

Abstract
The Parkinson's disease (PD) substantia nigra is characterized by the presence of Lewy bodies containing fibrillar α-synuclein. Early-onset PD has been linked to two point mutations in the gene that encodes α-synuclein, suggesting that disease may arise from accelerated fibrillization. However, the identity of the pathogenic species and its relationship to the α-synuclein fibril has not been elucidated. In this in vitro study, the rates of disappearance of monomeric α-synuclein and appearance of fibrillar α-synuclein were compared for the wild-type (WT) and two mutant proteins, as well as equimolar mixtures that may model the heterozygous PD patients. Whereas one of the mutant proteins (A53T) and an equimolar mixture of A53T and WT fibrillized more rapidly than WT α-synuclein, the other (A30P) and the corresponding equimolar mixture with WT fibrillized more slowly. However, under conditions that ultimately produced fibrils, the A30P monomer was consumed at a comparable rate or slightly more rapidly than the WT monomer, whereas A53T was consumed even more rapidly. The difference between these trends suggested the existence of nonfibrillar α-synuclein oligomers, some of which were separated from fibrillar and monomeric α-synuclein by sedimentation followed by gel-filtration chromatography. Spheres (range of heights: 2–6 nm), chains of spheres (protofibrils), and rings resembling circularized protofibrils (height: ca. 4 nm) were distinguished from fibrils (height: ca. 8 nm) by atomic force microscopy. Importantly, drug candidates that inhibit α-synuclein fibrillization but do not block its oligomerization could mimic the A30P mutation and thus may accelerate disease progression.
Keywords: amyloid, aggregation, protofibril, atomic force microscopy (AFM)
Parkinson's disease (PD) is an age-related neurodegenerative disorder characterized by difficulty in initiating movements, rigidity, and resting tremor (1). In PD, neuronal death is localized to dopaminergic neurons in the substantia nigra region of the brain stem and precedes appearance of symptoms; ca. 70% of neurons may have died by the time symptoms become apparent (1). The postmortem PD substantia nigra is characterized by sporadic intraneuronal cytoplasmic inclusions known as Lewy bodies (LB), the fibrous portion of which contains the protein α-synuclein (2, 3). Lewy bodies themselves could be neurotoxic, analogous to the proposed toxicity of amyloid plaques in Alzheimer's disease (AD) (4); the frequency of cortical Lewy bodies correlates with the severity of the AD-like dementia diffuse Lewy body disease (5). Alternatively, Lewy bodies may be an epiphenomenon, induced by neuronal death. Finally, it is possible that Lewy bodies are an inert end point of a process that, early on, produces a neurotoxic species. This scenario would predict that Lewy body formation may protect against neuronal death by sequestering the toxic species. Two neuropathological observations may be relevant to this issue. First, inclusion-bearing neurons appear to be more healthy than neighboring neurons (6–8). Second, the postmortem brains of asymptomatic-aged individuals often contain Lewy bodies (9).
Some involvement of Lewy bodies in PD pathogenesis, albeit not necessarily causative, is supported by the connections between the α-synuclein gene and PD. A polymorphism in the α-synuclein promoter is reported to be a susceptibility factor for idiopathic PD, suggesting that its expression level may be critical (10). This finding is consistent with the premise that α-synuclein oligomerization and/or fibrillization is pathogenic. Furthermore, rare, early-onset forms of PD, which otherwise are indistinguishable from idiopathic PD, are linked to two autosomal dominant point mutations in the gene encoding α-synuclein (11–13). The mutations, A30P and A53T, do not affect the conformational behavior of the monomeric protein; wild-type α-synuclein (WT), A53T, and A30P are all “natively unfolded” at low concentration (14, 15). At higher concentrations, WT, A53T, and A30P all form amyloid fibrils of similar morphology (15–17, 45) by what appears to be a nucleation-dependent mechanism (18). All three proteins also produce nonfibrillar oligomers (15, 45) that may be assembly intermediates, analogous to the Aβ protofibril (19, 20). Fibrillization of α-synuclein is clearly accelerated by the A53T mutation (15–17) but the effect of the A30P mutation on fibril formation has not been determined, although A30P accelerates the formation of nonfibrillar oligomers (15) and the disappearance of unsedimentable protein from solution (17). Furthermore, the behavior of mixtures of the mutant proteins and WT, which are directly relevant to the early-onset PD patients, all of whom are heterozygotes, has not been determined.
