


Abstract
Hepatitis delta virus RNAs possess self-cleavage activities that produce 2′,3′-cyclic phosphate and 5′-hydroxyl termini (i.e. cis-acting delta ribozyme). Trans-acting delta ribozymes have been engineered by removing a junction from the cis version, thereby producing one molecule possessing the substrate sequence and the other the catalytic domain. According to the pseudoknot model, the secondary structure of the delta ribozyme includes a pseudoknot (i.e. P1.1 stem) formed by two base pairs from residues of the L3 loop and J1/4 junction. A collection of 48 P1.1 stem mutants was synthesized in order to provide an original characterization of both the importance and the structure of this pseudoknot in a trans-acting version of the ribozyme. Several structural differences were noted compared to the results reported for cis-acting ribozymes. For example, a combination of two stable Watson–Crick base pairs composing the essential P1.1 stem was demonstrated to be crucial for a significant level of activity, while the cis version required only one base pair. In addition, we present the first physical evidences revealing that the composition of the P1.1 stem affects the substrate specificity for ribozyme cleavage. Depending on the residues forming the J1/4 junction, non-productive ribozyme–substrate complexes can be observed. This phenomenon is proposed to be important for further development of a gene-inactivation system based on delta ribozyme.
INTRODUCTION
Hepatitis delta virus (HDV) possesses a single-stranded circular RNA genome of 1.7 kb that replicates by a double rolling-circle mechanism in human cells (reviewed in 1). Both genomic and antigenomic HDV RNAs have self-cleavage activities that produce 2′,3′-cyclic phosphate and 5′-hydroxyl termini (i.e. delta ribozyme) (reviewed in 2). Delta ribozyme has several unique features, all of which are related to the fact that it is a catalytic RNA motif that has been discovered in human cells. For example, it is efficient at low Mg2+ concentrations (∼1 mM, i.e. human cell-like conditions) (3), and it exhibits an outstanding stability in human cells (4). Moreover, delta ribozyme has a highly ordered catalytic center whose existence is revealed by a number of unusual properties reported for its cis-acting versions as compared to other self-cleaving RNA motifs (reviewed in 5). For example, it is extremely stable, with an optimal reaction temperature of ∼65°C, and retains its activity at temperatures as high as 80°C and in buffer containing 5 M urea or 18 M formamide.
According to the pseudoknot model, which is well supported by experimental data, the secondary structure of the cis-acting delta ribozyme consists of one stem (P1), one pseudoknot (P2), two stem–loops (P3–L3 and P4–L4) and three single-stranded junctions (J1/2, J1/4 and J4/2) (Fig. 1) (1,2,5). The P2 stem, and the P4–L4 hairpin that is invariably preceded by a homopurine base pair, are structural motifs located each side of the catalytic center. The P3 stem length (i.e. a 3 bp region with a preference for GC pairing) is critical, suggesting that it positions the L3 loop in a precise conformation in the catalytic center. The single-stranded L3 and J4/2 regions are essential for catalysis, while the J1/4 junction is single-stranded at the initial stages of folding, but then is involved in the formation of a pseudoknot, referred to as the P1.1 stem (Fig. 1). This additional pseudoknot, which is formed by two GC base pair interactions, was initially observed in the crystal structure of a self-cleaved product of the genomic version of delta ribozyme (6,7). Its existence was further supported by a mutational analysis of cis-acting constructs (8,9).
Figure 1.

Secondary structure and nucleotide sequences of a trans-acting delta ribozyme. S and Rz represent the substrate and ribozyme, respectively. The dotted line represents the J1/2 single-stranded region, which joins the S and Rz molecules in the cis-acting version. The pseudoknot P1.1 is formed by the base pairs G27C12 and G28C11, and is boxed. The stems are numbered P1 to P4. The homopurine base pair (i.e. GG) at the top of the P4 stem is indicated by two large dots, whereas the wobble base pair (i.e. GU) is indicated by a single large dot. The bold arrow indicates the cleavage site.
Trans-acting delta ribozymes have been engineered by removing the J1/2 junction, thereby producing one molecule possessing the substrate (S) sequence and the other the catalytic domain (Rz) (2). We have derived a trans-acting delta ribozyme of 57 nt from the antigenomic HDV genome (Fig. 1) (10,11). In this model system, an 11 nt substrate is cleaved into RNA products of 4 and 7 nt. Several studies, including Fe(II)-EDTA protection assays, nuclease probing and cross-linking experiments, suggested that cis- and trans-acting delta ribozymes of both genomic and antigenomic polarities have similar global tertiary structures, and that only subtle differences exist between the various forms (12,13). However, the presence of the P1.1 pseudoknot in a trans-acting delta ribozyme has not yet been unambiguously proven using the base pair compensation approach. We demonstrate here the necessity of the P1.1 pseudoknot for the formation of an active tertiary RzS complex. Clearly, this is a key motif in a trans-acting ribozyme whose contribution to the folding pathway is even more significant than in the cis-acting version. In addition, the composition of the P1.1 stem plays a significant role in determining the catalytic efficiency of the ribozyme.
MATERIALS AND METHODS
DNA constructs
Two procedures were used to synthesize a collection of ribozymes with mutations in the P1.1 stem. The first approach was based on the use of randomized DNA oligonucleotides (Invitrogen) that encode the P1.1 stem. In this approach an oligonucleotide of 74 nt corresponding to the T7 RNA promoter followed by the full-length ribozyme in which the four positions with the potential to form the P1.1 stem were randomized (5′-TAATACGACTCACTATAGGGTC CACCTN11N12TCGCGGTCCGACCTN27N28GCATGCGG CTTCGCATGGCTAAGGGACCC-3′, where N is equally likely to be A, C, G or T) served as the first DNA strand. A second oligonucleotide complementary to the 3′ end region (5′-GGGTCCCTTAGCCATGCGAAGCCGC-3′) served as primer for the second strand synthesis. The two oligonucleotides were annealed, and the second strand synthesized by adding 2.5 U of Pwo DNA polymerase (Roche Diagnostic) in a final volume of 100 µl containing 200 µM dNTP, 10 mM Tris–HCl pH 8.9, 25 mM KCl, 5 mM (NH4)2SO4 and 2 mM MgSO4. The resulting mixture was purified by extracting twice with phenol and chloroform, and then the nucleic acid precipitated with ethanol. After dissolving in loading buffer, the nucleic acids were fractionated through a 2% agarose gel and the bands corresponding to the correct sizes cut out and passed through Spin-X columns (Costar). The DNA was precipitated from the eluates with ethanol, washed with 70% ethanol and dried. After dissolving in water, the DNA fragments were cloned into SmaI digested pUC19, yielding a plasmid that contained only one of the 256 (i.e. 44) possible variants of delta ribozyme. Several colonies were selected and DNA sequenced in order to determine the identity of the four positions that composed the P1.1 stem.
