

Abstract
Osteoarthritis (OA), one of the most common age-related chronic disorders of articular cartilage, joints, and bone tissue, represents a major public health problem. Genetic studies have identified multiple gene variations associated with an increased risk of OA. These findings suggest that there is a large genetic component to OA and that the disorder belongs in the multigenetic, multifactorial class of genetic diseases. Studies of chondrodysplasias and associated hereditary OA have provided a better understanding of the role of structural genes in the maintenance and repair of articular cartilage, in the regulation of chondrocyte proliferation and gene expression, and in the pathogenesis of OA.
Keywords: cartilage, chromosomes, genetics, linkage, osteoarthritis
Introduction
Osteoarthritis (OA), the most common form of arthritis, is no longer regarded as a simple consequence of age-related cartilage degeneration, but rather is regarded as the result of an active process, which may be regenerative rather than degenerative in nature. Furthermore, OA is probably not a single disorder, but rather a group of overlapping distinct diseases. These diseases are the consequences of mechanical or biological events that destabilize the normal coupling of synthesis and degradation of extracellular matrix in articular cartilage and subchondral bone. It is commonly assumed that multiple factors, including genetic and developmental, metabolic, and traumatic factors can trigger osteoarthritic disease. At later stages, the disease is characterized by molecular, morphological, and biomechanical changes which lead to softening, fibrillation, ulceration, and loss of articular cartilage, eburnation of subchondral bone, osteophytes, and subchondral bone cysts [1]. Extracellular matrix molecules play a critical role in the normal maintenance of articular cartilage structure, regulation of chondrocyte proliferation and gene expression, and cartilage aging and repair, and they are important in the pathophysiology of OA.
Articular cartilage is composed of an extracellular matrix designed to resist tensile and compressive forces and provide a smooth surface to permit low-friction movement in joints. These properties are the result of the interactions between a large number of proteins and proteoglycans present in the matrix (Fig. 1). Many of these components are listed in Table 1; this list, which is by no means exhaustive, includes those components that have been studied the most. Type II collagen is the major collagenous component, but collagens III, VI, IX, X, XI, XII, and XIV also contribute to the mature cartilage matrix. Noncollagenous components include large amounts of the hyaluronate-binding proteoglycan aggrecan and its associated link protein, as well as other collagen-binding proteoglycans, such as decorin, fibromodulin, and lumican, and proteins such as PRELP (proline/arginine-rich and leucine-rich repeat protein) and cartilage oligomeric matrix protein (COMP) [2]. The structure and abundance of these components change with age because of a combination of changes in both synthetic and degradative events [3]. The effects of mutations in the genes encoding these structural components of the matrix have provided insight into the function of the individual gene products in the pathogenesis of OA.
Figure 1.
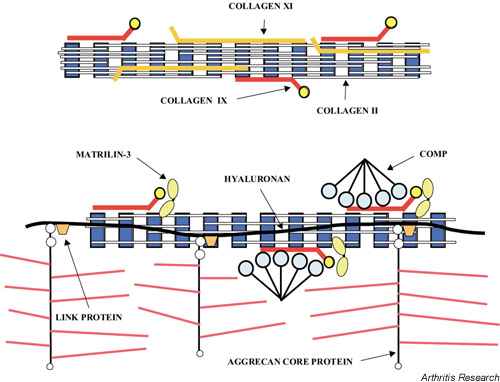
Diagram showing the collagen components (collagens II, IX and XI) of cartilage fibril (top) and the association between the fibril and noncollagenous components of cartilage, such as matrilin-3, COMP, and complexes of aggrecan, link protein and hyaluronan (bottom). COMP, cartilage oligomeric matrix protein.
Table 1.