We report herein that A30P and an equimolar mixture of A30P and WT fibrillize more slowly than WT alone. However, during the course of incubation, the A53T and A30P monomers are consumed more rapidly than WT, demonstrating that acceleration of oligomerization, rather than acceleration of fibrillization, is a shared property of both mutant proteins. This finding suggests that formation of a nonfibrillar oligomer is likely to be critical in pathogenesis. Three classes of nonfibrillar α-synuclein oligomers, two of which have been observed previously and one that is novel, were separated from fibrils and imaged by atomic force microscopy (AFM).
Materials and Methods
Expression and Purification of Recombinant Proteins.
Preparation of α-Synuclein Incubations for Aggregation Studies.
Samples were dissolved in PBS, pH 7.4, and filtered through a Millipore Microcon 100K MWCO filter; sodium azide (0.02%) was added to the 200-μM incubations only. Protein concentrations were determined by quantitative amino acid analysis. Samples were incubated at 37°C without agitation.
Thioflavin T Fluorescence Assay for Fibril Formation.
A 100-μM aqueous solution of Thioflavin T (Thio T; Sigma) was prepared and filtered through a 0.2-μm polyether sulfone filter. At various time points, aliquots of the α-synuclein incubations were diluted to 10 μM in H2O. Fluorescence measurements for the 300-μM α-synuclein incubations were performed in a 1-cm fluorescence cuvette as described elsewhere; the fluorescence maximum of 100,000 is a function of the fluorometer emission parameters (45). Fluorescence measurements for the 200-μM α-synuclein incubations were performed in a 384-well microplate. To each well was added 40 μl of 10 μM α-synuclein and 50 μl of 10 μM Thio T in 90 mM glycine-NaOH, pH 8.5 (prepared from 100 μM Thio T). The microplate was centrifuged briefly (ca. 1 min) at 500 × g to remove air pockets and equilibrated for 3–5 min. Fluorescence at 490 nm was measured with a Wallac Victor2 1420 multilabel counter (excitation at 450 nm, bandwidth 30; emission at 490 nm, bandwidth 10; courtesy of the Harvard Institute of Chemistry and Cell Biology). There may be differences in fluorescence intensity of Thio T attributable to WT, A53T, or A30P fibrils. Differences are not directly measurable because we have not obtained a pure fibrillar sample. However, our experiments indicate that, if there is a difference in fluorescence intensity, that difference does not qualitatively change the interpretations below.
Congo Red Absorbance Assay for Fibril Formation.
A 20-μM solution of Congo red in PBS, pH 7.4, was prepared (extinction coefficient at 498 nm ≈ 3.7 × 104 M−1) (21) and filtered through a 0.2-μm polyether sulfone filter. At various time points, 5-μl aliquots of the α-synuclein incubations were added to 250 μl of 20 μM Congo Red in PBS, pH 7.4, in a 96-well microplate. The microplate was incubated on a rotary shaker at room temperature for 30 min. Absorbance data were collected at 540 nm with a Molecular Devices Thermomax microplate UV/VIS spectrophotometer.
Circular Dichroism Spectroscopy of Protein Incubations.
At various time points, aliquots of the α-synuclein incubations were removed and diluted to 10 μM in PBS, pH 7.4. Circular dichroism (CD) spectra were collected on an AVIV 62A DS spectrophotometer using a 0.1-cm cell at room temperature.
Sedimentation and Gel-Filtration Separation of Monomeric and Oligomeric α-Synuclein from Fibrillar Material.