Secondly, an approach of individual ribozyme cloning was adopted in order to complete the collection because we wanted a greater diversity of P1.1 stem composition. These constructions were synthesized as described previously (10). Briefly, pairs of complementary and overlapping DNA oligonucleotides corresponding to the T7 RNA promoter followed by the full-length ribozyme were synthesized, annealed and cloned into PstI and SphI co-digested pUC19. The sequences of all ribozyme genes were confirmed by DNA sequencing.
RNA synthesis
Ribozymes. Ribozymes were synthesized by in vitro run-off transcription in 100 µl reactions using linearized recombinant plasmids (10 µg) in the presence of 27 U of RNA Guard (Amersham Biosciences), 80 mM HEPES–KOH pH 7.5, 24 mM MgCl2, 2 mM spermidine, 40 mM DTT, 4 mM of each NTP, 0.01 U of yeast pyrophosphatase (Roche Diagnostic) and 10 µg of purified T7 RNA polymerase at 37°C for 3 h. The reactions were stopped by adding 5 U of DNase I (RNase free; Promega) and incubating at 37°C for 30 min. The mixtures were extracted twice with both phenol and chloroform, and the nucleic acid precipitated with ethanol. After dissolution in equal volumes of water and formamide dye buffer (95% formamide, 10 mM EDTA, 0.025% bromophenol blue and 0.025% xylene cyanol) the ribozymes were fractionated by 8% denaturing (7 M urea) polyacrylamide gel electrophoresis (PAGE; 19:1 ratio of acrylamide to bisacrylamide) in buffer containing 45 mM Tris-borate pH 7.5 and 1 mM EDTA. The reaction products were visualized by UV shadowing and the bands corresponding to the ribozymes cut out. The transcripts were eluted from the gel slices overnight at room temperature in a solution containing 0.1% SDS and 0.5 M ammonium acetate. The resulting mixtures were passed through G-50 Sephadex spun columns (Amersham Biosciences), and the RNA precipitated by the addition of 0.1 vol of 2 M sodium acetate pH 4.5 and 2.5 vol of ethanol. After washing in 70% ethanol and drying, the pellets were resuspended in ultrapure water, and the quantity of RNA determined by spectrophotometry at 260 nm.
Substrates and analogs. The 11 nt substrate was synthesized by in vitro transcription as described previously (10). Briefly, complementary and overlapping oligonucleotides were annealed in 20 µl of buffer containing 10 mM Tris–HCl pH 7.5, 10 mM MgCl2 and 50 mM KCl by incubating at 95°C for 2 min and then allowing the solution to slowly cool to 37°C. The resulting DNA duplexes (500 pmol) were then used as templates for in vitro transcription reactions, and the RNA purified through a 20% PAGE as described above.
The mutated substrate, the SdC4 and the P2 analogs were chemically synthesized using 2′-ACE chemistry (Dharmacon). The deprotection of the polymers was performed according to the manufacturer’s recommendations in 400 µl of buffer (100 mM acetic acid with the pH adjusted to pH 3.8) for 30 min at 60°C, and lyophilized. The polymers were then resuspended in ultrapure water, purified by denaturing 20% PAGE, and recovered as described above.
Labeling of RNA with [γ-32P]ATP
Either purified substrates or ribozymes (20 pmol) were dephosphorylated in a final volume of 20 µl containing 200 mM Tris–HCl, pH 8.0, 10 U of RNA Guard and 0.2 U of calf intestinal alkaline phosphatase (Roche Diagnostic) at 37°C for 30 min. The reactions were purified by extracting twice with phenol and chloroform, and the RNA precipitated with ethanol, washed with 70% ethanol and dried. Dephosphorylated RNA molecules (5 pmol of substrates, ribozymes or analogs) were 5′ end-labeled in a final volume of 10 µl containing 3.2 pmol of [γ-32P]ATP (6000 Ci/mmol; Amersham Biosciences), 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 50 mM KCl and 3 U of T4 polynucleotide kinase (Amersham Biosiences) at 37°C for 45 min. The reactions were stopped by the addition of 5 µl of formamide dye buffer, and the mixtures were fractionated through denaturing 8 or 20% PAGE. The bands containing the appropriate 5′ end-labeled RNA were excised, and the nucleic acid recovered as described above.
Cleavage reactions and kinetic assays
Unless otherwise stated, cleavage reactions were carried out in 20 µl reaction mixtures containing 50 mM Tris–HCl pH 7.5, and 10 mM MgCl2 at 37°C. Prior to the reaction, trace amounts of 5′ end-labeled substrates (<1 nM) and non-radioactive ribozymes (200 nM) were mixed together, heated at 95°C for 2 min, snap-cooled on ice for 1 min and then incubated at 37°C for 5 min. Following this pre-incubation step, the cleavage reactions were initiated by the addition of Tris–HCl pH 7.5 and MgCl2. Aliquots (2–3 µl) were removed at various times up to 4 h, or until the end-point of the cleavage was reached, and were quenched by the addition of 8 µl of ice-cold formamide dye buffer. The mixtures were fractionated on denaturing 20% PAGE and exposed on PhosphorImager screens (Molecular Dynamics). The extent of cleavage was determined from measurements of the radioactivity present in the substrate and the 5′ product bands at each time point using the ImageQuant software. The fractions of cleaved substrate were determined, and the rate of cleavage (kobs) obtained by fitting the data to the equation At = Ao(1 – e–kt), where At is the percentage of cleavage at time t, Ao is the maximum percent cleavage (or the end-point of cleavage), and k is the rate constant (kobs). Each rate constant was calculated from at least two independent measurements. Kinetic assays were performed under single turnover conditions as described above (14,15). Trace amounts of 5′ end-labeled substrate (<1 nM) were mixed with various concentrations of ribozyme (5–400 nM). The values of kobs obtained were plotted as a function of ribozyme concentration in order to determine the other kinetic constants (k2, KM and k2/KM).
Metal ion-induced cleavage
The metal ion-induced cleavage experiments were performed as described previously (16). Trace amounts (∼0.01 pmol) of 5′ end-labeled ribozyme were resuspended in 3 µl of H2O, and then were pre-incubated at 70°C for 1 min in either the presence or the absence of a substrate or 3′ end product (10 pmol). After cooling on ice for 2 min, the mixtures were equilibrated at 37°C for 5 min. The cleavage reactions were initiated by adding 250 mM CHES–NaOH, pH 9.5 (1 µl) and 100 mM MgCl2 (1 µl), and allowed to proceed by incubating at 37°C. Aliquots were removed after 15 and 120 min. The reactions were stopped by adding 250 mM EDTA, pH 8.0 (1 µl), and then 6 µl of stop solution (95% formamide, 10 mM EDTA, 0.05% xylene cyanol) was added prior to fractionation on 10% PAGE. In order to identify the cleavage sites, the reaction products of an RNA ladder produced by both alkaline hydrolysis and ribonuclease T1 digestion were fractionated on the gels. The resulting gels were dried and exposed to an X-ray film and/or a PhosphorImager (Molecular Dynamics) screen when quantification was required.