The most-studied components of cartilage matrix
| Collagens |
| Types IIa and IIb |
| Type III |
| Type VI |
| Type IX |
| Type X |
| Type XI |
| Type XII |
| Type XIV |
| Proteoglycans |
| Aggrecan |
| Versican |
| Link protein |
| Biglycan (DS-PGI) |
| Decorin (DS-PGII) |
| Epiphycan (DS-PGIII) |
| Fibromodulin |
| Lumican |
| Perlecan |
| Proteoglycan 4 |
| Noncollagenous proteins |
| COMP (cartilage oligomeric matrix protein; thrombospondin-5) |
| CMP (cartilage matrix protein; matrilin-1) |
| Matrilin-3 |
| CILP (cartilage intermediate layer protein) |
| Fibronectin |
| PRELP (proline/arginine-rich and leucine-rich repeat protein) |
| Chondroadherin |
| Fibrillin |
| Tenascin-C |
| Elastin |
| MGP (matrix Gla protein) |
| Chondromodulin-I |
| Chondromodulin-II |
| CD-RAP (cartilage-derived retinoic acid-sensitive protein) |
| ANKH |
| Matrix metalloproteinases and related enxymes |
| Tissue inhibitors of metalloproteinases |
Large genetic component in osteoarthritis
Twin studies and cohort studies have highlighted a surprisingly large genetic component to OA [4-6]. These findings have prompted the search for predisposing genes using parametric linkage analyses of rare families in which OA segregates as a Mendelian trait, model-free linkage analysis of affected sibling pairs, and association analysis of known candidate genes. Linkage studies have highlighted chromosomes 1, 2, 4, 6, 7, 9,11–13,16,19, and X as potential chromosomes with OA susceptibility genes [7-15]. Chromosomes 2,4,7,11, and 16 have been identified in multiple genome-wide scans and are therefore the most likely candidates. In addition, association analysis of candidate genes suggests that COL2A1 (chromosome12), COL1A1 (chromosome17), COL9A1 (chromosome6), COL11A2 (chromosome6), CMP (cartilage matrix protein) (chromosome1), VDR (vitamin D receptor) (chromosome12), ER (estrogen receptor) (chromosome6), IGF-1 (insulin-like growth factor 1) (chromosome12), aggrecan (chromosome15), and TGFβ1 (chromosome19) may represent 'osteoarthritic genes' [16-25]. These findings support the notion that there is a large genetic component to OA and that the disease can be classified as a multigenetic, multifactorial genetic disease.
Hereditary osteoarthritis
Mutations in genes encoding molecules expressed in cartilagenous tissue lead to hereditary OA [26,27]. The phenotypic spectrum is quite broad, encompassing very severe forms, which become manifest early in life, to mild disorders, which become clinically evident only late in life. Hereditary OA can be subdivided into conditions such as early-onset OA associated with an underlying familial osteochondrodysplasia, conditions associated with metabolic joint diseases including crystal-associated arthropathies (familial calcium pyrophosphate deposition disease and familial hydroxyapatite deposition disease), and primary generalized osteoarthritis (PGOA) with mild dysplasia [26,27]. The familial osteochondrodysplasias represent a heterogeneous group of disorders characterized by abnormalities in the development and growth of articular and growth-plate cartilages. The term 'chondrodysplastic rheumatism' has been used to refer to these forms of OA. They include the type II and type XI collagenopathies, the multiple epiphyseal dysplasias (MEDs), and the metaphyseal chondrodysplasias. They are usually inherited as fully penetrant autosomal dominant disorders. The expression of the OA phenotype occurs early in life, with distinct radiographic changes that are different from the nondysplastic forms of inherited OA.
Families with PGOA without dysplasia exhibit a higher incidence of OA than is seen in the general population, with premature development of Heberden's and Bouchard's nodes and cartilage degeneration at multiple joints [28]. Early studies showed that first-degree relatives of PGOA probands were twice as likely to have radiographically visible generalized disease as a control population [29].
Structural genes responsible for familial osteochondrodysplasias were initially considered strong candidates also for PGOA, on the basis that mutations in such genes could cause a spectrum of phenotypes from the most severe chondrodysplasia with associated OA to a very mild phenotype of OA only. For example, several point mutations in the COL2A1 gene have been identified as causes of severe forms of type II collagenopathies [30], but mutations in this gene are also associated with milder phenotypes. Thus, mutations in the COL2A1 gene have been identified in PGOA/late-onset spondyloepiphyseal dysplasia (SED). Mutations resulting in an Arg519Cys substitution have been identified in several unrelated families with PGOA and mild chondrodysplasia [31-34]. The Arg75Cys change has also been identified in a large family with severe OA, crystal deposition disease, and late-onset SED [35]. It is likely that the chondrocalcinosis phenotype in this family is a secondary consequence of the advanced and severe OA, since structural changes in the articular cartilage extracellular matrix may predispose to crystal formation [36,37]. Other heterozygous substitutions in type II collagen, Gly976Ser [38] and Gly493Ser [39], have been identified in other kindreds with mild dysplasia and early-onset OA.
