

Abstract
The distinct electronic states of assemblies of metallic quantum dots are discussed in a simple approximation where each dot is mimicked as an “atom” that carries one valence electron. Because of their large size, the charging energy of the dots, I = energy required to add another electron, is much smaller than for ordinary atoms. The Coulomb blocking of charge migration is therefore easier to overcome. For the theory, however, this is a challenge, because ionic states, which are typically higher in energy, come down, so the density of electronic states is high, and special methods need to be adapted. Quantum dots are prepared by wet chemical methods and accordingly are not quite identical. They will have a size distribution that can be narrow (when the dots can be assembled into an ordered array) or broad. Other sources of disorder are packing imperfections, which are characteristic of a wider size distribution, ligand deformations, and chemical unevenness. Two experimental control parameters are the size of the dots and the spacing between them. We discuss the combined effects of the low charging energy and disorder and examine the distinct electronic phases that can be realized.
Quantum dots are clusters of atoms (or molecules) that are small enough that their electronic states are discrete (1–4); they can be prepared with a variety of compositions and covering ligands. The study of the properties of individual dots is an active subject in its own right. Here we examine the electronic structure of assemblies of metallic dots, where the dots are packed close enough that they are interacting (see ref. 5 for a general overview of building assemblies with quantum dots). For the purpose of our discussion, the dots are regarded as “atoms.” The key point is that they are “designer” atoms, because their electronic properties can be controlled through the synthetic method that is used to prepare the dots. Of direct concern to us are the size of the dot and the nature of the ligands used to passivate the dots so that they do not coalesce. The energy required to remove or add an electron to the dot is determined by its size. The ligands control how closely the dots can be packed and hence the strength of the coupling, because of electron transfer, between adjacent dots. An important parameter is the energy cost, I, of adding an electron to a dot. The large size of the dots means that the Coulomb repulsion of the added electron is low. Unlike most ordinary atoms, dots have a high capacity for accommodating an additional electron. Another experimental control parameter is the ability to compress an assembly of dots (4, 6) and thereby change the distance between them.
Quantum dots are prepared by wet chemical methods and so are not quite identical. In particular, the dots will have a variable size. The size distribution can be made narrow (e.g., 10%) but can also be broad. Designer atoms are therefore different than ordinary atoms; the latter are identical and in particular have the same ionization potential. A lattice of quantum dots is inherently disordered in that, because the dots are not identical and have a variable ionization potential, the sites of the lattice are not equivalent. This can be the case even when a scanning tunneling microscopy (STM) probe of such a lattice shows an ordered array (6). When the size distribution of the dots is broad, the STM scan of the lattice shows it to be geometrically distorted (7). Other sources of disorder that can be introduced are chemical unevenness (8), in that the dots do not have an identical chemical composition or in that the ligands are not the same. The essential point is that there is always minimal disorder because of distribution in size, and that additional disorder can be built in.
We discuss the electronic structures possible for an array of atoms with the essential properties that we indicated: the sites of the array are not quite equivalent, and the charging energy of a site is atypically low. In other words, we examine the electronic isomerism made possible by the special properties of the dots.
A simple approximation is to regard each dot as having one valence orbital. In the isolated metallic dot, the least-bound electron occupies this orbital. Because this electron is largely confined to the volume of the dot, its energy is expected to scale roughly with the size of the dot, as for a particle in square-well potential. When another dot is nearby, the valence electron can tunnel from a given dot to its near neighbor and thereby couple the dots together. All the other electrons of the dot are placed in its core, and the core is frozen. This approximation is equivalent to the π electron approximation of molecular orbital theory, which has been successfully used to discuss extended structures (9). It is also equivalent to the “tight-binding” approximation of solid-state theory (10). When the dots are fairly compressed and therefore are strongly interacting, this picture offers a starting point, because the coupling of the dots is strong enough to overcome the charging energy. We have used this approximation before (11) to demonstrate that, on compression, an assembly of designer atoms can undergo a transition from a localized to a delocalized electronic state. We have further suggested that this transition can be seen in a spectroscopic experiment (4, 5, 11). The theoretical variable that controls this transition is the strength of the coupling between the dots vs. the inherent disorder in the energy of the valence orbital (which is caused by the fluctuation in their size). The corresponding experimental variable is the separation between the dots, which determines the strength of the coupling. In this paper, we add one more physical variable, the charging energy, which must be taken into account once the dots are further apart and therefore are only weakly coupled. By preparing dots of different sizes and of different metallic elements, the measured charging energy (12) can be varied by over an order of magnitude, e.g., from 0.5 eV to 0.025 eV (1 eV = 1.602 × 10−19 J).