Samples of α-synuclein (total protein concentration = 200 μM) were incubated at 37°C. The samples consisted of (i) individual proteins (WT, A30P, or A53T) or (ii) equimolar mixtures (A30P/WT and A53T/WT). A 50-μl aliquot was removed from each sample at various times and cleared of insoluble material by sedimentation for 5 min at 16,000 × g in a benchtop centrifuge. The supernatant from the spin was then eluted from a Superdex 200 column (1 cm × 30 cm) in PBS, pH 7.4/0.02% NaN3 (0.5 ml/min), monitoring at 254 nm. The column was calibrated with the following protein molecular mass standards: Blue Dextran 2000 (≈2,000 kDa), albumin (67 kDa), ovalbumin (43 kDa), and ribonuclease A (13.7 kDa). The elution volume of Blue Dextran 2000 was taken to be the void volume. As reported, WT migrated anomalously, due to its “natively unfolded” structure, as a ca. 57 kDa species (14). Peak contents were quantified in two ways: by cutting and weighing the peak area of the chart and by measuring peak height (the two measurements were indistinguishable as pertains to the Discussion).
Preparation of Circularized Protofibrils.
The soluble portion of an equimolar mixture of WT and A53T, incubated for 36 days at 37°C, was eluted from a Superdex 200 column as described above. The void-volume elution peak was collected and concentrated ca. 10-fold by centrifugation at 14,000 × g in a Millipore Microcon 100K MWCO concentrator. Aliquots were removed periodically from the concentrated protein solution (stored at 4°C) and analyzed by AFM.
AFM of Protein Incubations.
Incubations were mixed gently to suspend any aggregates. Aliquots of 2–3 μl were placed on freshly cleaved mica (Ted Pella, Redding, CA) and incubated for 60 s, after which mica was gently rinsed twice with 50 μl of filtered, deionized water to remove salt and loosely bound protein. Excess water was removed with a stream of filtered, compressed trifluoroethane. Images were obtained with a Nanoscope IIIa Multimode scanning probe workstation (Digital Instruments, Santa Barbara, CA) operating in Tapping Mode by using etched silicon NanoProbes (model FESP; Digital Instruments). Scanning parameters varied with individual tips and samples. Typical values were: free oscillation amplitude, 0.55–0.9 V; setpoint, 0.3–0.8 V; drive (tapping) frequency 60–70 kHz; and scan rate, 1.1–1.49 Hz.
Results
Fibril Formation Is Nucleation-Dependent and Can Be Seeded by Exogenous Fibrils [See also Wood et al. (18)].
Fibril formation was followed by two independent and complementary dye-binding assays (these two dyes do not compete for the same binding site on the amyloid fibril): a Thio T assay based on a fluorescence enhancement of the bound dye and a UV/VIS assay of bound Congo red, based on a shift in the λmax of the absorption spectrum (22, 45). Incubations were not stirred to allow the observation of prefibrillar intermediates, should they be populated (stirring greatly accelerated fibril formation). Both methods of fibril quantitation showed a distinct lag phase for fibril formation, which is typical of amyloid fibrillizations and suggests that fibril formation may be nucleation-dependent (Fig. 1) (22–27). Consistent with that proposal, addition of preformed α-synuclein fibrils comprising WT, A53T, or A30P (1–2% by moles) to an incubation containing the identical protein resulted in a significant reduction in the lag time for appearance of a Thio T signal (lag time for A53T at 200 μM is ca. 35–42 days; WT, 45–49 days; A30P, 52–63 days; in each case addition of the homologous seed induced significant fibrillization within 15–30 days). This is consistent with the report of Wood et al. (18). Heterologous seeding (e.g., seeding of WT with A30P fibrils) of the type reported by Wood et al. (18) was not attempted.
Figure 1.

Fibrillization and β-sheet formation of WT, A53T, and A30P α-synuclein (300 μM), followed by three complementary methods. (A) Thio T fluorescence assay shows that A53T fibrillizes most rapidly (red), followed by WT (white) and then A30P (green); signal of 100,000 is maximum (refer to Materials and Methods). (B) Congo red-binding assay also shows that A53T fibrillizes most rapidly (red), followed by WT (white) and then A30P (green). (C) CD spectroscopy detects the random-coil (monomer)-to-β-sheet (fibril) transition. At day 0, all three variants are predominantly random coil. At day 31, the transition to β-sheet structure is most complete in the case of A53T (red), less in the case of WT (black), and has not begun in the case of A30P (green).
