

Abstract
The second and third modules of human decay accelerating factor (DAF) are necessary and sufficient to accelerate decay of the classical pathway (CP) convertase of complement. No structure of a mammalian protein with decay-accelerating activity has been available to date. We therefore determined the solution structure of DAF modules 2 and 3 (DAF∼2,3). Structure-guided analysis of 24 mutants identified likely contact points between DAF and the CP convertase. Three (R96, R69, and a residue in the vicinity of L171) lie on DAF∼2,3's concave face. A fourth, consisting of K127 and nearby R100, is on the opposite face. Regions of module 3 remote from the semiflexible 2–3 interface seem not to be involved in binding to the CP convertase. DAF thus seems to occupy a groove on the CP convertase such that both faces of DAF close to the 2–3 junction (including a positively charged region that encircles the protein at this point) interact simultaneously. Alternative pathway convertase interactions with DAF require additional regions of CCP 3 lying away from the 2–3 interface, consistent with the established additional requirement of module 4 for alternative pathway regulation.
Keywords: CD55, structure, complement control protein module
Human decay-accelerating factor (DAF, CD55), is a 70-kDa glycophosphatidylinositol-anchored glycoprotein that regulates complement activation in a species-restricted manner. Its key role is to function intrinsically on the plasma membranes of self cells to dissociate autologous C3 convertases that assemble on their surfaces. In cooperation with membrane cofactor protein, it prevents amplification of C3b-deposition and propagation of the complement cascade, thereby protecting cells from autologous complement-mediated damage (1, 2).
The physiological and potentially therapeutic significance of DAF is broadly established. Knockout mice deficient in DAF are markedly more sensitive to autoantibody-induced glomerulonephritis (3), experimental autoimmune myasthenia gravis (4), and dextran sodium sulfate-induced inflammatory bowel disease.∥ DAF supplied alone, or together with membrane cofactor protein, to the endothelial cells of a xenograft suppresses hyperacute rejection (5). Incorporation of DAF into the envelope of baculovirus generates complement-resistant gene transfer vehicles (6). DAF is reportedly a functionally critical component of a lipopolysaccharide receptor complex (7). Pathology in paroxysmal nocturnal hemoglobinuria is secondary to the combined deficiency of DAF and CD59 (8).
A second important function of DAF is to serve as a binding partner with the seven-transmembrane domain protein CD97 (9). In yet another capacity, DAF is the target of pathogenic organisms including Dr+ Escherichia coli, coxsackieviruses B1, B3, and B5, echovirus 7, and enteroviruses EV70 and CAV21 (10).
Human DAF has four N-terminal complement control protein modules (CCPs) followed by an O-linked oligosaccharide-rich cushion that is glycophosphatidylinositol-anchored to the plasma membrane (11–13). CCPs contain 60–70 amino acid residues (aa) and occur between 1 and ≈40 times in each of ≈50 human proteins (http://smart.embl-heidelberg.de/). The complement-regulatory activity of DAF resides within its CCPs 2–4 (DAF∼2–4) (14). Modules 2 and 3 of DAF (DAF∼2,3) are necessary and sufficient to accelerate decay of the classical pathway (CP) C3 convertase (14, **). An N-glycosylation site between CCPs 1 and 2 of DAF is not required (15). Module 4 is additionally necessary for decay acceleration of the alternative pathway (AP) convertase (14).
Little structural information exists about DAF, or any other mammalian protein with decay-accelerating activity, on which to base an understanding of function. Crystallization of a DAF fragment, consisting of CCPs 3 and 4 has been reported (16). This is not, however, a biologically active portion of DAF. DAF∼2,3, on the other hand, contains the entire CP regulatory activity of the full-length protein.** A site-directed mutagenesis study, guided by a homology-derived model of DAF∼2–4 (17), probed the functional roles of 24 substitutions in the second and third modules of DAF (18). Thus, DAF∼2,3 (as a compact, well-characterized protein fragment with a single, well defined biological activity) represents an attractive target for structural studies. We report here the solution structure and dynamics of DAF∼2,3 providing a characterized structure of a mammalian protein with decay-accelerating activity.