More recent studies have not supported these initial ideas. In fact, the loci identified on chromosomes 2, 4, 7, 11, and 16 as the most likely candidates for OA susceptibility genes do not in general correspond to structural matrix genes. Furthermore, although a recent cohort study found that a specific COL2A1 haplotype seemed to predispose to generalized radiographic OA [23], linkage analysis of 14 candidate genes in OA kindreds resulted in the exclusion of 10 important cartilage genes, including COL2A1 [40]. Analysis of 47 families with the phenotype of early-onset primary OA without an SED phenotype failed to detect mutations in the COL2A1 in most families, with the exception of one case [41]. Other studies using multiallelic polymorphism and genetic analysis of sibling pairs failed to identify COL2A1 as the disease locus in families with features of PGOA in the absence of SED [42,43]. Therefore, a majority of patients with PGOA are likely to have mutations in genes other than COL2A1 and other structural matrix genes.
Familial osteochondrodysplasias
OA associated with familial osteochondrodysplasias caused by mutations in (mostly) structural cartilage matrix genes represents a subset of secondary OA. Studies of this form of OA, however, are important as they provide insights into the molecular processes that lead to articular cartilage destruction in all forms of OA.
Table 2 summarizes genetic defects in structural macromolecules of cartilage that lead to alterations in chondrogenesis, skeletal malformations, reduced skeletal function, or predisposition to injury and OA. Mutations in type II collagen cause a spectrum of diseases known as 'type II collagenopathies'. The severity ranges from developmental lethality (achondrogenesis type II, hypochondrogenesis), to moderately severe dwarphism (SED, Kniest dysplasia), to normal stature with premature OA. In the case of lethal mutations with absence of type II collagen, some embryonic cartilage develops with type I collagen susbstituted for type II collagen, and bone is formed [44]. Mutations that cause a moderately severe phenotype (SED, Kniest dysplasia) generally result from reduced secretion of typeII collagen or reduced content of this collagen in cartilage [45-47]. The mildest phenotype (Stickler syndrome, typeI) is caused by premature stop codons resulting in a null-allele mosaicism [47,48] and is associated with early-onset OA. Several mouse models of the human type II collagenopathies exist and resemble, depending on the type and position of the mutation in the COL2A1 gene, a lethal or a mild human phenotype [49-52].
Table 2.
Structural gene mutations in cartilage that result in abnormal cartilage matrix
| OMIM# | Gene name | Gene symbol | Diseases and disorders |
| 12140 | Collagen, type II α1 | COL2A1 | Achondrogenesis, type II |
| Achondrogenesis-hypochondrogenesis, type II | |||
| Epiphyseal dysplasia, multiple, with myopia and conductive deafness | |||
| Hypochondrogenesis | |||
| Kniest dysplasia | |||
| Osteoarthritis with mild dysplasia | |||
| Spondyloepiphyseal dysplasia, congenital type | |||
| Spondyloepiphyseal dysplasia, Namaqualand type | |||
| Spondyloepiphyseal dysplasia, Strudwick type | |||
| Spondyloepiphyseal dysplasia, various types | |||
| Spondyloepiphyseal dysplasia with precocious OA | |||
| Spondyloperipheral dysplasia | |||
| Stickler syndrome, type I | |||
| Wagner syndrome | |||
| 120180 | Collagen, type III α1 | COL3A1 | Arterial and aortic aneurysm |
| Ehlers–Danlos syndrome, types III and IV | |||
| 120220 | Collagen, type VI α1 | COL6A1 | Bethlem myopathy |
| 120240 | Collagen, type VI α2 | COL6A2 | Bethlem myopathy |
| Ullrich scleroatonic muscular dystrophy | |||
| 120250 | Collagen, type VI α3 | COL6A3 | Bethlem myopathy |
| 120210 | Collagen, type IX α1 | COL9A1 | Epiphyseal dysplasia, multiple, type 1 |
| Intervertebral disk disease | |||
| 120260 | Collagen, type IX α2 | COL9A2 | Epiphyseal dysplasia, multiple, type 2 |
| Intervertebral disk disease | |||
| 120270 | Collagen, type IX α3 | COL9A3 | Epiphyseal dysplasia, multiple, type 3 |
| Epiphyseal dysplasia, multiple, with myopathy | |||
| 120110 | Collagen, type X α1 | COL10A1 | Metaphyseal chondrodysplasia, Schmid type |
| Spondylometaphyseal dysplasia, Japanese type | |||
| 120260 | Collagen, type XI α1 | COL11A1 | Stickler syndrome, type II |
| Marshall syndrome | |||
| 120290 | Collagen, type XI α2 | COL11A2 | Sensorineural deafness, autosomal dominant nonsyndromic |
