and closed states, respectively. R denotes the ribose ring, P denotes the phosphate groups, and the waves depict the floppy binding and inaccurate positioning of GTP within the nucleotide-binding site in the open state. The dashed lines depict the hydrogen bonding interaction between Asp-449 and N2 of guanine and possible additional interactions formed between active site residues and GTP in the closed conformation.
Several previous studies also suggest the presence of a nonspecific GTP-binding site in FtsY. Crosslinking and protease protection studies of E. coli FtsY suggested that GDP can interact with an FtsY mutant, FtsY(D449A), at least as efficiently as wild-type FtsY (30). The mammalian homolog of FtsY, SRα, retains an affinity for GTP within 2-fold of that of wild-type SRα on mutation of the conserved Asp (31). Thus, it appears that promiscuous nucleotide recognition is a conserved property among bacterial FtsY and its eukaryotic homologs. On the other hand, reduced interaction with GTP and increased interaction with XTP on mutation of the Asp have been reported in two cases (16, 32). Interestingly, the FtsY construct used in one of these studies contains only the NG domain of FtsY (16), raising the possibility that the A domain may play a role in modulating the GTPase site of FtsY.
These structural and biochemical features of free FtsY are distinct from those observed with many classical GTPases, for which, in all structures examined to date, a network of hydrogen bonding and salt bridge interactions within the nucleotide-binding pocket provide a snug fit for GDP or GTP (18–21). This leads to a large penalty in binding energy when modifications are made on GTP or on active site residues to disrupt favorable interactions and introduce unfavorable electrostatic and/or steric interactions, as is the case for the Asp → Asn mutation and the guanine → xanthine base substitution. The snug binding of the guanine ring in classical GTPases also leads to slow GDP dissociation, which becomes the rate-limiting step in the functional cycles of these GTPases and thus to the requirement for nucleotide exchange factors to convert from their GDP- to GTP-bound forms. Thus, the floppy nucleotide-binding pocket of FtsY rationalizes the absence of corresponding nucleotide exchange factors for the SRP family of GTPases. The open GTPase site of FtsY provides an alternative view to a previously proposed model (16) in which the low affinity of FtsY for nucleotides was attributed to a built-in exchange factor domain that, as an added-on feature, actively stabilizes the nucleotide-free state of FtsY.
Conformational Change on Complex Formation Between FtsY and SRP.
In contrast to the low nucleotide specificity in its free form, FtsY acquires 103-fold enhanced specificity when it forms a complex with SRP, allowing it to discriminate strongly between cognate and noncognate nucleotides. Our data provide strong evidence that during formation of the SRP⋅FtsY complex, Asp-449 [or Asn-449 in mutant FtsY(D449N)] forms specific hydrogen bonds with the exocyclic amine of guanine (or the O2 of xanthine; Fig. 1). Thus, significant conformational changes must occur in the GTP-binding site of FtsY on complex formation.
The model in Fig. 6 accounts for the observed increase in the nucleotide specificity of FtsY on binding SRP. We propose that FtsY alters between two conformational states, an “open” and a “closed” state [Fig. 6, (FtsY)o and (FtsY)c, respectively]. In the open state, the nucleotide-binding pocket is oversized, and thus interactions of GTP with Asp-449 and possibly with other active site residues are not formed. In this state, FtsY binds nucleotides weakly and with little discrimination. In the “closed” state, GTP is more precisely positioned in the GTPase site, forming specific interactions with Asp-449 and possibly also with other catalytic residues at the active site. Because free FtsY exhibits little discrimination between guanine and xanthine and shows no effect of the D449N mutation, it would exist predominantly in the open state (Fig. 6, KΔ < 1). On complex formation with SRP, FtsY displays nucleotide recognition properties expected for the closed state (Fig. 6, KΔ′ > 1). Thus, SRP must bind preferentially to the population of FtsY molecules that are in the closed state (Fig. 6, Kb,closed > Kb,open), thereby driving the open → closed conformational change.
A large body of data supports the presence of a conformational change on FtsY during complex formation. Conformational changes in the GTPase sites of FtsY and Ffh have been proposed based on biochemical data that suggest that active site contacts with the γ-phosphate of GTP are formed only in the SRP⋅FtsY complex but not in the free proteins (26). In addition, proteolysis studies suggested conformational changes in the N domain of FtsY (33), and site-directed mutagenesis provided evidence for conformational rearrangements at the NG interface of Ffh and FtsY (34). It remains to be determined whether the previous results and the data presented herein report on the same or different conformational rearrangements that occur during complex formation.