Oligonucleotide hybridization assays
Oligonucleotide hybridization assays were performed as described previously (17). Briefly, end-labeled ribozyme (0.2 pmol) and SdC4 analog (20 pmol) in RNA dissolving buffer (4 µl containing 1 mM Tris–HCl pH 8.0 and 0.1 mM EDTA) were denatured at 95°C for 2 min and then pre-incubated at 37°C for 2 min. Ribozyme and SdC4 analog folding were initiated by the addition of 4 µl of 2× folding buffer (100 mM HEPES pH 8.0, 40 mM KCl, 0.1 mM EDTA, 2 mM dithiothreitol and 20 mM MgCl2). The mixtures were incubated at 37°C, and aliquots (1 µl) removed after 0, 1 and 5 min were added to RNase H cleavage mixture [4 µl containing 0.5 U/µl RNase H from Ambion in folding buffer that included an oligonucleotide (10 µM) complementary to the L3 loop from either the original ribozyme (5′-GCGAGGA-3′) or the mutant ribozyme with the P1.1 stem composed of G27C12/C28G11 (5′-GCGAGCA-3′)]. The RNase H hydrolyses were stopped after 30 s by the addition of ice-cold formamide dye buffer (6 µl), and then kept on ice until fractionation on 10% denaturing PAGE. The gels were then exposed to either Instant Imaging (Packard) or PhosphorImager.
RESULTS
Engineering of a collection of P1.1 stem mutants
In order to evaluate the importance of the P1.1 stem in a trans-acting version of delta ribozyme for which the thermodynamic behavior has been extensively characterized under both single- and multiple-turnover conditions (10,11), a collection of mutants was constructed using two strategies. Initially, an approach of randomized nucleotides was used. This involved the use of an oligonucleotide in which the four positions that form the P1.1 stem (i.e. positions 11,12, 27 and 28; see Fig. 1) were randomized (i.e. 25% of each residue A, C, G and T). The corresponding second strands were synthesized by primer extension and the resulting small genes cloned. Thirty-nine different P1.1 stem mutants were identified by DNA sequencing. Alternatively, some desired P1.1 mutants were also constructed using oligonucleotides with specific sequences at the appropriate positions. A total of 48 mutants were synthesized, including versions with the potential to form zero, one or two Watson–Crick base pairs as the P1.1 stem (see Table 1).
Table 1. Cleavage assays of P1.1 mutants.
| Base pair | P1.1 identity 27-12/28-11 | H-bonds | End-point (%) |
|---|---|---|---|
| 0 | UU/UU | 0 | <0.1 |
| CU/UU | 0 | <0.1 | |
| GG/GG | 0 | <0.1 | |
| UU/AG | 0 | <0.1 | |
| UC/UU | 0 | <0.1 | |
| GA/UC | 0 | <0.1 | |
| UC/UC | 0 | <0.1 | |
| GA/UU | 0 | <0.1 | |
| UU/GG | 0 | <0.1 | |
| CC/GG | 0 | <0.1 | |
| AA/AA | 0 | <0.1 | |
| 1 | GU/UC | 2 | <0.1 |
| GU/UU | 2 | <0.1 | |
| GU/AA | 2 | <0.1 | |
| UG/UU | 2 | <0.1 | |
| UG/UC | 2 | <0.1 | |
| UU/GU | 2 | <0.1 | |
| AU/GA | 2 | <0.1 | |
| AU/CA | 2 | <0.1 | |
| AU/AA | 2 | <0.1 | |
| CU/AU | 2 | <0.1 | |
| UU/AU | 2 | <0.1 | |
| UU/UA | 2 | <0.1 | |
| CU/UA | 2 | <0.1 | |
| GC/CC | 3 | 3.0 | |
| CG/CC | 3 | 2.5 | |
| CC/GC | 3 | 2.3 | |
| CC/CG | 3 | 3.2 | |
| 2 | UG/GU | 4 | <0.1 |
| GU/UG | 4 | <0.1 | |
| GU/AU | 4 | 1.2 | |
| UA/GU | 4 | 1.2 | |
| UA/AU | 4 | 1.3 | |
| GU/GU | 4 | 1.8 | |
| GU/UA | 4 | 1.8 | |
| CG/UA | 5 | 2.2 | |
| GU/CG | 5 | 4.1 | |
| UA/GC | 5 | 4.8 | |
| CG/AU | 5 | 12.3 | |
| UA/CG | 5 | 15.7 | |
| GC/UA | 5 | 29.1 | |
| GC/AU | 5 | 52.9 | |
| AU/GC | 5 | 58.7 | |
| AU/CG | 5 | 74.6 | |
| CG/CG | 6 | 4.5 | |
| GC/CG | 6 | 18.2 | |
| CG/GC | 6 | 33.9 | |
| a | GC/GC | 6 | 89.6 |
aIndicates wild-type sequence.
The P1.1 stem requires two Watson–Crick base pairs
The cleavage activity of all mutants was determined using a small model substrate under single-turnover conditions. Trace amounts of 32P 5′ end-labeled substrate (<1 nM), which produces RNA products of 4 and 7 nt, were incubated in the presence of an excess of ribozyme (200 nM). A typical autoradiogram is shown in Figure 2A, while Figure 2B provides a graphical representation of the cleavage activity. Both the original ribozyme (G27C12/G28C11, i.e. the version derived from the wild-type cis-acting ribozyme) and the A27U12/C28G11 mutant formed a P1.1 stem that included 2 bp and exhibited a relatively efficient cleavage. However, the original ribozyme is a lot more efficient than the mutant (i.e. approximately one order of magnitude better when their kobs are compared). In fact, the original ribozyme was the most efficient member of the collection. The mutant C27G12/C28C11, which has the potential to form a 1 bp P1.1 stem, exhibited only a moderate cleavage activity (i.e. ∼two orders of magnitude slower than the original ribozyme). Finally, the mutant U27U12/U28U11, in which no Watson–Crick base pair can be formed as a P1.1 stem, exhibited no detectable activity even after 4 h of incubation. Prolonged incubation (up to 24 h) with this mutant did not result in the detection of any cleavage product (data not shown). These results clearly show that the presence of the base pairs forming the P1.1 stem is a critical factor in observing a good level of cleavage activity.
Figure 2.

Cleavage activities of the P1.1 stem mutants as compared to that of the original delta ribozyme. (A) Typical autoradiogram of a 20% denaturing gel for the analysis of the cleavage reaction of three P1.1 stem mutants and the original delta ribozyme. For each ribozyme, aliquots were recovered from the reaction pool after 0, 60 and 120 min. The positions of the bromophenol blue (BPB), the 11 nt substrate (S) and the 4 nt product (P) are indicated. (B) Graphical representation of time courses for the cleavage reactions catalyzed by the original ribozyme (i.e. G27C12/G28C11, closed circles), the A27U12/C28G11 mutant (i.e. forming a 2 bp P1.1 stem, open circles), the C27G12/C28C11 mutant (i.e. forming a 1 bp P1.1 stem, closed squares), and the U27U12/U28U11 mutant (i.e. forming 0 bp and therefore no P1.1 stem, open squares).