Methods
Expression, Purification, and Sample Preparation.
Human DAF∼2,3 (amino acids 61–189) was expressed, and it was 13C,15N-labeled, as described (19, 20). For nuclear Overhauser effect (NOE) and relaxation experiments, samples contained 0.9 mM protein in 550 μl of H2O/2H2O (90:10) and 50 mM δ3-sodium acetate (pH 5.0). For measurement of 1H—15N one-bond residual dipolar couplings (RDCs), partial alignment of a 0.3-mM sample of [15N]DAF∼2,3 was induced by addition of cetylpyridinium bromide:hexanol in a ratio of 1:1.33 (wt/wt) to a final concentration of 4% total cosolvent (wt/vol) (21).
NMR Experiments.
Spectra were recorded (37°C) on Varian INOVA-600 and -800 spectrometers. Assignment of 1H, 13C, and 15N nuclei has been described (20). To obtain 1H—1H NOEs, the following were assigned manually: 15N-edited and 13C-edited NOESY spectra (100-ms mixing times) at 600 MHz; an additional 100-ms mixing time 13C-edited NOESY spectrum at 800 MHz; and two experiments designed for CH3-containing residues: NOESY-heteronuclear sequential quantum correlation (HSQC)-CH3NH and HSQC-CHNH-NOESY-HSQC-CH3NH (22). H-bonds were identified from relatively slowly exchanging (>10 min) amides in H2O/D2O-exchange [15N,1H]-HSQC spectra; and proton acceptors inferred with the support of NOEs. JHNHα values came from a 600-MHz HNHA experiment. RDCs were derived from a pair of in-phase, antiphase [15N,1H]-HSQC experiments (23).
Steady-state 1H—15N NOEs (Fig. 1a) and 15N T1 and T2 (Fig. 1 b and c) were measured at 600 MHz. T1 and T2 were estimated from data acquired at seven relaxation times. 1H—15N NOEs were calculated from the ratio of the intensities of the cross-peaks in the reference spectra to those recorded with saturation of the 1H signal.
Fig 1.

Relaxation measurements and distance restraints. (a–c) 15N,1H NOEs, 15N T1s and 15N T2s, respectively, vs. aa number. Filled and open symbols with error bars derive from 800- and 600-MHz spectra, respectively. Black horizontal bars show extent and annotation of β-strands. Larger filled circles correspond to prolines. (d) Number of NOEs (left axis) as a stack plot vs. aa number. Vertical filled bars = (i − I + >4); hashed bars = (i − I + (2–4); gray bars = sequential; and open bars = intraresidue NOEs. Open symbols (right axis) show rmsds, based on Cαs of the structure closest to the mean, vs. aa number calculated for each module separately. Horizontal filled bars summarize 2° structure, with location of Cys and disulfides shown as arrows and dotted lines.
Structure Calculations and Analysis.
Doubled resonances occurred in the [15N,1H]-HSQC spectra for some CCP 2 residues. Resolved pairs of cross-peaks (with a 3:2 ratio of intensities) were assigned for I80-Y84. Partially overlapped cross-peaks occurred for V66 and T68, and for G88-V91. Pairs of cross-peaks were observed for many of the corresponding side-chains. The presence of multiple isoforms potentially complicates conversion of NOE cross-peaks to distance constraints. Cross-peaks were therefore calibrated according to ref. 24: intensities of nondegenerate signals were scaled to compensate for the differences in population of the two putative forms; the intensity of the major form of the doubled peaks was multiplied by the inverse of its corresponding intensity proportion (i.e., 5/3). Cross-peaks due to the minor form were ignored.
A total of 2,440 unique NOEs (Table 1, Fig. 1d) were classified as follows: strong (<2.8 Å), medium (<3.7 Å), weak (<4.8 Å), or very weak (<5.8 Å). Distance restraints representing 14 inferred H-bonds, 54 3JHNHα, and 59 RDCs that restrained parts of the backbone not implied in local motion on the time-scale of the experiment were also used as input, as were four pairs of disulfides defined by homology with other CCPs.
Table 1.