| Otospondylomegaepiphyseal dysplasia | |||
| Stickler syndrome, type III | |||
| Weissenbacher–Zweymuller syndrome | |||
| 600310 | Cartilage oligomeric matrix protein | COMP | Pseudoachondroplasia |
| Epiphyseal dysplasia, multiple, Fairbanks type | |||
| Epiphyseal dysplasia, multiple, type 1 | |||
| 602109 | Matrilin-3 | MATN3 | Multiple epiphyseal dysplasia, MATN3-related |
| Epiphyseal dysplasia, multiple, type 5 | |||
| 134797 | Fibrillin | FBN1 | Marfan syndrome, various type |
| Ectopia lentis, familial | |||
| Marfanoid skeletal syndrome | |||
| MASS syndrome | |||
| Shprintzen–Goldberg syndrome | |||
| 154870 | Matrix γ-carboxyglutamic acid protein | MGP | Keutel syndrome |
| 142461 | Perlecan | PLC | Schwartz–Jampel syndrome, type 1 |
| Dyssegmental dysplasia, Silverman–Handmaker type | |||
| Chondrodystrophic myotonia | |||
| 222600 | Diastrophic dysplasia sulfate transporter | DTDST | Achondrogenesis IB |
| Atelostogenesis type II | |||
| Diastrophic dysplasia | |||
| Epiphyseal dysplasia, multiple type 4 | |||
| Diastrophic dysplasia, broad-bone–platyspondylic variant | |||
| 604283 | Proteoglycan 4 | PRG4 | Camptodactyly–arthropathy–coxa vara–pericarditis syndrome |
| 605145 | ANK | ANKH | Craniometaphyseal dysplasia, autosomal dominant |
| Chondrocalcinosis 2 |
MASS, mitral valve, aorta, skeleton, skin; OA, osteoarthritis; OMIM, Online Mendelian Inheritance in Man™.
Mutations in genes for type IX collagen result in MED. This clinically heterogeneous disorder is characterized by mild short stature and early-onset OA. In some families, splice-site mutations in COL9A2 and COL9A3 causing skipping of exon 3 in α2(IX) and α3(IX) transcripts, respectively [53-58], are associated with a phenotype characterized by normal to near-normal height, epiphyseal dysplasia of several joints during childhood, and OA of the knees in adulthood. A complex splicing defect of exon 8 and/or exon 10 in the COL9A1 gene has been identified in other families [59]. Consistent with these findings, transgenic mice overexpressing a truncated α1(IX) chain (exerting a dominant negative effect on assembly of collagen IX molecules) exhibited mild chondrodysplasia and progressive OA [60]. A similar phenotype was seen in mice carrying two null alleles for COL9A1 [61]. Type IX collagen molecules are heterotrimers composed of three different polypeptide chains, α1(IX), α2(IX), and α3(IX), which are localized on the surface of collagen-II-containing fibrils, where they get cross-linked to residues within type II collagen molecules and may help stabilize the fibrillar network [62]. The absence of α1(IX) chains or expression of a dominant negative form results in the absence or reduced amounts of collagen IX in cartilage, and this may destabilize the collagen network.
Mutations in the COL11A1 and COL11A2 genes, encoding two of the polypeptide subunits of heterotrimeric collagen XI molecules, give rise to the 'type XI collagenopathies'. COL11A1 mutations are associated with Marshall or Stickler syndrome, characterized by severe myopia, vitreoretinal degeneration, cleft palate, midfacial hypoplasia, early-onset OA, and sensorineuronal hearing loss [63-65]. Extensive genotype–phenotype correlations of patients with Stickler, Stickler-like, or Marshall syndrome have suggested that null-allele mutations in COL2A1, encoding the polypeptide chains of collagen II molecules as well as one of the chains in collagen XI, cause the typical Stickler phenotype (in which vitreoretinal degeneration is common and hearing loss is less common), while splicing mutations in COL11A1 are responsible for the Marshall syndrome (in which hearing loss is common and vitreoretinal degeneration is less common). Patients with glycine substitutions or small deletions in COL11A1 (dominant negative mutations) may have a mixed phenotype characteristic of both syndromes [66]. Mutations in COL11A2 are associated with a nonocular Stickler-like syndrome, otospondylomegaepiphyseal dysplasia [67-70], Weissenbacher–Zweymuller syndrome [69], or nonsyndromic forms of deafness called DFNA13 (deafness, autosomal dominant nonsyndromic sensorineural 13) [71]. The explanation for the lack of an ocular phenotype in these syndromes is the presence of a unique form of type XI collagen in the vitreous. In cartilage, collagen XI molecules are heterotrimers of the products of COL11A1, COL11A2, and COL2A1, but in the vitreous the COL11A2 chain is replaced by a chain encoded by COL5A2 [72].