The maximal cleavage percentage (i.e. the end-point of cleavage) was determined for all P1.1 stem mutants (Table 1). All mutants lacking the ability to form at least one Watson–Crick base pair were devoid of any detectable cleavage activity. Among the mutants that supported the formation of only 1 bp, only those with either a GC or CG base pair exhibited cleavage activity, and only did so at a reduced level (i.e. the maximal end-point observed was 3.2%). Using several 1 bp mutants and various reaction conditions (e.g. in the presence of 100 mM NaCl in order to stabilize helix conformation), we were unable to rescue cleavage activity. The situation was a lot more complicated for the mutants allowing the formation of 2 bp. Two clusters of ribozymes can be formed with respect to the number of hydrogen bonds (H-bonds) involved in the Watson–Crick base pairs. The first cluster is composed of the mutants that include two Watson–Crick base pairs involving four H-bonds (i.e. AU, UA, GU or UG base pairs). These ribozymes exhibited a very low cleavage activity. More specifically, the two mutants with a UG base pair were deprived of any detectable activity, while the five other ribozymes (including AU, UA or GU base pairs) had maximal cleavage percentages that varied between 1.2 and 1.8%. It is noteworthy that the presence of a uridine residue at position 27 (either in a UA or UG base pair) results in a ribozyme with lesser catalytic activity. The second cluster is composed of the mutants that include two Watson–Crick base pairs involving either five or six H-bonds (i.e. contain at least one GC or CG base pairs). These ribozymes exhibited activity levels that ranged from 2.2 to 89.6%. With the exception of the C27G12/G28C11 mutant, all mutants possessing a pyrimidine (i.e. C or U) at position 27 exhibited a relatively low activity level (between 2.2 and 15.7%). In addition, with the exception of one ribozyme that includes a wobble base pair in position 27-12 (i.e. G27U12/C28G11), all other mutants have a purine at position 27 and exhibited a relatively efficient cleavage activity (i.e. between 18.2 and 74.6%). The presence of a stable base pair at position 27-12 appears to be critical for a good level of cleavage activity. Most likely, the stability of this top base pair, which is the first one of the helix formed by stacking the P1.1 stem over the P4 stem, is of primary importance. In general, it is preferable to have a purine–pyrimidine base pair to initiate the stacking of a helix (18). In contrast, the identity of the base pair in the position 28-11 appears not crucial for the level of cleavage of a trans-acting ribozyme, rather it is just important for a base pair to be present in order to ensure efficient catalysis. Finally, the original ribozyme, which has a guanosine residue in position 27, is the member of the collection with the greatest catalytic activity, exhibiting a maximal cleavage percentage of 89.6%. It is well known that end-point do not reach the 100% most likely due to the presence of small fraction of misfolded ribozymes (11).
Formation of the P1.1 stem is critical for the catalytic activity
In order to accurately assess the importance of the base at each position of the P1.1 stem, kinetic analyses were performed using an excess of various mutated ribozymes (5–800 nM), and the pseudo-first-order cleavage rate constants determined (k2, KM and k2/KM; Table 2). The original ribozyme had k2 and KM values of 0.23 min–1 and 17.6 nM, respectively, which are virtually identical to those reported for this ribozyme in independent experiments [0.23 min–1 and 9.3 nM (11); 0.29 min–1 and 16 nM (17)]. Only the P1.1 stem mutants that were sufficiently active could be characterized. Among the P1.1 stem mutants, two populations, based on the kinetic parameters of the ribozymes, were observed. The first population of four mutants had k2 and KM values reduced by ∼4- and ∼2-fold, respectively, as compared to the original ribozyme. The apparent second-order rate constants (i.e. k2/KM) of these mutants were virtually identical, suggesting that they cannot be differentiated based on this parameter. However, these mutants had k2/KM values 2–3-fold smaller than that of the original ribozyme (i.e. 4.5–6.8 × 106 min–1M–1 compared to 1.3 × 107 min–1M–1; Table 2).
Table 2. Kinetic parameters of P1.1 mutants.
| 27-12/28-11 | k2 (min–1) | KM (10–9 M) | k2/KM (min–1 M–1) | Fold decrease |
|---|---|---|---|---|
| GC/GCa | 0.23 ± 0.01 | 17.6 ± 2.9 | 1.3 × 107 | – |
| AU/GC | 0.048 ± 0.007 | 7.1 ± 1.2 | 6.8 × 106 | 1.9 |
| AU/CG | 0.047 ± 0.006 | 7.2 ± 1.5 | 6.5 × 106 | 2.0 |
| GC/AU | 0.041 ± 0.009 | 7.2 ± 1.2 | 5.6 × 106 | 2.3 |
| CG/GC | 0.051 ± 0.005 | 11.3 ± 2.7 | 4.5 × 106 | 2.9 |
| UA/CG | 0.040 ± 0.003 | 81.5 ± 9.6 | 4.9 × 105 | 26.5 |
| GC/UA | 0.017 ± 0.002 | 80.9 ± 12.9 | 2.1 × 105 | 61.9 |
| GC/CG | 0.024 ± 0.002 | 126.5 ± 38.4 | 1.9 × 105 | 68.4 |
aIndicates wild-type sequence.
The second population of three mutants possessed KM values that were 5–8-fold larger than that of the original ribozyme, while their k2 values were 5–10-fold smaller. Overall the k2/KM values of these mutants were 26–68-fold smaller than that of the original ribozyme (i.e. 1.9–4.9 × 105 min–1M–1 compared to 1.3 × 107 min–1M–1). These mutants exhibited a drastically reduced cleavage activity.
Base pairings between residues of the substrate and the J1/4 junction prevent formation of P1.1 stem
The kinetic parameters reported in Table 2 revealed the existence of two populations of mutants (see above). One of these is composed of mutants that have k2/KM values at least 25-fold smaller than the original ribozyme. One possible explanation for this lack of activity is the formation of base pairs between the single-stranded 5′ end region of the substrate (i.e. positions –1 to –4) and the J1/4 region. This would have the effect of preventing the formation of the P1.1 pseudoknot, thereby inhibiting the cleavage activity. This hypothesis is supported by a study of the substrate specificity of delta ribozyme cleavage (14). Using a 5′-extended substrate (i.e. 14 nt long), the nucleotides in positions –1 to –4 have been shown to contribute to the substrate specificity. The presence of two consecutive pyrimidines (i.e. either C or U) in positions –1 and –2, which have the potential of base pairing with the guanosines of the J1/4 junction (i.e. G27G28), has been shown to be detrimental to the cleavage activity (14).
In order to investigate this possibility, we initially verified if these results are consistently observed with a ribozyme cleaving an 11 nt substrate. These experiments were performed using a trace amount of 5′ labeled substrate in the presence of 200 nM of ribozyme. A substrate with the sequence –4GGGC–1 was efficiently cleaved (Fig. 3A). In contrast, a substrate with the sequence –4GGCC–1, which allows for the formation of 2 bp between the 5′ end region of the substrate and the J1/4 junction (i.e. C–1G27/C–2G28), was cleaved at least one order of magnitude less than the original substrate (Fig. 3A). Finally, a substrate with the sequence –4CUAA–1 [i.e. that of the optimal 14 nt substrate (14)], was cleaved the most efficiently by the original ribozyme (Fig. 3A). Thus, these results with an 11 nt substrate are in good agreement with those reported for 14 nt substrates.