NMR structure determination statistics for DAF∼2,3
| Module 2 | Module 3 | |
|---|---|---|
| No. of NOEs used for structure calculation† | 1162 | 1278 |
| Sequential | 366 | 347 |
| Short-range 2 < |i − j| < 4 | 91 | 56 |
| Long-range |i − j| > 4 | 401 | 507 |
| Linker ↔ module‡ | 15 | 12 |
| Module ↔ module§ | 6 | |
| J couplings (HN—HA) | 31 | 23 |
| RDCs (1H—15N) | 28 | 31 |
| Phi vs. Psi (33)¶ | ||
| Residues, % | ||
| “Most favored” regions | 55.7 | |
| “Additional allowed” regions | 35.8 | |
| “Generously allowed” regions | 7.5 | |
| “Disallowed” regions | 0.9 | |
| RDC tensors | ||
| 2*Da (mean ± SD) | −21.4 ± 4.7 | −17.9 ± 5.3 |
| Rhombicity | 0.90 ± 0.07 | 0.79 ± 0.14 |
| RMS deviations from the experimental restraints | ||
| NOEs, Å | 0.044 ± 0.002 | |
| J restraints, ° | 0.96 ± 0.12 | |
| RDC (M2 and M3) | 0.641 ± 0.113 | 0.552 ± 0.128 |
| rmsd from idealized geometry | ||
| Bond lengths, Å | 0.0030 ± 0.0001 | |
| Bond angles, ° | 0.435 ± 0.016 | |
| Dihedrals, ° | 43.55 ± 0.27 | |
| rmsd from average module structures, Å | ||
| Backbone heavy atoms 65–120, 130–185 | 0.775 | 0.725 |
In total (for 42 structures), there were three NOE violations >0.7 Å.
Linker defined as residues 125–128.
97Hα & Hβ* − 172Hγ2*; 97Hβ* − 172Hδ1*; 98Hα* − 172Hδ1* & Hγ11; 99Hɛ* − 172Hγ2*.
All statistics for 42 “final” structures from 220 calculated.
Structure calculation was performed within “Crystallography and NMR systems” (25) with the PARALLHDG-UCL force field based on PARALLHDG5.1 (26). Stereochemistry was dealt with as described (27). Atom positions were randomized, and then 10 steps of simulated annealing were performed at 2,000 K, followed by cooling to 100 K over 40 steps. The resulting structures were reheated to 2,000 K and cooled in 20 steps to 1,000 K, and then in 20 further steps to 100 K. The RDC restraints were used only in the refinement cooling stage, by using the TENSO energy term within CNS (25). Because of the possibility of intermodular flexibility, a separate alignment tensor (AT) was defined for each module. A harmonic potential was used for the RDC restraints, with a force constant that was geometrically increased during the first cooling stage of refinement, from 0.001 to 0.3 kcal·(mol·Hz2)−1, and fixed at 0.3 kcal⋅(mol⋅Hz2)−1 during the second cooling stage. Statistics are summarized in Table 1.
Results and Discussion
Module Structure and Dynamics.
The structure of each module is generally well defined by the data with an indication of a flexible intermodular orientation (Figs. 1d and 2 a and b). Each module has an elongated structure (Fig. 2c) with five β-strands (B, D, E, G, and H; see ref. 28) aligned approximately with the long module axis, forming small areas of antiparallel β-sheets. Residues corresponding to β-strands A, C, and F in other CCPs (28, 29), mainly form short elongated segments in DAF∼2,3 that are also aligned approximately with the long axis, but do not participate in H-bonded 2° structure. A hydrophobic core includes a consensus Trp and is bounded at either end by the disulfides (Fig. 2c). Few residues within the strands and extended regions show significant mobility.
Fig 2.

The solution structure of DAF∼2,3. (a) Overlay (backbone trace) of 42 lowest energy structures on selected CCP-3 Cαs of structure closest to the mean. (b) As in a but structures overlaid on selected Cαs of CCP-2. (c) molscript trace (showing Cys and Trp) of structure with intermodular angles closest to average; hv, hypervariable loop; loops, turns, and bulges are labeled with aa numbers; strands are annotated as in Fig. 1.