Fibrillization of A53T Is Accelerated Relative to Wild-Type α-Synuclein, but Fibrillization of A30P Is Slowed.
An initial experiment, using complementary dye-binding assays ([α-synuclein] = 300 μM), demonstrated that A53T had a shorter lag time than WT but A30P had a longer lag time than either of the other two variants (Fig. 1 A and B). CD spectra collected at several time points demonstrated that the conversion of “random coil” structure to β-sheet structure was most extensive for A53T, followed by WT, then A30P at each time analyzed. This trend paralleled the relative rates of fibril formation (Fig. 1C). Subsequent experiments utilized lower α-synuclein concentrations (200 μM) and were analyzed exclusively by the Thio T assay, because that method was more sensitive and less variable than other analytical methods. Equal volume incubations were used for direct comparisons. Under these optimized assay conditions, the lag time for fibril formation by A53T again was significantly shorter than that of WT (35–42 days vs. 45–49 days; see Fig. 2), consistent with what we and others had observed regarding the relative rates of fibrillization measured by other methods (15–17, 28). Furthermore, the lag time of A30P was longer than that of WT or A53T under all conditions (Figs. 1A, 1B, and 2). Equimolar mixtures of α-synuclein (200 μM total), containing A53T or A30P and WT, fibrillized at a rate intermediate between the individual components (Fig. 2).
Figure 2.

Fibrillization (Thio T fluorescence assay) of the three α-synuclein variants, WT, A53T, and A30P (200 μM), and relevant A53T/WT and A30P/WT equimolar mixtures (200 μM total protein). (A) Fibrillization of (left to right) A53T (red), 1:1 A53T/WT (pink), and WT (white). (B) Fibrillization of (left to right) WT (white), 1:1 A30P/WT (light green), and A30P (green). Note expanded y axis relative to A (identical WT run is plotted in both A and B).
Consumption of A30P Monomer Is Comparable or Slightly Faster than WT, Whereas A53T Monomer Is Consumed Most Rapidly.
As we had observed previously in brief incubations at a low concentration (14), monomeric α-synuclein, sampled during fibrillization, eluted from gel-filtration chromatography as would be expected for a ca. 57-kDa globular standard, because of its extended, natively unfolded structure. There was no evidence for transient population of a species of the appropriate molecular mass for a globular form of α-synuclein (14 kDa) in any of the incubations, even after long incubation times.
Unfolded, monomeric α-synuclein thus could be separated from oligomeric forms and quantified. In the 200-μM incubations, loss of 10–20% of monomer was measured in some incubations before a Thio T fluorescence signal was detected (Fig. 3), suggesting that nonfluorogenic (or weakly fluorogenic), nonfibrillar oligomers were being populated (15). As the Thio T signal became apparent, loss of monomer continued. In the case of A30P, however, the emergent Thio T signal did not fully account for the loss of monomer (see days 44–45 in Figs. 2B and 3B), suggesting that a nonfibrillar oligomer may be populated. In equimolar mixtures of A53T with WT (also at 200 μM total protein), monomer was consumed at a rate intermediate between that observed for the pure incubations (Fig. 3A), similar to the case for fibrillization (Fig. 2A). However, in an incubation containing equimolar amounts of WT and A30P, monomeric α-synuclein was consumed at a comparable rate or slightly more slowly than both pure A30P and WT (Fig. 3B). This contrasts with the case of fibrillization, where the A30P/WT mixture fibrillized at an intermediate rate (Fig. 2B).
Figure 3.

Consumption of natively unfolded α-synuclein during fibrillization. Aliquots were removed from incubations run in parallel to those shown in Fig. 2. Quantitation is based on gel-filtration peak weight, but peak height data were comparable. (A) Consumption of A53T (red), 1:1 A53T/WT (pink), and WT (white) during fibrillization (parallels Fig. 2A). (B) Consumption of WT (white), 1:1 A30P/WT (light green), and A30P (green) during fibrillization (parallels Fig. 2B). Where bars are missing, no aliquot was analyzed.