Figure 3.

Graphical representation of the cleavage activity over time of various substrates by a P1.1 stem mutant and the original delta ribozyme. (A) The ribozyme with the original sequence. (B) The G27C12/C28G11 P1.1 mutant. Both ribozymes were incubated in the presence of the –4GGGC–1 (which is the original sequence; circles), the –4GGCC–1 (squares) and the –4CUAA–1 (diamonds) substrates.
The mutant ribozyme with the P1.1 stem composed of G27C12/C28G11 cleaved the original substrate (i.e. –4GGGC–1) relatively inefficiently (see Fig. 3B and Table 2). This low level of activity might result from the formation of 2 bp between the 5′ end region of the substrate and the J1/4 junction (i.e. C–1G27/G–2C28,). This new stem consists of two GC base pairs, as is observed for the P1.1 stem. Therefore, the observation of larger KM and smaller kcat values, as compared to the original ribozyme (Table 2), would be characteristic of a phenomenon of non-productive RzS complex formation (19). If this is the case, changing the sequence of the substrate in positions –1 and –2 should allow us to retrieve the cleavage activity. As expected, the substrate with the sequence –4GGCC–1 was cleaved a lot more efficiently (Fig. 3B), although at a reduced level as compared to the original ribozyme. In addition, the substrate with the sequence –4CUAA–1 was the most efficiently cleaved by this ribozyme (Fig. 3B), as was the case with the original one.
In summary, these experiments show that mutations for the positions –1 and –2 in the substrate can compensate for some P1.1 inactive mutants. The substrate with –4GGCC–1 was a poor one for the original ribozyme, but a good one for the G27C12/C28G11 P1.1 mutant. Conversely, the –4GGGC–1 substrate was a good one for the original ribozyme, but a poor one for the G27C12/C28G11 P1.1 mutant. The two other P1.1 mutants exhibited a limited level of cleavage as compared to the original ribozyme. These mutants also have the potential to form base pairs with the substrate. The G27C12/U28A11 mutant has the potential to base pair with positions –1 and –2 (i.e. forming C–1G27/G–2U28), but less stably than the G27C12/C28G11 mutant. The U27A12/C28G11 mutant has the potential to form base pairs with positions –2 and –3 (i.e. forming G–2U27/G–3C28). When these P1.1 stem mutants are incubated with substrates that prevent the formation of base pairs between the substrate and the J1/4 junction, higher cleavage levels are observed. Taken together, these results strongly support the hypothesis that any base pair formed between the single-stranded 5′ end region of the substrate and the J1/4 junction prevents the formation of the P1.1 stem, and is therefore detrimental to the cleavage activity of some ribozymes with the original substrate.
Probing the accessibility of the L3 loop
The hypothesis that base pairings occur between the substrate and the J1/4 junction required additional support. Since nuclease probing has been shown to be of limited use in achieving this task (14), we adopted the approach of oligonucleotide hybridization assays (using differential oligonucleotide hybridization and specific RNase H cleavage) that has been described previously (17). This method relies on the following two premises: (i) the selective susceptibility of an RNA:DNA duplex to RNase H cleavage; (ii) the accessibility of DNA oligonucleotides to various exposed single-stranded domains on the RNA molecules (20). This type of experiment has been used to deduce the minimal folding pathways for both the cis- and trans-acting forms of delta ribozyme (17). Furthermore, it has been shown that the L3 loop becomes inaccessible in the presence of magnesium, most likely because the formation of the P1.1 stem (i.e. involving the L3 loop and the J1/4 junction) is Mg2+ dependent.
Probing of the L3 loop structure was performed as described previously (17). 32P-end-labeled ribozyme (with either the original or G27C12/C28G11 P1.1 stem) and the SdC4 analog were incubated together at 95°C for 2 min, and then cooled to 37°C. The SdC4 analog is an 11 nt RNA identical to the substrate except for the presence of a deoxyribose residue at position 4 (i.e. the cleavage site), which renders it uncleavable. It has been shown in inhibition experiments that the use of the SdC4 analog mimics the formation of the P1 stem in the RzS complex (11). Once at 37°C, MgCl2 was added to the reactions so as to allow the folding to continue. Aliquots were recovered at various time points, and treated with RNase H in the presence of the appropriate oligonucleotide for the L3 loop in question. A typical autoradiogram is illustrated in Figure 4. Following the initiation of the folding (i.e. the addition of MgCl2), the level of RNase H hydrolysis within the L3 loop of the original ribozyme is reduced with time (i.e. from 75% at time zero to 14% after 5 min; Fig. 4A). In contrast, the G27C12/C28G11 P1.1 stem mutant conserved almost the same accessibility regardless of the incubation time (i.e. from 34% at time zero to 21% after 5 min; Fig. 4B), suggesting that the P1.1 stem is not formed (or is formed only in a very small proportion of the RzS complexes). Most likely this occurs because the residues of the J1/4 loop are already engaged in base pairing with positions –1 and –2 of the substrate. In both cases, the hydrolysis occurs at the same position, showing that the oligonucleotides bound to the same place. At time zero, we observed that the level of hydrolysis of the G27C12/C28G11 P1.1 stem mutant is ∼2-fold less than that of the original ribozyme (i.e. 34% compared to 75%). This difference is indicative of a variable accessibility as binding shift assays using an equal concentration of the appropriate ribozymes displaced one-half less of the 32P-labeled oligonucleotide in the case of the G27C12/C28G11 P1.1 stem mutant as compared to what is observed with the original sequence. Thus, if the reduction in the accessibility of the L3 loop results from P1.1 stem formation, these results indicate that this pseudoknot is formed within the RzS complex that includes the original ribozyme upon the addition of MgCl2, but not in the G27C12/C28G11 P1.1 stem mutant. This interpretation received support from similar experiments performed using various P1.1 stem mutants. RNase H cleavage rates of mutants with good activity were considerably higher than for the three ribozymes with k2/KM reduced by 26- to 68-fold (data not shown).
Figure 4.