The cho/cho mouse, which is homozygous for a premature stop codon in the amino-terminal region of α1(XI) collagen [73], has provided important insights into the role of collagen XI in skeletal development. Heterozygous animals are relatively unaffected; however, with age they develop ostearthritis. Homozygous animals die at birth, with short limbs, short snout, and cleft palate. Growth-plate cartilages show a disorganized structure with thick collagen fibrils. The presence of thick fibrils, providing direct evidence of a role of type XI collagen in regulating fibril diameters, leads to the formation of a large-pore network of fewer fibrils in cho/cho cartilage than the small-pore network of thin fibrils found in wild-type cartilage. The changes in pore size causes the proteoglycan aggregates to be more loosely entrapped within the mutant matrix than in the wild type. Mice with Col11a2 null alleles are phenotypically comparable with patients who have otospondylomegaepiphyseal dysplasia, and their phenotype suggests that the α2(XI) chain may be required for correct fibril assembly or lateral association between individual collagen fibrils [74].
Mutations that prevent glycosaminoglycan sulfation of aggrecan cause chondrodysplastic phenotypes associated with achondrogenesis, atelostogenesis, diastrophic dysplasia, and autosomal recessive MED [75]. MED and pseudoachondroplasia can also be the result of mutations in COMP, a pentameric molecule belonging to the thrombospondin family of matrix molecules and which is localized in the pericellular, territorial matrix of chondrocytes. The protein contains several repeat domains, including eight calcium-binding, calmodulin-like repeats. Most COMP mutations identified in patients with MED or pseudoachondroplasia are amino acid substitutions that may disturb calcium binding [76-78]. Mutations in the gene encoding matrilin-3 [79] can also give an MED clinical phenotype, a phenomenon which provides genetic evidence for a functional interaction between aggrecan, COMP, collagen IX, and matrilin-3 in cartilage [80].
Mutations in a gene encoding the core protein of a proteoglycan associated with articular cartilage cause camptodactyly–arthropathy–coxa vara–pericarditis syndrome [81]. This chondroitin sulfate proteoglycan, called superficial zone protein, lubricin, or proteoglycan 4, is produced by the superficial articular chondrocytes and synovial cells. It is responsible for lubrication of the cartilage surface [82]. The synthesis is impaired in arthritic joints and downregulated by inflammatory cytokines such as IL-1.
Mutation at the progressive ankylosis (ank) locus in the mouse causes a progressive form of arthritis with deposition of apatite crystals, formation of bony outgrowths, joint destruction, and ankylosis [83]. In humans, mutations in the ank gene, ANKH, have been linked to craniometaphyseal dysplasia [84,85]. In addition, analysis of two families from England and Argentina with familial calcium pyrophosphate deposition disease have identified mutations in ANKH [86,87]. An early-onset form of this disease with severe PGOA has been linked also to a region on chromosome 8q [88].
Interactions between structural genes and environment
In addition to specific structural gene mutations, well-established risk factors for OA include trauma, aging, obesity, and gender [89,90]. This raises the question of whether OA caused by mutations in structural genes is the result of changes in articular cartilage that are fundamentally very different from those that underlie the age-related, obesity-related, or trauma-related disease. We do not believe this is the case and argue that the clearly defined genetic abnormalities in structural components of articular cartilage result in OA simply because they lower the threshold at which biomechanical stress on the joint induces the cascade of cellular and molecular events that define this disease. For example, mutations in the structural components of cartilage may alter matrix-cell interactions and thereby cellular responses to cytokines, leading to apoptosis and matrix destruction. Alternatively, the chondrocyte response to mechanical stress in an abnormal matrix structure may result in different patterns of structural protein expression, dedifferentiation, hypertrophy, regeneration, and an abnormal pyrophosphate synthesis [91]. In addition, mutations in structural genes may lead to changes in molecular interactions between various components of the extracellular matrix, altering the thickness and three-dimensional organization of the cartilage collagen fibrils and destabilizing the cartilaginous matrix [92-94]. One of the curious features of OA associated with mutations in genes encoding structural matrix proteins is the selectivity with which different joints may be affected. Thus, affected members of families with OA as a result of mutations in COL9A2 show primarily knee OA, while mutations in COL11A2 may affect hip joints. Since collagens IX and XI are coexpressed with collagen II in the articular cartilage of all joints, this joint selectivity is quite puzzling. We suggest that the answer may be found in a more careful examination of the interactions between intrinsic and extrinsic factors that contribute to joint development and function.