In contrast to FtsY, its binding partner Ffh has a high specificity for GTP over noncognate nucleotides, and this GTP specificity is switched 104-fold to that for XTP by mutation of the corresponding Asp residue to Asn (S.S. and P.W., unpublished results). Consistent with the biochemical data, the crystal structure of Ffh revealed that the conserved Asp is positioned close to the N2 of guanine ring to make a good hydrogen bond (35). Thus, if the principles of our model also apply to Ffh, then Ffh and FtsY lie on different points of equilibrium for the open → closed conformational change. In this respect, therefore, Ffh and FtsY do not behave in a perfectly symmetric fashion as was previously thought.
Once the SRP⋅FtsY complex is formed, Ffh and FtsY act as GTPase activating proteins (GAPs) for one another, resulting in reciprocal stimulation of GTP hydrolysis. One possible mechanism for this activation is that Ffh inserts a missing catalytic residue(s) into the GTPase active site of FtsY (and vice versa), as proposed for the mechanism of action of GAPs for Ras and other classical G proteins (21, 36–42). Alternatively, the stimulation of GTPase activity could arise from conformational changes, whereas the essential catalytic groups are provided by the GTPase itself. The increased nucleotide specificity in FtsY shown here suggests that additional interactions between the nucleotide and active site residues are formed on complex formation. We suggest that these additional binding interactions can lead to better positioning of the γ-phosphate group with respect to catalytic residues in FtsY. This would reduce the rearrangements that need to occur in going from the ground state to the reaction's transition state, thereby effecting an acceleration of the GTP hydrolysis reaction (43). Thus, FtsY may use a mechanism for GTPase activation that is distinct from those discussed for other GTPases interacting with their respective GAPs. In the most extreme form of this model, these conformational changes account for all of the observed GAP activity without insertion of a catalytic residue from GAP. In principle, however, GAP activity could result from a combination of these effects.
The “open” state of FtsY defined here explains previous observations that FtsY (or its mammalian homolog, SRα) is in an “empty site” form before its interaction with SRP (31). This notion was based on the observation that, in contrast to the SRP–SR complex, GTP could not be copurified with SRα, and GppNHp prebound to SRα rapidly exchanges with GTP and GDP (31). Thus a model was proposed in which FtsY would be in a nucleotide-free state before interaction with SRP. However, the concept of a nucleotide-free state for FtsY before binding SRP is difficult to reconcile with affinity measurements of free FtsY for GTP and GDP (Kd = 15 and 30 μM, respectively; Table 2). On the basis of these affinities, free FtsY should always be saturated with GTP at physiological concentrations of GTP and GDP (900 and 100 μM, respectively; ref. 44) even before SRP binding. We propose that the previous observations can be better explained by the “open” state of FtsY, in which the nucleotide is bound but not well enough positioned and thus exchanges rapidly with the solution (koff = 15 s−1; ref. 15).
Biological Implications.
The switch of FtsY from the “open” to the “closed” conformation provides an attractive point of regulation by additional components of the protein targeting reaction. As shown here, free FtsY exists predominantly in the “open” state in which it is less active in interacting with SRP or in binding and hydrolyzing GTP. Thus, any factor that shifts the conformational equilibrium of FtsY toward the “closed” state would facilitate its interaction with SRP. Phospholipids in the membrane and/or the translocon, for example, are potential candidates to play such a regulatory role. In this way, FtsY molecules bound to the membrane and in the vicinity of available protein translocation sites would be selectively predisposed to interact with SRP. Reciprocally, the GTPase domain of Ffh might be similarly controlled, possibly involving other rearrangements in the active site that are less apparent as a gain in nucleotide specificity. For Ffh, potential regulators would include the ribosome and signal sequence. In this way, both SRP and FtsY would each be “primed” on interaction with their respective cargo molecules, so that the resulting SRP–FtsY interaction can efficiently deliver the nascent polypeptide chains to the translocation machinery. This model also explains why the substantial fraction of free FtsY normally present in the cytosol (45) does not lead to futile cycles of GTP hydrolysis or nascent chain release from SRP. Thus, the SRP–FtsY interaction, which functions at the very heart of SRP-dependent protein targeting, would be regulated not only by occupancy of either binding partner with nucleotide triphosphate per se but also by conformational switches that are controlled by their respective cargo molecules.