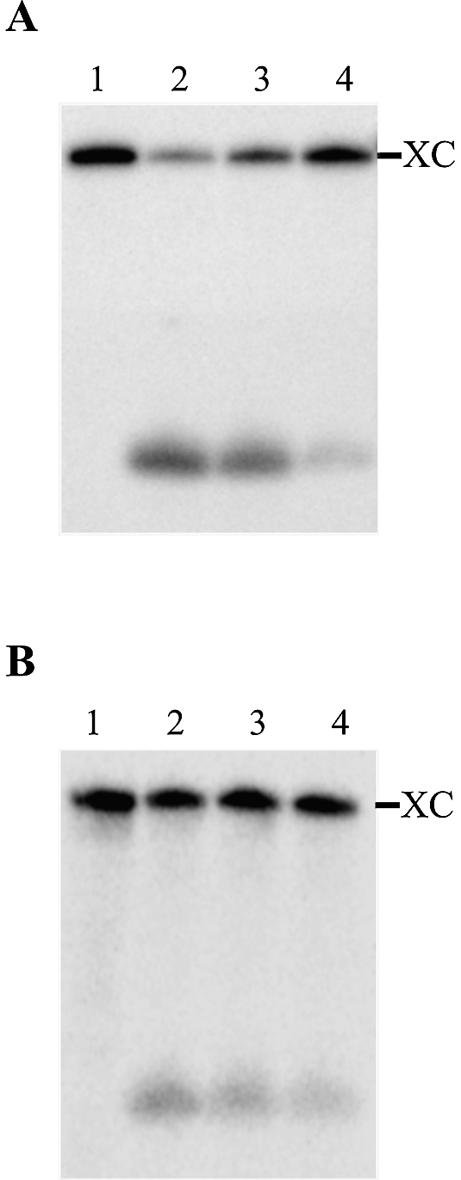
Typical autoradiograms of a 10% PAGE of the L3 loop probing. (A) The ribozyme with the original sequence. (B) The G27C12/C28G11 P1.1 mutant. Both ribozymes were pre-incubated in the presence of the SdC4 analog prior to the addition of MgCl2. Lanes 1 are aliquots of ribozyme and SdC4 analog removed at 0 min and treated with RNase H in the absence of any oligonucleotide. Lanes 2, 3 and 4 are aliquots removed at 0, 1 and 5 min, respectively, and treated with the RNase H in the presence of an oligonucleotide. The position of the xylene cyanol (XC) is indicated.
Magnesium-induced cleavage
An absolute requirement for the presence of divalent metal ions was shown for delta ribozyme catalysis at physiological pH (21). A Mg2+ ion was shown to specifically cleave the phosphodiester backbone at position G52, which is at the bottom of the P2 stem, solely within catalytically active RzS complexes according to metal-induced cleavage experiments (16). In order to establish whether or not the magnesium cation located near the bottom of the P2 stem is involved in both the reduction of accessibility of the L3 loop and the formation of P1.1 stem, experiments of Mg2+-induced cleavage at position G52 were performed using several P1.1 stem mutants.
Typical Mg2+-induced cleavage patterns of 5′ end-labeled ribozymes are shown in Figure 5. Figure 5A is an autoradiogram of an experiment performed using the original ribozyme. Briefly, in the absence of either magnesium (lanes 2 and 3) or substrate (lanes 4 and 5) the induced cleavage at position G52 was not detected. In contrast, in the presence of either the substrate (lanes 6 and 7) or the 3′ end product, which base pairs with the ribozyme (lanes 8 and 9), an induced cleavage at position G52 was observed (greatest after 120 min). Over-exposure was required to confirm the detection of the G52 band after only 15 min of incubation (lanes 6 and 8). These results are in good agreement with those reported previously (16); specifically that formation of a properly folded P1 stem is required in order to observe the Mg2+-induced cleavage at position G52. In contrast, the mutant that includes four uridines in the positions composing the P1.1 stem, and therefore cannot form the pseudoknot, did not result in the detection of any Mg2+-induced cleavage at position 52 regardless of the conditions tested (Fig. 5B). However, the balance of the pattern of bands was shared between this mutant and the original ribozyme. Similar experiments were performed with several P1.1 mutants (data not shown). The induced cleavage at position G52 was detected in all mutants that support the formation of a 2 bp P1.1 stem, although at a reduced level as compared to the original ribozyme. In contrast, no mutant with a 0 or 1 bp P1.1 stem allowed the detection of the G52-induced cleavage. No correlation between the amount of Mg2+-induced cleavage at position G52 (i.e. intensity of this band) and the kinetic parameters has been deduced. However, we observed that the presence of a G27C12, as compared to any other base pair at this position of the P1.1 stem, provided the most stable helix and resulted in a more intense induced cleavage at position 52. Together, these data suggest that the binding of the Mg2+ ion responsible for the G52-induced cleavage is a result of the formation of P1.1 stem, rather than a requirement for the formation of this pseudoknot. Most likely it either binds at the bottom of the P2 stem after the formation of the P1.1 pseudoknot, or becomes reactive only after formation of the P1.1 stem occurs. More importantly, it is not this magnesium ion that is involved in the reduction of the accessibility of the L3 loop.
Figure 5.
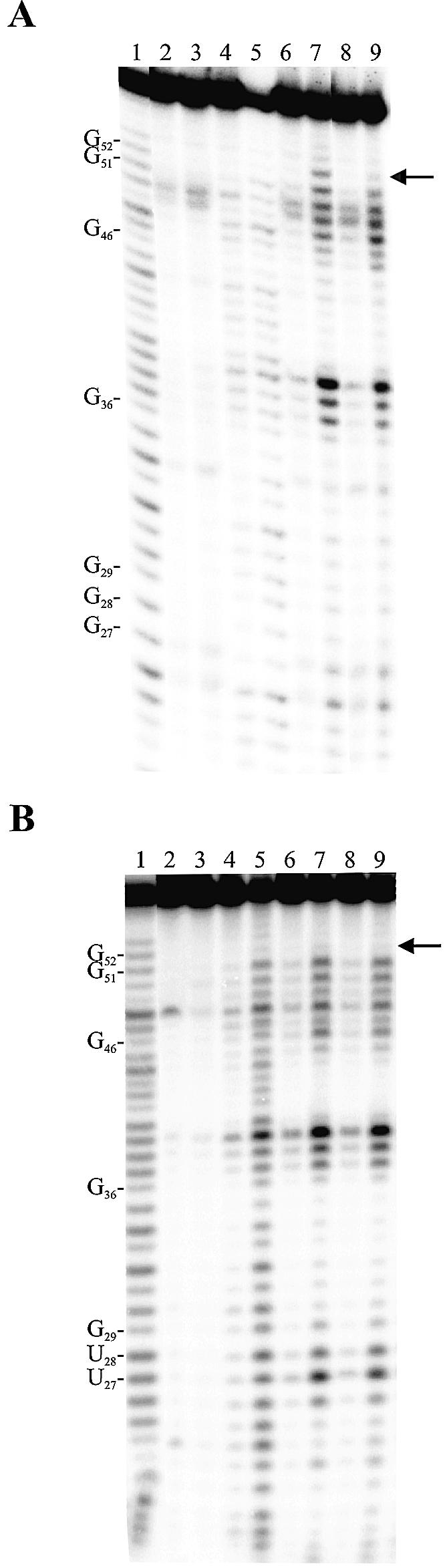
Typical autoradiograms of 10% PAGE of Mg2+-induced cleavage experiments. (A) The ribozyme possessing the original sequence (i.e. G27C12/G28C11). (B) The ribozyme possessing a P1.1 stem mutated to U27U12/U28U11. The 5′ end-labeled ribozymes were incubated in the presence of substrate, but in the absence of Mg2+ (lanes 2 and 3), or in the absence of substrate, but in the presence of Mg2+ (lanes 4 and 5). The remaining lanes contain the ribozymes, Mg2+ and either the substrate (lanes 6 and 7) or the 3′ product (lanes 8 and 9). Lanes 2, 4, 6 and 8 are incubations of 15 min, while lanes 3, 5, 7 and 9 are incubations of 120 min. Alkaline hydrolysis (lane 1) and RNase T1 hydrolysis (data not shown) of the 5′ end-labeled ribozymes were performed in order to determine the location of the metal ion-induced cleavage products. The locations of the RNase T1 major cuts are indicated on the left. The arrow on the right indicates the location of the G52.