One can identify the two coupling regimes that are discussed above with the names of Anderson (13, 14) and Mott (10), respectively. However, these labels refer to limiting behaviors where only one factor is important. The fascination of designer arrays is that typically they are in an intermediate regime and, most importantly, that the strength of different couplings can be tuned. Under the best of circumstances, this can be done continuously, as when compressing the array (4, 5). Otherwise it can be done in discrete steps, e.g., by changing the ligands or by changing the extent of packing disorder.
Theory
Three energies are used to characterize electronic states of the array of quantum dots, and all three are subject to fluctuations. (i) The energy α of the valence orbital of an isolated dot. α depends on the size of the dot and hence will vary from site to site of the array. The mean value of α can be taken as the zero of energy, so that what really matters is the size, Δα, of the fluctuations. (ii) The repulsion, I, of two valence electrons (of opposite spins) when they are on the same site. The magnitude of this repulsion depends on the radius R of the dots, and values all the way down to a few millivolts have been measured. Taking a full account of this term makes a demand on the computational machinery, because this is a term that depends on where two electrons are rather than on the mean field that a valence electron sees because of all other valence electrons. In principle, there is also an electrostatic repulsion between electrons that are on different sites. The same formalism that allows for the charging capacity of a dot can incorporate this term, too, but it is smaller and so will not be explicitly discussed. (iii) The strength β of the interdot coupling caused by the overlap of the wave function of adjacent sites. β increases exponentially as the interdot distance D decreases, and so it is experimentally continuously tunable when the lattice can be compressed or by discrete amounts when the size of the ligands is changed.
The experimental reality that the mean values of the three energetic parameters, α, β, and I, can be changed by design and, furthermore, that the actual values are subject to size-related fluctuations, Fig. 1, makes the simple model system rather rich in the nature of the electronic states that can be probed. In different types of experiments, these can be the ground and low-lying excited states or higher excited states. Furthermore, by using an STM setup, one can also add electrons to or withdraw them from the system. We will consider experiments for both a fixed and a variable number of electrons.
Figure 1.

The three energies that determine the electronic structure and response of an array of nanodots, plotted vs. D/2R. D is the spacing of the dots in the array and the variable of the abscissa, and 2R is the diameter of the dots. Fluctuations in size and packing disorder, Δ(D/2R)/(D/2R) = ΔD/D + ΔR/R, make the values of the energies fluctuate. The coupling, β, of adjacent dots saturates when the dots nearly touch and then decreases exponentially with D/2R. The midpoint, D0, and the range of β have been previously determined (11) from the measured nonlinear optical response (5) of an array. The charging energy I is relatively small (hundreds to tens of meV), so it does not fluctuate by much. (I scales as 1/R, so ΔI = (ΔR/R)I). The fluctuations, Δα, in the energy of the dots can cover a wider range. The result is that for a wider size distribution, it can be the case that I ≤ Δα.
We allow random fluctuations, within a specified range, in both the radius, R, of the dots and their packing distance D. The fractional range, e.g., ΔR/R, will be specified. The fluctuations in α, β, and I are given in terms of their functional dependence on R and D. α is expected to scale as R−2. I is inversely proportional to the capacity of the dot, which scales with R. The coupling energy β decreases, essentially exponentially, with D/2R (see Eq. 2 below).