Extrinsic factors such as physical activity, occupation, and trauma, along with intrinsic factors such as abnormal joint alignment, joint hypermobility, decreased muscle strength, and varus–valgus deformity, may alter the biomechanics and loading stress conditions of various joints in different ways [95] and play a critical role in the selection of genetically susceptible joints.
Conclusion
OA is a genetically complex disorder. Mutations in genes encoding structural components of articular cartilage give rise to rare forms of highly penetrant inherited diseases that are associated with early-onset OA, whereas the more common forms of the same disease that occurs with increased frequency at an older age are associated with genetic risk factors in the form of common population polymorphisms. As more mutations in the structural genes of articular cartilage are identified, careful clinical analyses will be required to understand the genotype–phenotype spectrum of these forms of hereditary OA. As with other multifactorial diseases, the initiation, progression, and severity of the OA disorder may be influenced by multiple environmental, hormonal, and intrinsic and extrinsic factors, with multiple genes in any given individual. The identification of the genetic pathways will be difficult and will represent a great challenge in the near future. Of critical importance are studies to understand the gene–gene and gene–environment interactions, using animal models. These efforts should provide a better understanding of the pathogenesis of OA as well as a basis for developing earlier preventive strategies and providing targets for the development of new forms of treatment.
Abbreviations
ANKH = human homologue of the mouse progressive ankylosis (ank) gene; COMP = cartilage oligomeric matrix protein; CMP = cartilage matrix protein; IL = 1-interleukin 1; MED = multiple epiphyseal dysplasia; OA = osteoarthritis; PGOA = primary generalized osteoarthritis; SED = spondyloepiphyseal dysplasia; TGFβ1 = transforming growth factor beta 1.
Acknowledgments
Acknowledgements
We thank Mrs Y Pittel for patient and expert editorial assistance. This work was supported by grants from the National Institutes of Health AR36819 and AR36820 (to BR Olsen) and by an Arthritis Foundation Arthritis Investigator Award (to AM Reginato).
References
- Poole AR, Kojima T, Yasuda T, Mwale F, Kobayashi M, Laverty S. Composition and structure of articular cartilage: a template for tissue repair. Clin Orthop. 2001;391:S153–S160. doi: 10.1097/00003086-200110001-00004. [DOI] [PubMed] [Google Scholar]
- Poole AR. Cartilage in health and disease. In: Koopman W, editor. In Arthritis and Allied Conditions A Textbook of Rheumatology. 14. New York: Lippincott Williams and Wilkins; 2001. pp. 2260–2284. [Google Scholar]
- Roughley PJ. Age-associated changes in cartilage matrix: implications for tissue repair. Clin Orthop. 2001;391:S153–160. doi: 10.1097/00003086-200110001-00015. [DOI] [PubMed] [Google Scholar]
- Spector TD, Cicuttini F, Baker J, Loughlin J, Hart D. Genetic influences on osteoarthritis in women: a twin study. BMJ. 1996;312:940–943. doi: 10.1136/bmj.312.7036.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felson DT, Couropmitree NN, Chaisson CE, Hannan MT, Zhang Y, McAlindon TE, LaValley M, Levy D, Myers RH. Evidence for a Mendelian gene in a segregation analysis of generalized radiographic osteoarthritis: the Framingham Study. Arthritis Rheum. 1998;41:1064–1071. doi: 10.1002/1529-0131(199806)41:6<1064::AID-ART13>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Hirsch R, Lethbridge-Cejku M, Hanson R, Scott WW, Jr, Reichle R, Plato CC, Tobin JD, Hochberg MC. Familial aggregation of osteoarthritis: data from the Baltimore Longitudinal Study on Aging. Arthritis Rheum. 