Loops, bulges, and turns constitute ≈50% of the structure (Fig. 2c). Many of these regions exhibit mobility, probably contributing to their generally higher rms deviations (rmsd) (Fig. 1). In module 2, amino acids 75–82 form the “hypervariable” (hv) loop, a region of sequence variation amongst CCPs and a site for insertions and deletions. This region projects prominently from module 2 and is a probable site of structural divergence between the conformers discussed below. The hv loop seems relatively well structured within itself but is mobile with respect to the remainder of the module on a range of time-scales. There is also evidence for 103 to 106 s−1 backbone motion of amino acids 144,145 in module 3's relatively short hv loop. Residues 173,174, which lie at the beginning of an inserted sequence that forms a pronounced loop (amino acids 173–176) of module 3 close to the intermodular interface (Fig. 2c), show evidence of rapid motion (lower heteronuclear NOEs). This region packs down against the body of module 3 and does not contact module 2.
The DAF∼2 module adopts two or more conformations that do not readily interconvert, as indicated by (i) doubled HSQC cross-peaks for certain residues, (ii) differences in 1H—15N RDCs between major and minor conformers, and (iii) consistency (based on data for the major form) of 3JHNHA values with φ torsion angles obtained from structure calculations. The structure of the major form was calculated whereas insufficient data were available for the minor form(s). The main structural differences likely occur amongst residues with nondegenerate shifts, e.g., amino acids 66–68 and amino acids 80–84, neither of which region is well defined by the data (Fig. 1d). Judging from the sequence position of such residues, conformational heterogeneity likely corresponds to cis-trans isomerization of x-Pro bonds. Inspection of the major forms of P78 and P67 confirmed their respective cis and trans configurations; but lack or overlap of peaks prevented analysis of corresponding bonds in the minor form. Overlap with water prevented determination of the configuration of P86, whereas the remaining six x-Pro bonds are trans. Putative proline isomerization was observed in the second CCP of the γ-aminobutyric acid type B receptor type 1a (S. Blein & P.N.B., unpublished work) but not, to date, in other complement regulatory proteins. That this effect arises in DAF∼2,3 due to the lack of module(s) 1 (and/or 4) cannot be excluded.
Intermodular Orientation.
There are few direct interactions between modules. In nearly all calculated structures, the alkyl chain of linker residue K126 in strand H of module 2 lies between the side-chains of Y99 (in strand E) and F148 of module 3, although it does not make van der Waals' contact with the latter. The side-chain of I172 (in module 3, and in contact with F148) makes van der Waals' contact with the CH2 of G98 in module 2. The F148 side-chain thus contributes to a loose hydrophobic cluster that includes parts of P97, G98, S173, K126, and I172. An aa with an aromatic or large alkyl side-chain is found at the equivalent position (to F148) of nearly all CCPs and participates in a diffuse hydrophobic grouping in other junctions of known structure (28, 29).
The orientation of one module with respect to the other is not defined well by the data (Fig. 2). Fig. 3 summarizes the angles that characterize intermodular orientations among the ensemble. Structures are relatively elongated overall, with an end-to-end arrangement of modules that share only a small interface. In the case of the tilt angle, there seem to be two populations (Fig. 3a), but there is no correlation with experimental energies.
Fig 3.

Intermodular angles in DAF∼2,3. (a) Distribution of tilt (filled), twist (gray), and skew (open) angles for the 42 lowest energy structures [definition of angles (34) summarized in Inset]. (b) Angles plotted by using polar coordinates; each wedge summarizes mean ± SD for the angle indicated.