Prefibrillar, α-Synuclein Oligomers Were Separated from Monomer and Fibrils.
After sedimentation of fibrils from aliquots, the supernatant was subjected to gel-filtration chromatography, revealing primarily natively unfolded monomer (14) and several other minor peaks, none of which seemed to appear and disappear in a regular manner, as would be expected of a simple intermediate. The monomeric species represented ≥90% of unsedimented (nonfibrillar) α-synuclein at all times analyzed (through 66 days; see Fig. 3). The void volume eluate (≥600 kDa, assuming globular structure) was concentrated, adsorbed onto atomically smooth mica, and analyzed by AFM (see Fig. 4) (19, 20, 29). Only apparently spherical oligomers were observed in the void volume eluate; no elongated protofibrils or fibrils were present. Several distinct species, with heights between 2 and 6 nm, were detected (Fig. 4A). The dependence of the distribution of spherical species on the α-synuclein sequence and the “pure” vs. “mixed” nature of the incubation was not obvious and is the subject of ongoing studies.
Figure 4.
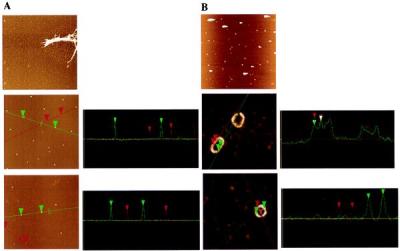
AFM images (and cross-sections) of oligomeric species from 200 μM α-synuclein incubations. (A Top) WT incubation (before sedimentation and gel filtration) showing fibrils and protofibril “spheres” (5-μm square). (Middle) 1:1 A53T/WT incubation after gel filtration showing “spheres” of 5.2–5.5 nm (green cursor and cross-section) and 2.6–3.6 nm (red cursor and cross-section) (2-μm square). (Bottom) 1:1 A30P/WT incubation after gel filtration showing spheres of 4.8–4.9 nm (green cursor and cross-section) and 3.3–3.5 nm (red cursor and cross-section) (2-μm square). (B Top) 1:1 A53T/WT (see A Middle), after incubation, showing rings (5-μm square; very bright features may be amorphous aggregates, with tails from AFM tip “skipping”). (Middle) Close-up of same sample (400-nm square) showing two ring types: circle and ellipse. The periodicity along the ellipse surface is shown in the cross-section to be regular (23 nm), with the maximum height of ca. 4 nm. (Bottom) Another circular ring in the same incubation (300-nm square), showing the diameter of ca. 50 nm and the difference between the maximum (3.6–4.1 nm) and minimum (2.1–2.2 nm) heights.
Incubation of “Spherical” α-Synuclein Oligomers Led to the Production of Ring-Like Structures.
The void volume from an equimolar A53T/WT incubation at 36 days contained primarily spherical oligomers (Fig. 4A). This eluate was concentrated (no immediate morphological change resulted; see Materials and Methods) and incubated for 72 h at 4°C. Analysis by AFM revealed the presence of ring-like structures (Fig. 4B). Analysis of the population of rings revealed the existence of two general classes: circular rings with diameters in the 35- to 55-nm range and elliptical rings (widths between 35 and 55 nm and lengths between 65 and 130 nm; Fig. 4B). Both species were characterized by regular height fluctuations, between ca. 2 and 4 nm, with a periodicity of ca. 23 nm (see cross-sections in Fig. 4B), corresponding to the measured diameter of the spherical oligomers.
Discussion
Many Neurodegenerative Diseases Are Characterized by Fibrous Protein Inclusions (30).