1998;41:1227–1232. doi: 10.1002/1529-0131(199807)41:7<1227::AID-ART13>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Wright GD, Hughes AE, Regan M, Doherty M. Association of two loci on chromosome 2q with nodal osteoarthritis. Ann Rheum Dis. 1996;55:317–319. doi: 10.1136/ard.55.5.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppavuori J, Kujala U, Kinnunen J, Kaprio J, Nissila M, Heliovaara M, Klinger N, Partanen J, Terwilliger JD, Peltonen L. Genome scan for predisposing loci for distal interphalangeal joint osteoarthritis: evidence for a locus on 2q. Am J Hum Genet. 1999;65:1060–1067. doi: 10.1086/302569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin J, Mustafa Z, Irven C, Smith A, Carr AJ, Sykes B, Chapman K. Stratification analysis of an osteoarthritis genome screen-suggestive linkage to chromosomes 4, 6, and 16. Am J Hum Genet. 1999;65:1795–1798. doi: 10.1086/302685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman K, Mustafa Z, Irven C, Carr AJ, Clipsham K, Smith A, Chitnavis J, Sinsheimer JS, Bloomfield VA, McCartney M, Cox O, Cardon LR, Sykes B, Loughlin J. Osteoarthritis-susceptibility locus on chromosome 11q, detected by linkage. Am J Hum Genet. 1999;65:167–174. doi: 10.1086/302465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roby P, Eyre S, Worthington J, Ramesar R, Cilliers H, Beighton P, Grant M, Wallis G. Autosomal dominant (Beukes) premature degenerative osteoarthropathy of the hip joint maps to an 11-cM region on chromosome 4q35. Am J Hum Genet. 1999;64:904–908. doi: 10.1086/302291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa Z, Chapman K, Irven C, Carr AJ, Clipsham K, Chitnavis J, Sinsheimer JS, Bloomfield VA, McCartney M, Cox O, Sykes B, Loughlin J. Linkage analysis of candidate genes as susceptibility loci for osteoarthritis-suggestive linkage of COL9A1 to female hip osteoarthritis. Rheumatology (Oxford) 2000;39:299–306. doi: 10.1093/rheumatology/39.3.299. [DOI] [PubMed] [Google Scholar]
- Loughlin J, Mustafa Z, Smith A, Irven C, Carr AJ, Clipsham K, Chitnavis J, Bloomfield VA, McCartney M, Cox O, Sinsheimer JS, Sykes B, Chapman KE. Linkage analysis of chromosome 2q in osteoarthritis. Rheumatology (Oxford) 2000;39:377–381. doi: 10.1093/rheumatology/39.4.377. [DOI] [PubMed] [Google Scholar]
- Ingvarsson T, Stefansson SE, Gulcher JR, Jonsson HH, Jonsson H, Frigge ML, Palsdottir E, Olafsdottir G, Jonsdottir T, Walters GB, Lohmander LS, Stefansson K. A large Icelandic family with early osteoarthritis of the hip associated with a susceptibility locus on chromosome 16p. Arthritis Rheum. 2001;44:2548–2555. doi: 10.1002/1529-0131(200111)44:11<2548::aid-art435>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Demissie S, Cupples LA, Myers R, Aliabadi P, Levy D, Felson DT. Genome scan for quantity of hand osteoarthritis: the Framingham Study. Arthritis Rheum. 2002;46:946–52. doi: 10.1002/art.10149. [DOI] [PubMed] [Google Scholar]
- Loughlin J, Irven C, Athanasou N, Carr A, Sykes B. Differential allelic expression of the type II collagen gene (COL2A1) in osteoarthritic cartilage. Am J Hum Genet. 1995;56:1186–1193. [PMC free article] [PubMed] [Google Scholar]
- Loughlin J, Sinsheimer JS, Mustafa Z, Carr AJ, Clipsham K, Bloomfield VA, Chitnavis J, Bailey A, Sykes B, Chapman K. Association analysis of the vitamin D receptor gene, the type I collagen gene COL1A1, and the estrogen receptor gene in idiopathic osteoarthritis. J Rheumatol. 2000;27:779–784. [PubMed] [Google Scholar]
- Ushiyama T, Ueyama H, Inoue K, Nishioka J, Ohkubo I, Hukuda S. Estrogen receptor gene polymorphism and generalized osteoarthritis. J Rheumatol. 1998;25:134–137. [PubMed] [Google Scholar]