The fluctuations in the energies, as shown in Fig. 1, mean that defining a coupling regime is not that obvious. For example, as discussed, a simple (Anderson-like) regime is where the dot–dot coupling β is the dominant effect. But Fig. 1 suggests that even a moderate (15%) size and packing disorder is enough to allow β to reach large values on a lattice which, on the average, is not closely packed.
For a compressed lattice, when the dot–dot coupling dominates, a Hückel-type Hamiltonian
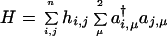 |
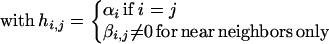 |
1 |
is a sufficient approximation. The Hamiltonian is written in terms of creation and annihilation operators for electrons of a given spin (μ) on any one of the possible n sites. The site energies αi are allowed to fluctuate by up to ±Δα about a mean value α0. In a computation, we draw values of the site energies within the range specified by the size distribution of the dots. The transfer integral β determines how facile it is to remove an electron from site i and move it to a neighboring site j. This coupling is governed by the extent of overlap of the orbital wave function of the valence electron on two adjacent dots. We take it to saturate at the value β0 when the dots contact and to exponentially decrease when the lattice is expanded, as shown in Fig. 1.
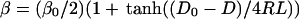 |
2 |
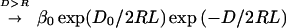 |
D0 is the separation between the dots when their wave functions become significantly overlapping. L determines how fast the dot–dot coupling decreases with D/2R. The magnitude of L, 1/5.5, which we obtain by a fit to the optical nonlinear response experiment (8), is comparable to what we compute in terms of how much the wave function of a dot leaks out (11). In other words, the available evidence for metallic dots is that the coupling is through space. It will be interesting to examine whether one can choose such ligands that participate in the dot–dot coupling, aiding or reducing the facility for electron transfer. Unlike the dot energies, the transfer integral β can fluctuate in its value because of both size and packing disorder. As seen in Fig. 1, the fractional change, Δβ/β, can therefore be higher than Δα/α.
There are two effects that can be demonstrated easily in the Hückel limit. One is the breaking of the symmetry of a lattice because of the fluctuations. In a perfect array of identical atoms, all the lattice sites are equivalent. It is then the case that the energies of the molecular orbitals have a mirror symmetry about the mean value, αo. In the ground state, for each occupied molecular orbital with an energy below α0 there will be an orbital with the same energy above α0. In the language of the Hückel approximation, one can say that the lattice has “alternate symmetry” (15). The fluctuation breaks this symmetry of the excitation energies and also of the molecular orbitals themselves. We have previously shown (11) how the breaking of the symmetry of the molecular orbitals makes otherwise forbidden optical transitions allowed. Here we discuss the breaking of this mirror symmetry, as seen in an experiment with a variable number of electrons. The results shown in Fig. 2 are obtained by computing, separately, the energies of the states of n − 1, n, and n + 1 electrons in an n-site hexagonal array. A familiar approximation (sometimes known as Koopman's theorem and often used in photoelectron spectroscopy) is that the ionization energy of an electron is the energy of the occupied molecular orbital from whence it came. Similarly, the energy cost of attaching an extra electron is the energy of the unoccupied molecular orbital it is to go into. If there is a mirror symmetry, the density of states available to accept an electron should be equal to the density of states available to donate an electron. As seen in Fig. 2, this is the case when the dot–dot coupling is strong enough to overcome the fluctuations in the site energies, but this is not the case at a higher value of D/2R .
Figure 2.