More generally, regardless of the mutant analyzed, the banding patterns were virtually identical with only minor variations being observed between the various mutants. Most of the time these variations were located in either the L3 loop or at the top of the P4 stem (i.e. specifically positions 26–28), both regions that are in close proximity to the bases involved in the formation the P1.1 stem. This observation supports the idea that the reduction of cleavage activity of the various mutants is not the result of folding into alternative structures, which reduce the concentration of productive ribozymes.
DISCUSSION
The P1.1 stem is critical for the activity of the trans-acting delta ribozyme
Evaluation of the cleavage activity of a collection of P1.1 stem mutants revealed that the presence of this pseudoknot is crucial for the catalysis to occur (see Table 1). More specifically, these assays demonstrated that an active ribozyme requires that not only two Watson–Crick base pairs forming the P1.1 stem, but also that these base pairs must be stable (i.e. at least one of the two base pairs must contain the three H-bonds found in GC and CG pairings). These results contrast with some of the data reported for a cis-acting delta ribozyme (9). In that case, some mutants possessing P1.1 stems composed of only one canonical base pair exhibited a relatively good level of activity. In cis-acting delta ribozymes, the stability of the P1.1 stem might be less critical in obtaining a good level of self-cleavage because additional interactions contribute to the adoption of an active tertiary structure. It is noteworthy that the rate constant for the chemistry in the trans reaction is ∼40-fold slower than that of self-cleavage under the same reaction conditions (2). This difference in rate constant was attributed to a higher entropic penalty for the bimolecular reaction as compared to the unimolecular one, and to a kinetically less stable catalytic center resulting from the absence of the J1/2 crossover, which could add rigidity to the tertiary structure (6).
According to the minimal kinetic pathway, the first step is the binding of the substrate to the ribozyme (i.e. formation of the P1 stem). This is followed by a relatively slow internal conformation rearrangement yielding the active RzS complex (11) (see Fig. 6). Kinetic intermediates determined in the folding of a trans-acting delta ribozyme using oligonucleotide hybridization assays coupled with RNase H activity have shown that the P1.1 stem appears after the formation of an appropriate P1 stem (17). This step appears to be relatively slow, and is a good candidate to be the rate-limiting step of the folding pathway. Preliminary RNase H probing experiments of the L3 loop accessibility of various P1.1 stems support the hypothesis that the formation of the P1.1 stem might be the rate-limiting step in the kinetic pathway (J.Perreault, unpublished data).
Figure 6.

Schematic representation of the minimal folding and catalytic pathway of delta ribozyme. This representation includes the formation of either productive (i.e. with P1.1 stem) or non-productive (i.e. with base pairs between positions –1 and –2 of the substrate and positions 27 and 28 of the J1/4 region) RzS′ complexes. The base pairs that are formed after the formation of the P1 stem are indicated by dotted lines.
It has been proposed that Mg2+ was required for the formation of the P1.1 stem (9,17). In this study Mg2+-induced cleavage experiments indicated that the binding of the magnesium ion responsible for the specific hydrolysis of the phosphodiester bond at position G52 most likely resulted from the formation of P1.1 stem, rather than being required for the formation of this pseudoknot. Another Mg2+ ion was located within the bottom of the P1 stem, adjacent to the scissile phosphate, based on nuclear magnetic resonance analysis (22). As this magnesium cation is in close proximity to the P1.1 stem, it might be the one required for observation of the formation of the pseudoknot. However, this does not exclude the involvement of an additional magnesium ion specific for the formation of the P1.1 stem. The P1.1 stem is most likely critical for, and central to, the conformational transition that produces the active ternary complex (RzS′), but it is not the only interaction that has to take place within this highly ordered catalytic center. For example, several 2′-hydroxyl groups specific to the ribose moiety were involved in a network of H-bonds within the catalytic center (6). The involvement of most of these 2′-hydroxyl groups was confirmed by a systematic analysis using 2′-H modifications (i.e. 2′-hydrogen) (15). Interestingly, this study also showed that the residues composing the P1.1 stem can be all deoxyribonucleotides and the cleavage activity of the resulting ribozyme is virtually identical to that of the original ribozyme (15). Thus, regardless of the composition of the P1.1 stem (i.e. either deoxyribo- or ribonucleotides), it seems to fold into an RNA A-helix. This might be a consequence of its stacking with the P4 stem, which is still composed of ribonucleotides, and therefore adopts a typical A-helix.
The composition of P1.1 stem contributes to the substrate specificity
The observation of a population of mutants having drastically reduced k2/KM values (i.e. 26–68-fold) as compared to the original ribozyme prompted experiments aimed at explaining this observation. Cleavage assays were performed using P1.1 stem mutants and substrates having different nucleotides in positions –4 to –1, an approach similar to that of compensatory mutations. The resulting data indicated that the formation of base pairs between the single-stranded 5′ end region of the substrate and the J1/4 junction of the ribozyme prevented the folding of the P1.1 stem, which in turn leads to a dramatic decrease in the level of cleavage. These results unambiguously prove that the single-stranded region adjacent to the cleavage site contributes to the substrate specificity.
The results presented here suggest that the single-stranded 5′ end region of the substrate has the potential to slow down the rearrangement of the RzS complex. Regardless of whether or not a substrate has a suitable sequence in positions –4 to –1, the P1 stem is formed. After formation of the RzS complex, two different pathways may be followed (see Fig. 6). In the presence of a suitable substrate, the P1.1 stem is efficiently formed. This results in the formation of productive RzS′ complexes and occurrence of the chemical step. Conversely, in the presence of a substrate that forms extra base pairs with the residues of the J1/4 junction, the formation of the P1.1 stem is slowed down, thereby reducing the proportion of ribozymes present in active RzS′ complexes. In other words, the RzS complexes become non-productive (see Fig. 6).