Supplementary Material
Acknowledgments
We thank A. E. Johnson for advice on fluorescence experiments and T. Powers, R. Gilmore, H. Bourne, R. Stroud, and members of the Walter and Stroud laboratories for comments on the manuscript. This work was supported by National Institutes of Health Grant GM 32384 (to P.W.). P.W. is an Investigator of the Howard Hughes Medical Institute, and S.S. is a Cancer Research Fund Fellow of the Damon Runyon–Walter Winchell Foundation.
Abbreviations
- SRP
signal-recognition particle
- XTP
xanthosine 5′-triphosphate
- GppNHp
5′-guanylylimido diphosphate
- GAP
GTPase-activating protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gilman A G. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 2.Bourne H R, Sanders D A, McCormick F. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 3.Walter P, Johnson A E. Annu Rev Cell Biol. 1994;10:87–119. doi: 10.1146/annurev.cb.10.110194.000511. [DOI] [PubMed] [Google Scholar]
- 4.Freymann D M, Keenan R J, Stroud R M, Walter P. Nature. 1997;385:361–364. doi: 10.1038/385361a0. [DOI] [PubMed] [Google Scholar]
- 5.Keenan R J, Freymann D M, Walter P, Stroud R M. Cell. 1998;94:181–191. doi: 10.1016/s0092-8674(00)81418-x. [DOI] [PubMed] [Google Scholar]
- 6.Montoya G, Svensson C, Luirink J, Sinning I. Nature. 1997;385:365–368. doi: 10.1038/385365a0. [DOI] [PubMed] [Google Scholar]
- 7.Freymann D M, Keenan R J, Stroud R B, Walter P. Nat Struct Biol. 1999;6:793–801. doi: 10.1038/11572. [DOI] [PubMed] [Google Scholar]
- 8.Zelazny A, Seluanov A, Cooper A, Bibi E. Proc Natl Acad Sci USA. 1997;94:6025–6029. doi: 10.1073/pnas.94.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein H D, Poritz M A, Strub K, Hoben P J, Brenner S, Walter P. Nature. 1989;340:482–486. doi: 10.1038/340482a0. [DOI] [PubMed] [Google Scholar]
- 10.Lütcke H, High S, Römisch K, Ashford A J, Dobberstein B. EMBO J. 1992;11:1543–1551. doi: 10.1002/j.1460-2075.1992.tb05199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zopf D, Bernstein H D, Johnson A E, Walter P. EMBO J. 1990;9:4511–4517. doi: 10.1002/j.1460-2075.1990.tb07902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Römisch K, Webb J, Lingelback K, Gausepohl H, Dobberstein B. J Cell Biol. 1990;111:1793–1807. doi: 10.1083/jcb.111.5.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batey R T, Rambo R P, Lucast L, Rha B, Doudna J A. Science. 2000;287:1232–1239. doi: 10.1126/science.287.5456.1232. [DOI] [PubMed] [Google Scholar]
- 14.Jagath J R, Rodnina M V, Lentzen G, Wintermeyer W. Biochemistry. 1998;37:15408–15413. doi: 10.1021/bi981523a. [DOI] [PubMed] [Google Scholar]
- 15.Jagath J R, Rodnina M V, Wintermeyer W. J Mol Biol. 2000;295:745–753. doi: 10.1006/jmbi.1999.3427. [DOI] [PubMed] [Google Scholar]
- 16.Moser C, Mol O, Goody R S, Sinning I. Proc Natl Acad Sci USA. 1997;94:11339–11344. doi: 10.1073/pnas.94.21.11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powers T, Walter P. Science. 1995;269:1422–1424. doi: 10.1126/science.7660124. [DOI] [PubMed] [Google Scholar]
- 18.Berchtold H, Reshetnikova L, Reiser C O, Schirmer N K, Sprinzl M, Hilgenfeld R. Nature. 1993;365:126–132. doi: 10.1038/365126a0. [DOI] [PubMed] [Google Scholar]
- 19.Jurnak F. Science. 1985;230:32–36. doi: 10.1126/science.3898365. [DOI] [PubMed] [Google Scholar]
- 20.Kjeldgaard M, Nyborg J. J Mol Biol. 1992;223:721–742. doi: 10.1016/0022-2836(92)90986-t. [DOI] [PubMed] [Google Scholar]