These inclusions can be either extracellular (e.g., amyloid plaques in AD), cytoplasmic [e.g., neurofibrillary tangles in AD and frontotemporal dementia (FTD)], or nuclear [e.g., nuclear inclusions in Huntington's disease (HD)] (4, 31, 32). Each inclusion comprises primarily insoluble ordered fibrils of a specific protein; for example, Aβ in AD amyloid plaque, tau in FTD neurofibrillary tangles, and huntingtin in HD nuclear inclusions. Mutations in the genes encoding the fibrillar proteins are associated with early-onset familial forms of these diseases, suggesting that protein unfolding, oligomerization, and/or fibril formation could be promoted by the mutations (31). Biophysical studies support this premise; for example, Aβ42, the form of Aβ that is increased by all early-onset AD mutations, is known to fibrillize more rapidly than the predominant form Aβ40 in vitro (24). In addition, expansion of a polyglutamine tract, the abnormality that gives rise to HD, increases the rate of huntingtin fibril formation (33, 34). However, the fibril has not been linked convincingly to neuronal death. In fact, a prefibrillar intermediate such as the Aβ protofibril (19, 20, 35, 36), an off-pathway species whose formation is linked to fibrillization (37), or the fibril itself may be the toxic species (38). Elucidation of this issue is critical, because many drug candidates have been selected on the basis of their ability to inhibit fibril formation, without consideration of prefibrillar intermediates (39–41). If such compounds allow intermediate formation and inhibit fibrillization of these intermediates, they could cause accumulation of the toxic species (38).
Comparative in Vitro Biophysical Studies of Wild-Type and Mutant Protein Fibrillization Processes Can Implicate Discrete Species in Disease Pathogenesis.
The step(s) that are affected by mutations are likely to induce accumulation of the pathogenic species (38). Several caveats to this type of modeling must be emphasized. First, our interpretation is based on the likely possibility that a shared molecular mechanism accounts for both forms of α-synuclein-linked PD. Of course, this may not be the case. Second, the comparisons made herein are not relevant if the expression levels of each α-synuclein variant are not identical, for example, if the mutations affect mRNA stability. Third, the cytoplasmic concentration of free α-synuclein may vary with the mutation, if lipid or protein binding (42, 43) or ubiquitin-dependent degradation are affected. For example, A30P seems to be defective in vesicle binding (43); thus, its “available” concentration may be highest. Despite similar caveats, the in vitro modeling approach has provided important insight into the etiologies of AD (4, 22) and HD (33, 34).
The Rate of α-Synuclein Amyloid Fibril Formation Is Not Accelerated by Both PD-Linked Mutations.
We currently are able to directly monitor two distinct species along the α-synuclein fibrillization pathway: soluble, natively unfolded and monomeric α-synuclein, a normal neuronal protein, and the α-synuclein amyloid fibril, the appearance of which, in the form of Lewy bodies, correlates with Parkinson's disease. The time-dependent behavior of other putative species in our in vitro experiments currently must be inferred. Under controlled conditions, soluble A30P monomer is consumed at a comparable rate or slightly more rapidly than WT monomer; however, WT fibrils appear more rapidly than A30P fibrils. This is consistent with our previous observation that nonfibrillar, apparently spherical, species are formed by A30P before fibrils can be detected (15). In mixed incubations containing both A30P and WT [relevant to the heterozygous PD patients (12)], both loss of monomer and appearance of fibrils are slowed relative to the pure WT incubation. It may be that the latter effect is of greater magnitude, so a prefibrillar intermediate may be populated to a greater extent in the A30P/WT incubation than in the WT incubation.
The differences in kinetic behavior between the pure and mixed incubations are mildly surprising, considering that mutant and wild-type fibrils are not easily distinguished (16, 45) and that A53T fibrils seed polymerization of WT α-synuclein (18). This raises the questions of whether the preferred intermediates contain one or both variants and whether one variant can inhibit fibrillization of another. This issue has been approached experimentally for other amyloidogenic proteins by using peptide models of Aβ variants and the polymorphic forms of the prion protein (27, 44).
Nonfibrillar α-Synuclein “Spheres,” “Chains,” and “Rings” May Be Sequential Species in a Pathway.