The lack of convergence among calculated structures occurs despite the use of RDCs (that effectively provide long-range structural restraints) and is a consequence of the paucity of NOEs (Table 1) between the bodies of the modules. To investigate whether this range of intermodular orientations reflects genuine flexibility, the RDCs were reexamined. Only if modules are moving effectively independently, with a flexible attachment between them, can each of the two modules align in the liquid crystalline medium differently, as reflected in their ATs. The ATs derived from RDCs measured within modules 2 and 3 (Table 1) lie within one another's error bounds. Whereas this observation is not informative with regard to intermodular flexibility, structure calculations carried out with a common AT for both modules resulted in a 50% increase of the energy associated with violations of the experimental couplings. The improvement of the fit when using two ATs as opposed to one was statistically significant. Thus, the RDCs measured for well-structured residues in module 2 and module 3 are not consistent with a rigid connection between the two modules. The limited range of variations obtained from structure calculations therefore reflects genuine flexibility. Residues within the intermodular linking sequence do not have unusual 15N relaxation properties such as might be expected if this region were mobile on the fast (109 to 1012 s−1) or intermediate (103 to 106 s−1) time-scales. As pointed out previously (29, 30), however, segmental motion on the 10−8 s time-scale is not necessarily manifested in 15N relaxation measurements.
The structure with lowest overall energy has tilt, twist, and skew angles of 39°, 125°, and 243°, respectively, whereas the structure with the second lowest overall energy exhibits equivalent angles of 35°, 131°, and 214°. The structure whose intermodular orientation was overall closest to the mean has tilt, twist, and skew angles of 44°, 125°, and 238°. Subsequent discussions apply equally well to all three of these structures, but Fig. 2c and Fig. 4 show the structure with angles closest to the mean values.
Fig 4.

Electrostatic surface representation of DAF∼2,3 and outcomes of mutagenesis. (a) Electrostatic surface representations: the Left view is the same as that used in Fig. 2 (but rotated −90° about an axis perpendicular to the page). Red is negative and blue is positive as indicated by the upper left bar (range 1.4–128 kT). (b) Surface views as in a to illustrate outcome of mutating the labeled individual residues (20). Percentage of wild-type AP regulatory activity remaining after substitution is color coded according to the color bar (lower right). (c) As in b, but this frame summarizes the effects of the same mutations on CP regulatory activity. All mutations were to Ala except N71K and S72F.
The intermodular angles conspire to create a concave surface (approximately the “front” face in Fig. 2c) composed of, in module 2, strand B and amino acids 63–70; and in module 3, the BDF face. In this representation, the two hv loops project to either side and forward of the concave face, and the intermodular linker lies closer to the “back” convex surface. A band of positive charge encircles the protein near the 2–3 interface (Fig. 4a). The intermodular angles observed amongst the ensemble of DAF∼2,3 structures do not resemble other intermodular angles determined in other structures of CCP pairs (28, 29). Therefore, whereas individual modules within homology-based models of DAF (17, 18) overlay reasonably well (rmsd = 2.3–2.7 Å; Cα) with their experimentally derived equivalents, intermodular angles differ significantly. Flexibility at the DAF∼2,3 junctions resembles that of the complement receptor type 1s (CR1s) CCP 16–17 (29) junction and the CCP 2–3 junction of vaccinia virus complement control protein (VCP) (28), but it differs from the better defined intermodular orientations of CR1s CCP 15–16 (29) and VCPs 3–4 module pairs (31).
Mutagenesis Data in Light of the Structure.
Fig. 4 b and c each shows two views of a surface representation that is color-coded according to the extent to which activity vs. the AP (Fig. 4b) or the CP (Fig. 4c) is lost (or gained) when a specific aa is mutated (20).
Of 24 such mutations in DAF modules 2 and 3, only 9 resulted in >50% loss of decay-accelerating activity directed toward the CP C3 convertase (Fig. 4c), and five yielded losses of >80%. The most striking results (<10% activity remaining) followed mutation of R69 and R96 in module 2, and F148 in module 3. Mutation of R100, in module 2, K127 in the linker and L171 in module 3 also severely curtailed CP decay-accelerating activity.
R96 is the last residue of strand D of module 2 (Fig. 2); its largely exposed side-chain lies on the concave face of the molecule close to (but not participating in) the intermodular junction. The side-chain of R69 is also exposed and lies ≈13 Å away on the same face of the molecule, toward its N terminus (Fig. 4). The side-chains of R100 and K127 lie close together and are exposed on the opposite face of the protein to the one that is shared by R69 and R96. None of these four positively charged aa plays any obvious structural role, and they likely represent points of direct contact during decay acceleration of the CP convertase.