The density of states for externally adding (positive energy) or removing an electron from the array, computed for a hexagonal lattice of seven sites at two different compressions, as shown. There is a 5% fluctuation in the size and another 5% fluctuation in packing. Coulombic effects are included at the Parr–Pariser–Pople level (I = 0.3 eV). This density of states is determined from the tunneling current in an STM experiment (7, 12), and the energy scale is shown in eV to conform to the experimental plots. For D/2R = 1.2, the dots are strongly coupled (cf. Fig. 1), the wave function is delocalized, the lattice is “conducting,” and the density of states (DOS) is symmetric with respect to electrons and holes. At a somewhat wider separation, shown is D/2R = 1.4, the wave function is localized (cf. Fig. 3), and the density of states is asymmetric. The symmetry of the DOS for a conducting lattice and the appearance of an asymmetric DOS for an expanded lattice are as seen in the experiments (7, 12). For even higher values of D/2R, the Hückel level computation is not reliable because the charging energy I becomes comparable with the dot–dot coupling. When D/2R is larger and the Coulomb blocking plays a more important role, the asymmetry is higher. This is understandable because the finite charging energy means that adding an electron is energetically not the mirror image of removing one, and it takes less energy to remove an electron.
The other effect of disorder that can be demonstrated already at the Hückel level is the transition from localized to delocalized molecular orbitals. In a perfect array of identical atoms, all the orbitals will be delocalized. This is why free electron models (15) are so useful. A simple proof is to write the Hückel Hamiltonian of the perfect array in matrix form, HHückel = α0 I + βM. Here I is the identity matrix, and M is the adjacency matrix; Mi,j = −1 if i and j are near neighbor sites, and it is zero otherwise. Because I commutes with M, the molecular orbitals are the eigenvectors of M. They are determined by the geometry and are delocalized. Once the site energies are allowed to fluctuate, the Hamiltonian is no longer of that simple form. However, as long as Δα is small compared with β, the role of the fluctuations is only to break the symmetry, but it remains the case that β can effectively couple any two neighboring sites. The molecular orbitals will then remain delocalized. When the dots have a wider size distribution, and/or when the lattice is expanded so that β is smaller, β can no longer bridge the gap in the site energies, and the molecular orbitals are localized. This is an example of the Anderson-like transition. Fig. 3 shows a computational example, where it is evident that the distinction between a localized state, where the wave function drops exponentially with distance away from its center, and a delocalized state, is quite sharp. We emphasize, however, that the scenario becomes richer when the role of charging energy is allowed for.
Figure 3.

The weight (= charge density) of the highest occupied molecular orbital (HOMO) and lowest unoccupied one (LUMO) on the different sites of a 91-dot array (computed for a closely packed and somewhat expanded lattice). The weight is plotted on a logarithmic scale so as to show that when the dot–dot coupling is no longer sufficient to overcome the effects of disorder, the localized molecular orbitals decrease exponentially with distance from their center.
The simplest Hamiltonian that includes the Coulomb blocking was derived by Hubbard (16).
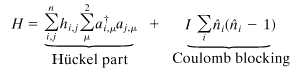 |
3 |
In the second term, we introduced the number (operator) n̂i ≡ Σμai,μ†ai,μ of electrons on the site i. One can add a third term, γ Σi,jn̂in̂j, to represent the polarization of a dot by an electron on a neighboring site. One expects γ to be smaller than I by at least D/2R, and it plays a more limited role. The Hamiltonian is then of the form known in quantum chemistry as formulated by Pariser, Parr, and Pople (17). To diagonalize exactly a Hamiltonian containing explicitly two electron operators we use the Unitary group approach (18, 19), as thoroughly discussed by Paldus (20). The one practical problem is the size of the basis set. For the simplest hexagonal array, which has seven sites (see Eq. 4 below) with one electron per site, there are 784 doublet states. The next completed hexagonal array has 19 sites, and for 19 electrons there are over two billion doublet states. One cannot use the hexagonal symmetry to reduce this number, because the whole point is that we do not have a hexagonal symmetry except on the average. We are working on possible schemes to simplify the handling of larger arrays.