The kinetic data discussed above could be reconciled with the genetic data by a mechanism in which a nonfibrillar pathogenic oligomer accumulates in both mixed incubations. To directly quantitate such a species and to determine the effects of the PD-linked mutations on the rate of its formation and its stability, new methods must be developed and applied. However, morphologically detailed “snapshots” of several species allow the construction of a testable model to guide those experiments. We have observed three discrete, nonfibrillar α-synuclein oligomers (Fig. 4) that seem to be related, because they can share a height (ca. 4–5 nm) and apparent diameter (ca. 20 nm). These are (i) “spheres” of several heights, some of which (4–5 nm) (15) resemble early Aβ protofibrils (Fig. 4A) (20), (ii) chains that appear to comprise linearly-associated 4–5 nm spheres, analogous to elongated Aβ protofibrils (45), and (iii) rings, apparently comprising circularized chains (Fig. 4B). The detailed mechanism of formation of these species and of their possible interconversions has not been determined. However, a sphere-to-chain-to-ring progression is one simple possibility that is consistent with our data. By analogy to the Aβ situation (20), the chain may be a direct precursor to the fibril; thus, its circularization may prevent fibril formation. Accordingly, fibrils and rings may arise from α-synuclein by a bifurcated pathway that diverges from a common protofibrillar intermediate.
Could α-Synuclein Fibrils and Lewy Bodies Be Harmless Epiphenomena of a Pathogenic Oligomerization Pathway?
The possibility that protofibrils and/or protofibril rings are actually the pathogenic species and that fibrils are innocuous (or less toxic) is consistent with pathological studies that have suggested that Lewy bodies may be neuroprotective (6–8). This proposal has several important consequences. First, the most pathogenic mutations, which presumably would be selected against by evolution, would promote protofibril formation and prevent fibrillization. This information could be useful in constructing animal models in which pathogenesis is accelerated. Second, animal models should be evaluated based on neuronal loss in the substantia nigra, rather than on the presence of Lewy bodies, because the two are correlated but not necessarily linked. Finally, compounds that inhibit α-synuclein fibrillization, but allow protofibril formation, may promote disease. This is an important consideration for high-throughput screening efforts in which compounds are selected for their ability to inhibit fibrillization. The experimental testing of this proposal is the subject of ongoing studies.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NS 34719 and AG 08470). J.-C.R. was supported by a postdoctoral fellowship from the Alberta Heritage Foundation for Medical Research.
Abbreviations
- PD
Parkinson's disease
- AD
Alzheimer's disease
- AFM
atomic force microscopy
- Thio T
Thioflavin T
- WT
wild type
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Dunnett S B, Bjorklund A. Nat Suppl. 1999;399:A32–A39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- 2.Baba M, Nakajo S, Pang-Hsien T, Tomita T, Nakaya K, Lee V M-Y, Trojanowski J Q, Iwatsubo T. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Spillantini M G, Crowther R A, Jakes R, Hasegawa M, Goedert M. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkoe D. Nat Suppl. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 5.Kosaka K, Iseki E. Curr Opin Neurol. 1996;9:271–275. doi: 10.1097/00019052-199608000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Forno L S. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Tompkins M M, Hill W D. Brain Res. 1997;775:24–29. doi: 10.1016/s0006-8993(97)00874-3. [DOI] [PubMed] [Google Scholar]
- 8.Tompkins M M, Basgall E J, Zamrini E, Hill W D. Am J Pathol. 1997;150:119–131. [PMC free article] [PubMed] [Google Scholar]
- 9.Forno L S, Langston J W. Neurodegeneration. 1993;2:19–24. [Google Scholar]
- 10.Kruger R, Vieira-Saecker A M, Kuhn W, Berg D, Muller T, Kuhnl N, Fuchs G A, Storch A, Hungs M, Woitalla D, et al. Ann Neurol. 1999;45:611–617. doi: 10.1002/1531-8249(199905)45:5<611::aid-ana9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 11.Polymeropoulos M H, Lavedan C, Leroy E, Ide S E, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 12.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen J T, Schols L, Riess O. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 13.Papadimitriou A, Veletza V, Hadjigeorgiou G M, Patrikiou A, Hirano M, Anastasopoulos I. Neurology. 1999;52:651–654. doi: 10.1212/wnl.52.3.651. [DOI] [PubMed] [Google Scholar]