The F148 side-chain is located on a loop between strands B and D of module 3 and is proximal to (but does not contact) R96. F148 is an interface residue (see above), and mutation F148A could weaken module–module interactions and increase flexibility. Thus, F148 may not represent a site of direct contact between DAF and the convertase.
One CH3 of L171 is exposed, the other makes van der Waals' contact with S180 whereas the γ proton contacts the β protons of F169. Mutation L171A would disrupt these contacts and cause a local structural perturbation. Thus, whereas L171 cannot be assigned unambiguously as a direct point of contact, there must be contact points on module 3 in this vicinity. The mutation F169A has only limited impact on CP activity, ruling out its exposed aromatic ring, which lies close to L171, as a contact point. It is interesting that the highly conserved F154 and Y160 are buried and play structural roles in the portion of module 3 away from the interface with module 2, but may be mutated with little effect on activity. In a similar vein, mutations of K161, F163, and S155, all located away from the 2–3 interface, have no detrimental effect. Finally, module 2's semiconserved L70 and F123 are substantially buried, but substitutions L70A and F123A have no or little effect on activity.
In summary, four probable contact points between DAF and the CP convertase have been identified. Three of these (R96, R69, and a residue in the neighborhood of L171) may be considered to lie on the “front,” concave face of the representative structure of DAF as shown in Fig. 4c. The fourth, consisting of K127 and nearby R100, lies on the opposite face (Fig. 4c); this arrangement is true for all calculated structures, not just the representative one. Regions of module 3 away from the intermodular interface seem not to be involved in binding. The lack of effect of the structurally perturbing L70A and F123A mutations implies that there is not a requirement for precise structural features in much of module 2 either; subsequent structure-guided mutations seem to confirm this finding (unpublished work). The emerging picture therefore is consistent with DAF lying within a groove on the CP convertase such that both faces of DAF close to the junction [including a large positive area encircling the protein at this point (Fig. 4a)] are involved in simultaneous interactions.
Fig. 4b shows that contact points identified in control of the CP also seem important in AP regulation. They are supplemented by F169, whose exposed aromatic ring is adjacent to the side-chain of L171, and possibly by K126, the NH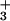 of which is proximal to R96 on the front face. Mutation of K126 (leaving 9 ± 5% activity) could, however, alter intermodular flexibility so it may not be a direct point of contact. Residues L147 and K125 (both of which are substantially exposed) could also contribute to binding. These residues lie between the R69/R96/F169/K126/L171 face and the K127/R100 face and, as in the CP convertase interaction, are consistent with this part of DAF∼2,3 fitting within a groove. For AP regulation, the structural residues F154, Y160, and L70 seem more important than they do in the case of the CP, consistent with a binding site that requires intact modules 2 and 3. The extension of a binding surface further toward the C terminus of module 3 (Fig. 4b) is consistent with the result that module 4 is additionally required for AP but not for CP regulation.
of which is proximal to R96 on the front face. Mutation of K126 (leaving 9 ± 5% activity) could, however, alter intermodular flexibility so it may not be a direct point of contact. Residues L147 and K125 (both of which are substantially exposed) could also contribute to binding. These residues lie between the R69/R96/F169/K126/L171 face and the K127/R100 face and, as in the CP convertase interaction, are consistent with this part of DAF∼2,3 fitting within a groove. For AP regulation, the structural residues F154, Y160, and L70 seem more important than they do in the case of the CP, consistent with a binding site that requires intact modules 2 and 3. The extension of a binding surface further toward the C terminus of module 3 (Fig. 4b) is consistent with the result that module 4 is additionally required for AP but not for CP regulation.