The physics of the Coulomb blocking term in the Hamiltonian (3) is that there is an energy cost, I, associated with placing two electrons (of opposite spins) on the same site. To see the implications, consider first the limit where the lattice is very expanded so that the coupling, β, of adjacent dots can be neglected. Then the Hamiltonian of the noninteracting dots has the form Hsite = Σi=1n αin̂i,i + I Σi=1nn̂i,i (n̂i,i − 1) and this Hamiltonian can be diagonalized analytically. For the simplest hexagonal array (seven sites), the states of the noninteracting dots fall into four bands. For each band, one can count the states using a unitary group formalism (20), and the numbers are given. Shown is a typical state of each band where a bar denotes an electron
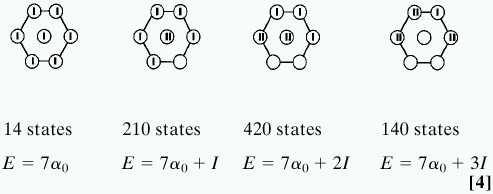 |
4 |
Below each state is the energy in the absence of fluctuations (and, of course, we neglected the dot–dot coupling β). The physics of the system is governed by the question of whether the bands overlap when we allow fluctuations. The first 14 states will remain in place with a sharp energy of seven α0, because variations of the individual site energies must average out to zero. The next 210 states will fan out into a band, primarily because of the variation in the site energies. The width of the band is therefore Δα, and, depending on whether the charging energy I is larger than Δα, the band will or will not overlap the first band. The question of band overlap is so critical because it determines whether there is energy penalty to charge migration. If the first 14 states are quasidegenerate with the next 210 states, then even a weak dot–dot coupling can induce charge reorganization, and there is no barrier to conduction. Otherwise, it is not until the array is compressed to the point where the dot–dot coupling β is comparable to the charging energy I that the array will become conducting.
The regime where the dot–dot coupling is not large is therefore split by disorder into two classes. One is that characteristic of no or of limited disorder in the site energies. Then the ground and low excited states are very localized, in that each electron is assigned to its own dot, and no dot has more than one electron. β cannot couple states of this band, because its action is to shift an electron from one dot to a neighboring one, and there are no states in this ground band where a dot is empty. For noninteracting dots, it takes a finite energy, I, to promote an electron to a higher-energy band of ionic states. There is, however, an energy shift of the covalent states, which by second-order perturbation theory is of the order of β2/I. This is because β couples a state of the ground band to a state of the next higher band, at an energy I higher, and then this state is coupled by β back to the ground band. Once β increases to the point where β2/I is comparable to I, the gap between the two bands will be closed. This is the Mott-type insulator (10) to metal transition on increasing the site–site coupling.
The other possible scenario is when there is more spread in the sizes of the dots and so more variations in their energies. Then, no matter how weak the dot–dot coupling, the ground state need not have a uniform charge distribution. A state with one empty and one doubly occupied dot can well be lowest in energy if the differences in the site energies of these two dots are larger than I. In the high-disorder regime, where I < Δα, there will not be a lowest energy band of localized insulator-type states. On the other hand, because by assumption we are in the range where β is weaker, the states will not be thoroughly mixed. If β is comparable to the typical spacing, the wave function will be delocalized over a limited domain. When β is even smaller, the ground state will be localized with some dots empty and others doubly occupied, as shown in Fig. 4.
Figure 4.

The weights of the ground electronic state on the states of the noninteracting dots. These zero-order states have each electron assigned to a particular site as, for example, in Eq. 4 above. Computed at large interdot separation (D/2r = 1.8) for a moderate, Δα < I (Upper), and a higher, Δα > I (Lower), disorder . I = 0.3 eV for both Upper and Lower. In Upper, there is 5% fluctuation in sizes and 5% in packing, whereas in Lower, we introduce 15% fluctuation in size and 30% in packing. In Upper, the ground state at large separation is a covalent state with one electron per site, whereas in Lower, where the fluctuations in sizes and in packing overcome the effect of the charging energy, the ground electronic state will be an ionic state. In this example, it is an ionic state with two doubly occupied sites (see Inset).