- 14.Weinreb P H, Zhen W, Poon A W, Conway K A, Lansbury P T., Jr Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 15.Conway K A, Harper J D, Lansbury P T. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 16.Giasson B I, Uryu K, Trojanowski J Q, Lee V M Y. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 17.Narhi L, Wood S J, Steavenson S, Jiang Y, Wu G M, Anafi D, Kaufman S A, Martin F, Sitney K, Denis P, et al. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 18.Wood S J, Wypych J, Steavenson S, Louis J C, Citron M, Biere A L. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- 19.Harper J D, Wong S S, Lieber C M, Lansbury P T., Jr Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 20.Harper J D, Wong S S, Lieber C M, Lansbury P T., Jr Biochemistry. 1999;38:8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 21.Zhen W, Han H, Anguiano M, Lemere C A, Cho C G, Lansbury P T., Jr J Med Chem. 1999;42:2805–2815. doi: 10.1021/jm990103w. [DOI] [PubMed] [Google Scholar]
- 22.Harper J D, Lansbury P T. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 23.Jarrett J T, Lansbury P T., Jr Biochemistry. 1992;31:12345–12352. doi: 10.1021/bi00164a008. [DOI] [PubMed] [Google Scholar]
- 24.Jarrett J T, Berger E P, Lansbury P T., Jr Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 25.Jarrett J T, Lansbury P T., Jr Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 26.Come J H, Fraser P E, Lansbury P T., Jr Proc Natl Acad Sci USA. 1993;90:5959–5963. doi: 10.1073/pnas.90.13.5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Come J H, Lansbury P T., Jr J Am Chem Soc. 1994;116:4109–4110. [Google Scholar]
- 28.El-Agnaf O M, Jakes R, Curran M D, Wallace A. FEBS Lett. 1998;440:67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 29.Wong S S, Harper J D, Lansbury P T, Jr, Lieber C M. J Am Chem Soc. 1998;120:603–604. [Google Scholar]
- 30.Koo E H, Lansbury P T, Jr, Kelly J W. Proc Natl Acad Sci USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lansbury P T. Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 32.Goedert M, Crowther R A, Spillantini M G. Neuron. 1998;21:955–958. doi: 10.1016/s0896-6273(00)80615-7. [DOI] [PubMed] [Google Scholar]
- 33.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates G P, Davies S W, Lehrach H, Wanker E E. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 34.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates G P, Lehrach H, Wanker E E. Proc Natl Acad Sci USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh D M, Lomakin A, Benedek G B, Condron M M, Teplow D B. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 36.Walsh D M, Hartley D M, Kusumoto Y, Fezoui Y, Condron M M, Lomakin A, Benedek G B, Selkoe D J, Teplow D B. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 37.Lambert M P, Barlow A K, Chromy B A, Edwards C, Freed R, Liosatos M, Morgan T E, Rozovsky I, Trommer B, Viola K L, et al. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lansbury P T. Proc Natl Acad Sci USA. 1999;96:3342–3344. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pallitto M M, Ghanta J, Heinzelman P, Kiessling L L, Murphy R M. Biochemistry. 1999;38:3570–3578. doi: 10.1021/bi982119e. [DOI] [PubMed] [Google Scholar]
- 40.Soto C, Sigurdsson E M, Morelli L, Kumar R A, Castano E M, Frangione B. Nat Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 41.Findeis M A, Musso G M, Arico-Muendel C C, Benjamin H W, Hundal A M, Lee J J, Chin J, Kelley M, Wakefield J, Hayward N J, Molineaux S M. Biochemistry. 1999;38:6791–6800. doi: 10.1021/bi982824n. [DOI] [PubMed] [Google Scholar]
- 42.Engelender S, Kaminsky Z, Guo X, Sharp A H, Amaravi R K, Kleiderlein J J, Margolis R L, Troncoso J C, Lanahan A A, Worley P F, et al. Nat Genet. 1999;22:110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 43.Jensen P H, Nielsen M S, Jakes R, Dotti C G, Goedert M. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 44.Jarrett J T, Costa P R, Griffin R G, Lansbury P T., Jr J Am Chem Soc. 1994;116:9741–9742. [Google Scholar]
- 45.Conway, K. A., Harper, J. D. & Lansbury, P. T., Jr. (2000) Biochemistry, in press. [DOI] [PubMed]