That a positively charged region close to the 2–3 interface likely contacts the convertases of the complement cascade in the case of DAF may be compared with the situation in C4b binding protein, where R39, R63, R64, and R66 are important for binding of C4b and for regulation (cofactor activity) of the CP C3 convertase (32). According to a model, these residues also form a positively charged region, close to the interface between modules 1 and 2. Positively charged residues within complement receptor type 1's cofactor site 2 have been implicated strongly in contacting C3b (29), but these do not align, or spatially coincide, with the DAF residues identified here. That the functionally critical region of DAF∼2,3 is flexible could underlie its ability to interact with different binding partners (e.g., the convertases of the CP and AP), or might be a requirement for DAF's ability to bring about decay acceleration of the convertases.
Note
After submission of this manuscript, a paper describing the structure of DAF CCPs 3 and 4 was published by Williams et al. (35). These authors surmised that both surfaces of CCP 3 are involved in interaction with the convertases.
Acknowledgments
We thank Dinesh Soares for help with Fig. 4. This work was supported in part by National Institutes of Health Grant AI 23598 (to M.E.M.) and the Wellcome Trust (Edinburgh Protein Interaction Centre).
Abbreviations
aa, amino acid residue
AT, alignment tensor
AP, alternative pathway
CCP, complement control protein module
CP, classical pathway
DAF, decay-accelerating factor
hv, hypervariable
rmsd, rms deviation
NOE, nuclear Overhauser effect
RDC, residual dipolar coupling
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code ).
Lin, F., Spencer, D., Levine, A. D. & Medof, M. E. (2002) Int. Immunopharmacol. 2, 1250 (abstr.).
Kuttner-Kondo, L., Dybvig, M., Mitchell, L., Atkinson, J. P., Medof, M. E. & Hourcade, D. (2002) Int. Immunopharmacol. 2,1275 (abstr.).
References
- 1.Medof M. E., Kinoshita, T. & Nussenzweig, V. (1984) J. Exp. Med. 160, 1558-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brodbeck W. G., Mold, C., Atkinson, J. P. & Medof, M. E. (2000) J. Immunol. 165, 3999-4006. [DOI] [PubMed] [Google Scholar]
- 3.Lin F., Emancipator, S. N., Salant, D. J. & Medof, M. E. (2002) Lab. Invest. 82, 563-569. [DOI] [PubMed] [Google Scholar]
- 4.Lin F., Kaminski, H. J., Conti-Fine, B., Wang, W., Richmonds, C. & Medof, M. E. (2002) J. Clin. Invest. 110, 1269-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auchincloss H. & Sachs, D. H. (1998) Rev. Immunol. 16, 433-470. [DOI] [PubMed] [Google Scholar]
- 6.Huser A., Rudolph, M. & Hofmann, C. (2001) Nat. Biotechnol. 19, 451-455. [DOI] [PubMed] [Google Scholar]
- 7.Heine H., Ulmer, A. J., El-Samalouti, V. T., Lentschat, A. & Hamann, L. (2001) J. Endotoxin Res. 7, 227-231. [PubMed] [Google Scholar]
- 8.Yomtovian R., Prince, G. M. & Medof, M. E. (1993) Transfusion 33, 852-873. [DOI] [PubMed] [Google Scholar]
- 9.Hamann J., Wishaupt, J. O., van Lier, R. A., Smeets, T. J., Breedveld, F. C. & Tak, P. P. (1999) Arthritis Rheum. 42, 650-658. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl G., Sjobring, U. & Johnsson, E. (2000) Curr. Opin. Immunol. 12, 44-51. [DOI] [PubMed] [Google Scholar]
- 11.Medof M. E., Kinoshita, T., Silber, R. & Nussenzweig, V. (1985) Proc. Natl. Acad. Sci. USA 82, 2980-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caras I. W., Davitz, M. A., Rhee, L., Weddell, G., Martin, D. W., Jr. & Nussenzweig, V. (1987) Nature 325, 545-549. [DOI] [PubMed] [Google Scholar]