The base identity at each position from –4 to –1 has been demonstrated to contribute differently to the substrate specificity (14). For these positions, a consensus sequence –4YHRH–1 (where H indicates U, C or A, Y indicates U or C, and R indicates G or A) has been derived. Although this portion of the substrate is not a part of the recognition domain (i.e. the P1 stem), it plays a crucial role in the ability of a substrate to be cleaved (14,23). This hypothesis received physical support from two different studies (24,25). First, a study that established the energetic contribution of non-essential 5′ sequences of small substrates to the catalysis of another version of the delta ribozyme revealed that the sequence composition of this region might influence the kinetic parameters by more than one order of magnitude (24). Secondly, a study aimed at developing delta ribozymes targeting the human hepatitis B virus showed that most of the efficiently cleaved sites did not possess consecutive pyrimidines at positions –1 and –2 (25). The latter result supports the idea that the rules established with small substrates are consistently respected in a long RNA target. However, it now appears possible to modify these rules. As shown in this study, some P1.1 stem mutants have different substrate specificities than the original ribozyme. However, the kinetic analysis performed with the collection of mutants revealed that the most active P1.1 stem mutants had k2/KM values only 2–3-fold smaller that the original ribozyme. The ribozyme with the G27C12/G28C11 P1.1 stem was the most efficient of the entire collection. It is notable that the composition of the P1.1 stem of the original ribozyme (i.e. G27C12/G28C11) is perfectly conserved in all known natural variants of the cis-acting delta ribozyme, suggesting that it is optimal for catalytic activity. Therefore, it might be possible to use a P1.1 stem mutant if a different substrate specificity is required in order to target a specific site. Clearly, the P1.1 pseudoknot is central to both the structure and mechanism of the delta ribozyme.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Mr Dominique Lévesque for technical assistance. This work was supported by a grant from the Canadian Institute of Health Research (CIHR) to J.-P.P. The RNA group is supported by grants from both the CIHR and Fonds FCAR (Québec). P.D. was the recipient of a pre-doctoral fellowship from CIHR-Health Canada, J.P. from Fonds FCAR-FRSQ (Québec), and J.O. from Fonds FCAR. J.-P.P. is an Investigator from the CIHR.
REFERENCES
- 1.Mercure S., Lafontaine,D., Roy,G. and Perreault,J.P. (1997) Le motif autocatalytique d’ARN du virus delta de l’hépatite humaine. Médecine/Science, 13, 662–667. [Google Scholar]
- 2.Shih I.H. and Been,M.D. (2002) Catalytic strategies of the hepatitis delta virus ribozymes. Annu. Rev. Biochem., 71, 887–917. [DOI] [PubMed] [Google Scholar]
- 3.Wu H.N., Lin,Y.J., Lin,F.P., Makino,S., Chang,M.F. and Lai,M. (1989) Human hepatitis δ virus RNA subfragments contain an autocleavage activity. Proc. Natl Acad. Sci. USA, 86, 1831–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lévesque D., Choufani,S. and Perreault,J.P. (2002) Delta ribozyme benefits from a good stability in vitro that becomes outstanding in vivo. RNA, 8, 464–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doherty E.A. and Doudna,J.A. (2000) Ribozyme structure and mechanism. Annu. Rev. Biochem., 69, 597–615. [DOI] [PubMed] [Google Scholar]
- 6.Ferré-D’Amaré A.R., Zhou,K. and Doudna,J.A. (1998) Crystal structure of a hepatitis delta virus ribozyme. Nature, 395, 399–404. [DOI] [PubMed] [Google Scholar]
- 7.Ferré-D’Amaré A.R. and Doudna,J.A. (2000) Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J. Mol. Biol., 295, 541–556. [DOI] [PubMed] [Google Scholar]
- 8.Wadkins T.S., Perrotta,A.T., Ferré-D’Amaré,A.R., Doudna,J.A. and Been,M.D. (1999) A nested double pseudoknot is required for self-cleavage activity of both the genomic and antigenomic hepatitis delta virus ribozymes. RNA, 6, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishikawa F. and Nishikawa,S. (2000) Requirement for canonical base pairing in the short pseudoknot structure of genomic hepatitis delta virus ribozyme. Nucleic Acids Res., 28, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ananvoranich S. and Perreault,J.P. (1998) Substrate specificity of delta ribozyme cleavage. J. Biol. Chem., 273, 13182–13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercure S., Lafontaine,D., Ananvoranich,S. and Perreault,J.P. (1998) Kinetic analysis of delta ribozyme cleavage. Biochemistry, 37, 16975–16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenstein S.P. and Been,M.D. (1996) Hepatitis delta virus ribozymes fold to generate a solvent-inaccessible core with essential nucleotides near the cleavage site phosphate. Biochemistry, 35, 11403–11413. [DOI] [PubMed] [Google Scholar]
- 13.Rosenstein S.P. and Been,M.D. (1991) Evidence that genomic and antigenomic RNA self-cleaving elements from hepatitis delta virus have similar secondary structures. Nucleic Acids Res., 19, 5409–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deschênes P., Lafontaine,D.A., Charland,S. and Perreault,J.P. (2000) Nucleotides –1 to –4 of hepatitis delta ribozyme substrate increase the specificity of ribozyme cleavage. Antisense Nucleic Acid Drug Dev., 10, 53–61. [DOI] [PubMed] [Google Scholar]
- 15.Fiola K. and Perreault,J.P. (2002) Kinetic and binding analysis of the catalytic involvement of ribose moieties of a trans-acting δ ribozyme. J. Biol. Chem., 277, 26508–26516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lafontaine D.A., Ananvoranich,S. and Perreault,J.P. (1999) Presence of a coordinated metal ion in a trans-acting antigenomic delta ribozyme. Nucleic Acids Res., 27, 3236–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ananvoranich S. and Perreault,J.P. (2000) The kinetics and magnesium requirements for the folding of antigenomic δ ribozymes. Biochem. Biophys. Res. Commun., 270, 600–607. [DOI] [PubMed] [Google Scholar]
- 18.Saenger W. (1983) Forces stabilizing associations between bases: hydrogen binding and base stacking. In Cantor,C.R. (ed.), Principles of Nucleic Acid Structure. Springer-Verlag, New York, NY, pp. 116–158.
- 19.Fersht A.R. (1985) The basic equations of enzyme kinetics. In Enzyme Structure and Mechanism, 2nd Edn. W.H. Freeman, New York, NY, pp. 98–120.
- 20.Zarrinkar P.P. and Williamson,J.R. (1994) Kinetic intermediates in RNA folding. Science, 265, 918–924. [DOI] [PubMed] [Google Scholar]
- 21.Murray J.B., Seyhan,A.A., Walter,N.G., Burke,J.M. and Scott,W.G. (1998) The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. (Lond.), 5, 587–595. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka Y., Hori,T., Tagaya,M., Sakamoto,T., Kurihara,Y., Katahira,M. and Uesugi,S. (2002) Imino proton NMR analysis of HDV ribozymes: nested double pseudoknot structure and Mg2+ ion-binding site close to the catalytic core in solution. Nucleic Acids Res., 30, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy G., Ananvoranich,S. and Perreault,J.P. (1999) Delta ribozyme has the ability to cleave in trans an mRNA. Nucleic Acids Res., 27, 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shih I.H. and Been,M.D. (2001) Energetic contribution of non-essential 5′ sequence to catalysis in a hepatitis delta virus ribozyme. EMBO J., 20, 4884–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergeron L. Jr and Perreault,J.P. (2002) Development and comparison of procedures for the selection of delta ribozyme cleavage sites within the hepatitis B virus. Nucleic Acids Res., 30, 4682–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]