- 21.Pai E F, Krengel U, Petsko G A, Goody R S, Kabsch W, Wittinghofer A. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bishop A, Buzko O, Heyeck-Dumas S, Jung I, Kraybill B, Liu Y, Shah K, Ulrich S, Witucki L, Yang F, et al. Annu Rev Biophys Biomol Struct. 2000;29:577–606. doi: 10.1146/annurev.biophys.29.1.577. [DOI] [PubMed] [Google Scholar]
- 23.Hwang Y W, Miller D L. J Biol Chem. 1987;262:13081–13085. [PubMed] [Google Scholar]
- 24.Weijland A, Parlato G, Armeggiani A. Biochemistry. 1994;33:10711–10717. doi: 10.1021/bi00201a019. [DOI] [PubMed] [Google Scholar]
- 25.Zhong X-M, Chen-Hwang M-C, Hwang Y W. J Biol Chem. 1995;270:10002–10007. doi: 10.1074/jbc.270.17.10002. [DOI] [PubMed] [Google Scholar]
- 26.Peluso P, Shan S, Nock S, Herschlag D, Walter P. Biochemistry. 2001;40:15224–15233. doi: 10.1021/bi011639y. [DOI] [PubMed] [Google Scholar]
- 27.Powers T, Walter P. EMBO J. 1997;16:4880–4886. doi: 10.1093/emboj/16.16.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sweet D J, Gerace L. J Cell Biol. 1996;133:971–983. doi: 10.1083/jcb.133.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peluso P, Herschlag D, Nock S, Freymann D M, Johnson A E, Walter P. Science. 2000;288:1640–1643. doi: 10.1126/science.288.5471.1640. [DOI] [PubMed] [Google Scholar]
- 30.Kusters R, Lentzen G, Eppens E, van Geel A, van der Weijden C C, Wintermeyer W, Luirink J. FEBS Lett. 1995;372:253–258. doi: 10.1016/0014-5793(95)00997-n. [DOI] [PubMed] [Google Scholar]
- 31.Rapiejko P, Gilmore R. Cell. 1997;89:703–713. doi: 10.1016/s0092-8674(00)80253-6. [DOI] [PubMed] [Google Scholar]
- 32.Moll R, Schmidtke S, Petersen A, Schafer G. Biochim Biophys Acta. 1997;1335:218–230. doi: 10.1016/s0304-4165(96)00141-9. [DOI] [PubMed] [Google Scholar]
- 33.Shepotinovskaya I V, Freymann D M. Biochim Biophys Acta. 2001;1597:107–114. doi: 10.1016/s0167-4838(02)00287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Qi H-Y, Hyndman J B, Ulbrandt N D, Teplyakov A, Tomasevic N, Bernstein H D. EMBO J. 2001;20:6724–6734. doi: 10.1093/emboj/20.23.6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Padmanabhan W, Freymann D M. Structure (Cambridge, UK) 2001;9:859–863. doi: 10.1016/s0969-2126(01)00641-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freissmuth M, Gilman A G. J Biol Chem. 1989;264:21907–21914. [PubMed] [Google Scholar]
- 37.Brownbridge G G, Lowe P N, Moore K J M, Skinner R H, Webb M R. J Biol Chem. 1993;268:10914–10919. [PubMed] [Google Scholar]
- 38.Tesmer J J, Berman D M, Gilman A G, Sprang S R. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 39.Scheffzek K, Mohammed R A, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 40.Rittinger K, Walker P A, Eccleston J F, Smerdon S J, Gamblin S J. Nature. 1997;389:758–762. doi: 10.1038/39651. [DOI] [PubMed] [Google Scholar]
- 41.Ahmadian M R, Stege P, Scheffzek K, Wittinghofer A. Nat Struct Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 42.Bourne H R. Nature. 1997;389:673–674. doi: 10.1038/39470. [DOI] [PubMed] [Google Scholar]
- 43.Jencks W P. Adv Enzymol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 44.Neuhard J, Nygaard P. In: Escherichia coli and Salmonella typhimurium. Neidhardt K C, Ingraham J L, Low K B, Magasanik B, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1987. pp. 447–473. [Google Scholar]
- 45.Luirink J, ten Hagen-Jongman C M, van der Weijden C C, Oudega B, High S, Dobberstein B, Kusters R. EMBO J. 1994;13:2289–2296. doi: 10.1002/j.1460-2075.1994.tb06511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}