The Different Electronic States
Fig. 3 showed the two regimes possible at higher compressions, when the dominant role is that of the interdot coupling β so that the charging energy can be neglected. There was a completely delocalized state at high β and a domain localized state when β is no longer larger than the range, Δα, of fluctuations in the site energies. Fig. 4 shows that at low compression, when the main role is that of the charging energy, the electronic ground state can be completely localized. It can be localized as a covalent state, with a uniform charge distribution, or, when I < Δα, it can become an ionic state. As the lattice is expanded, the complete sequence of transitions in the nature of the ground state is then delocalized → domain localized → site localized. This is shown in Fig. 5, which is obtained by diagonalizing the Hubbard Hamiltonian, Eq. 3, which includes both the transfer and the charging energies.
Figure 5.

A histogram of the weight, logarithmic scale, of the electronic ground state of the Hubbard Hamiltonian on the states of the noninteracting dots. Computed for a hexagonal array of seven sites, which has 784 states (ten percent wide-size distribution). The results are shown for the three electronic phases, delocalized, domain localized, and site localized, which can be successively accessed by expanding the lattice. The typical value of the weight is 1/(number of effectively participating states), and in the three regimes, this number is quite different. The value 1/784 is shown as a solid arrow in Top as it is a typical weight for the delocalized state, when all states are equally participating. The domain-localized state can be viewed as a linear combination of states of the noninteraction dots, with the number of states that contribute significantly substantially lower than the total number but somewhat larger than unity. A few weights are, therefore, large (right end of the abscicca), and most other weights are smaller than 1/784. The site-localized state can have a uniform charge distribution or a nonuniform one (see Fig. 4), but it is essentially a single state of the Hamiltonian of the noninteraction dots. One state has a weight of near unity, and all other weights are negligible.
To conclude: The possible electronic “isomers” of a lattice of quantum dots are shown in Fig. 6 as a function of reduced variables. What are the possible variables? In the Hückel (strong-coupling) limit, if we take the site energy α as the zero of energy, then all energies are determined by β alone. When we allow limited disorder, there will be two regions, a domain-localized and a delocalized phase, and the boundary between them is determined by the extent of disorder. Note, however, that in principle there are at least two not quite independent ways where the disorder comes in, namely in the size and in the spacing distributions. Therefore, even in the strong coupling limit, there really should be two axes of disorder. Once one allows for the effect of charging energy, the effective dot–dot coupling is determined by the dimensionless ratio, I/(I + β), of the charging energy to the strength of the dot–dot coupling. The strong coupling limit is I/(I + β) → 0. The weakly coupled lattice is at I/(I + β) → 1. In the absence of disorder, the transition-delocalized → site-localized will occur when I = β. Once there is a finite amount of disorder, there will also be a domain-localized phase, as shown in Fig. 5.
Figure 6.

The different possible coupling regimes for an array of quantum dots. The abscissa is the dimensionless ratio 0 ≤ I/(I + β) ≤ 1, and the ordinate is the fractional disorder. For a very disordered array, the state is localized, because β is far too small, and the variations in the site energies mean that an ionic state, where some sites are doubly occupied and others are empty, is lowest in energy. Reducing the disorder allows for a greater role of the dot–dot coupling. For very low disorder, the behavior is Mott like with a transition from a delocalized to a localized phase (shown as a solid line).