- 13.Medof M. E., Walter, E. I., Roberts, W. L., Haas, R. & Rosenberry, T. L. (1986) Biochemistry 25, 6740-6747. [DOI] [PubMed] [Google Scholar]
- 14.Brodbeck W. G., Liu, D., Sperry, J., Mold, C. & Medof, M. E. (1996) J. Immunol. 156, 2528-2533. [PubMed] [Google Scholar]
- 15.Brodbeck W. G., Kuttner-Kondo, L., Mold, C. M. & Medof, M. E. (2000) Immunology 101, 104-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lea S., Powell, R. & Evans, D. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 1198-1200. [DOI] [PubMed] [Google Scholar]
- 17.Kuttner-Kondo L., Medof, M. E., Brodbeck, W. & Shoham, M. (1996) Protein Eng. 9, 1143-1149. [DOI] [PubMed] [Google Scholar]
- 18.Kuttner-Kondo L. A., Mitchell, L., Hourcade, D. E. & Medof, M. E. (2001) J. Immunol. 167, 2164-2171. [DOI] [PubMed] [Google Scholar]
- 19.Lin F., Immormino, R. M., Shoham, M. & Medof, M. E. (2001) Arch. Biochem. Biophys. 393, 67-72. [DOI] [PubMed] [Google Scholar]
- 20.Uhrinova S., Lin, F., Uhrin, D., Medof, M. E. & Barlow, P. N. (2002) J. Biomol. NMR 23, 167-168. [PubMed] [Google Scholar]
- 21.Barrientos L. G., Dolan, C. & Gronenborn, A. M. (2000) J. Biomol. NMR 16, 329-332. [DOI] [PubMed] [Google Scholar]
- 22.Uhrin D., Bramham, J., Winder, S. J. & Barlow, P. N. (2000) J. Biomol. NMR 18, 253-259. [DOI] [PubMed] [Google Scholar]
- 23.Ottiger M., Delaglio, F. & Bax, A. (1998) J. Magn. Reson. 131, 373-378. [DOI] [PubMed] [Google Scholar]
- 24.Yuan X., Werner, J. M., Knott, V., Handford, P. A., Campbell, I. D. & Downing, K. (1998) Protein Sci. 7, 2127-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brünger A. T., Adams, P. D., Clore, G. M., Delano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Read, R. J., et al. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 26.Linge J. P. & Nilges, M. (1999) J. Biomol. NMR 13, 51-59. [DOI] [PubMed] [Google Scholar]
- 27.Bramham J., Hodgkinson, J. L., Smith, B. O., Uhrin, D., Barlow, P. N. & Winder, S. (2002) Structure 10, 249-258. [DOI] [PubMed] [Google Scholar]
- 28.Henderson C., Bromek, K., Smith, B. O., Uhrin, D. & Barlow, P. N. (2001) J. Mol. Biol. 307, 323-339. [DOI] [PubMed] [Google Scholar]
- 29.Smith B. O., Mallin, R. L., Krych-Goldberg, M., Wang, X., Hauhart, R. E., Bromek, K., Uhrin, D., Atkinson, J. P. & Barlow, P. N. (2002) Cell 108, 769-780. [DOI] [PubMed] [Google Scholar]
- 30.Copie V., Tomita, Y., Akiyama, S. K., Aota, S., Yamada, K. M., Venable, R. M., Pastor, R. W., Krueger, S. & Torchia, D. A. (1998) J. Mol. Biol. 277, 663-682. [DOI] [PubMed] [Google Scholar]
- 31.Murthy K., Smith, S. A., Ganesh, V. K., Judge, K. W., Mullin, N., Barlow, P. N., Ogata, C. M. & Kotwal, G. (2001) Cell 104, 301-311. [DOI] [PubMed] [Google Scholar]
- 32.Blom A. M., Kask, L. & Dahlback, B. (2001) J. Biol. Chem. 276, 27136-27144. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski R., MacArthur, M., Moss, D. & Thornton, J. (1993) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 34.Barlow P. N., Steinkasserer, A., Norman, D. G., Kieffer, B., Wiles, A. P., Sim, R. B. & Campbell, I. D. (1993) J. Mol. Biol. 232, 268-284. [DOI] [PubMed] [Google Scholar]
- 35.Williams P., Chaudhry, Y., Goodfellow, I. G., Billington, J., Brad Spiller, O., Evans, D. J. & Lea, S. (2003) J. Biol. Chem. 278, 10691-10696. [DOI] [PubMed] [Google Scholar]