Should one speak of Fig. 6 as showing a transition between phases or between isomers? We do not know a simple overall answer. One can say that a transition is phase-transition like if the ground state is not well separated from the excited states, but that depends on the coupling range and on the extent of disorder. One can say that a phase transition is a manifestation of a collective behavior, but this criterion also is not useful throughout the diagram. The transition to a fully delocalized state involves many sites and, as such, it is possibly deserving of that label, which is why we show it as a boundary in Fig. 6. The transition-site-localized → domain-localized involves only a small subset of sites. Also, it is not clear that the regions shown in Fig. 6 are exhaustive, and they certainly are not once we allow, say, an external magnetic field. There is clearly scope for much more work.
We thank James Heath and Stan Williams for many useful discussions and insights, and Jim Kinsey for a critical review of the original version. Sonderforschungsbereich 377 and the Alexander von Humboldt Foundation supported this work. F.R. is a Chercheur Qualifié from the Fonds National de la Recherche Scientifique, Belgium. F.R. acknowledges support by the Action de Recherche Concertée, Liège University, Liège, Belgium.
Abbreviation
- STM
scanning tunneling microscopy
References
- 1.Bawendi M G, Steigerwald M L, Brus L E. Annu Rev Phys Chem. 1990;41:477–496. [Google Scholar]
- 2.Murray C B, Kagan C R, Bawendi M G. Science. 1995;270:1335–1338. [Google Scholar]
- 3.Alivisatos A P. Science. 1996;271:933–937. [Google Scholar]
- 4.Collier C P, Vossmeyer T, Heath J R. Annu Rev Phys Chem. 1998;49:371–404. doi: 10.1146/annurev.physchem.49.1.371. [DOI] [PubMed] [Google Scholar]
- 5.Markovich G, Collier C P, Henrichs S E, Remacle F, Levine R D, Heath J R. Acc Chem Res. 1999;32:415–423. [Google Scholar]
- 6.Collier C P, Saykally R J, Shiang J J, Henrichs S E, Heath J R. Science. 1997;277:1978–1981. [Google Scholar]
- 7.Kim S-H, Meideiros-Ribeiro G, Ohlberg D A A, Williams R S, Heath J R. J Phys Chem B. 1999;103:10341–10347. [Google Scholar]
- 8.Remacle F, Collier C P, Markovitch G, Heath J R, Banin U, Levine R D. J Phys Chem B. 1998;102:7727–7734. [Google Scholar]
- 9.Hoffmann R. Solids and Surfaces: A Chemist's View of Bonding in Extended Structures. New York: VCH; 1988. [Google Scholar]
- 10.Mott N F. Metal-Insulator Transitions. London: Taylor & Francis; 1990. [Google Scholar]
- 11.Remacle F, Collier C P, Heath J R, Levine R D. Chem Phys Lett. 1998;291:453–458. [Google Scholar]
- 12.Medeiros-Ribeiro C, Ohlberg D A A, Williams R S, Heath J R. Phys Rev B. 1999;59:1633–1636. [Google Scholar]
- 13.Anderson P W. Concepts in Solids. Palo Alto: Benjamin; 1971. [Google Scholar]
- 14.Logan D E, Wolynes P G. Phys Rev B. 1987;36:4135–4147. doi: 10.1103/physrevb.36.4135. [DOI] [PubMed] [Google Scholar]
- 15.Platt J R. Free Electron Theory of Conjugated Molecules. New York: Wiley; 1964. [Google Scholar]
- 16.Hubbard J. Proc R Soc London. 1963;276:238–257. [Google Scholar]
- 17.Parr R G. Quantum Theory of Molecular Electronic Structure. New York: Benjamin; 1963. [Google Scholar]
- 18.Remacle F, Levine R D. J Chem Phys. 1999;110:5089–5099. [Google Scholar]
- 19.Remacle F, Levine R D, Schlag E W, Weinkauf R. J Phys Chem A. 1999;103:10143–10158. [Google Scholar]
- 20.Paldus J. In: The Unitary Group for the Evaluation of Electronic Energy Matrix Elements. Hinze J, editor. Vol. 22. Berlin: Springer; 1981. pp. 1–50. [Google Scholar]