- Uitterlinden AG, Burger H, Huang Q, Odding E, Duijn CM, Hofman A, Birkenhager JC, van Leeuwen JP, Pols HA. Vitamin D receptor genotype is associated with radiographic osteoarthritis at the knee. J Clin Invest. 1997;100:259–263. doi: 10.1172/JCI119530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitterlinden AG, Burger H, van Duijn CM, Huang Q, Hofman A, Birkenhager JC, van Leeuwen JP, Pols HA. Adjacent genes, for COL2A1 and the vitamin D receptor, are associated with separate features of radiographic osteoarthritis of the knee. Arthritis Rheum. 2000;43:1456–1464. doi: 10.1002/1529-0131(200007)43:7<1456::AID-ANR7>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Keen RW, Hart DJ, Lanchbury JS, Spector TD. Association of early osteoarthritis of the knee with a Taq I polymorphism of the vitamin D receptor gene. Arthritis Rheum. 1997;40:1444–1449. doi: 10.1002/art.1780400812. [DOI] [PubMed] [Google Scholar]
- Meulenbelt I, Bijkerk C, Miedema HS, Breedveld FC, Hofman A, Valkenburg HA, Pols HA, Slagboom PE, van Duijn CM. A genetic association study of the IGF-1 gene and radiological osteoarthritis in a population-based cohort study (the Rotterdam Study). Ann Rheum Dis. 1998;57:371–374. doi: 10.1136/ard.57.6.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenbelt I, Bijkerk C, De Wildt SC, Miedema HS, Breedveld FC, Pols HA, Hofman A, Van Duijn CM, Slagboom PE. Haplotype analysis of three polymorphisms of the COL2A1 gene and associations with generalised radiological osteoarthritis. Ann Hum Genet. 1999;63:393–400. doi: 10.1046/j.1469-1809.1999.6350393.x. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Okuizumi H, Miyauchi A, Takagi Y, Ikeda K, Harada A. Association of transforming growth factor beta1 genotype with spinal osteophytosis in Japanese women. Arthritis Rheum. 2000;43:452–460. doi: 10.1002/1529-0131(200002)43:2<452::AID-ANR28>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Horton WE, Jr, Lethbridge-Cejku M, Hochberg MC, Balakir R, Precht P, Plato CC, Tobin JD, Meek L, Doege K. An association between an aggrecan polymorphic allele and bilateral hand osteoarthritis in elderly white men: data from the Baltimore Longitudinal Study of Aging (BLSA). Osteoarthritis Cartilage. 1998;6:245–251. doi: 10.1053/joca.1998.0117. [DOI] [PubMed] [Google Scholar]
- Jimenez SA, Williams CJ, Karasick D. Hereditary osteoarthritis. In: Brandt KD, Doherty M, Lohmander LS, editor. In Osteoarthritis. Oxford: Oxford University Press; 1998. pp. 31–49. [Google Scholar]
- Jimenez SA, Williams CJ. Genetic and metabolic aspects. In: Reginster J-Y, Pelletier J-P, Martel-Pelletier J, Henrotin Y, editor. In Osteoarthritis: Clinical and Experimental Aspects. Berlin, New York: Springer; 1999. pp. 134–156. [Google Scholar]
- Kellgren JH, Moore R. Generalized osteoarthritis and Heberden's nodes. Brit Med J. 1952;1:181–187. doi: 10.1136/bmj.1.4751.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellgren JH, Lawrence JS, Bier F. Genetic factors in generalized osteoarthritis. Ann Rheum Dis. 1963;22:237–255. doi: 10.1136/ard.22.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuivaniemi H, Tromp G, Prockop DJ. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat. 1997;9:300–315. doi: 10.1002/(SICI)1098-1004(1997)9:4<300::AID-HUMU2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Ala-Kokko L, Baldwin CT, Moskowitz RW, Prockop DJ. Single base mutation in the type II procollagen gene (COL2A1) as a cause of primary osteoarthritis associated with a mild chondrodysplasia. Proc Natl Acad Sci USA. 1990;87:6565–6568. doi: 10.1073/pnas.87.17.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleasel JF, Holderbaum D, Brancolini V, Moskowitz RW, Considine EL, Prockop DJ, Devoto M, Williams CJ. Five families with arginine 519-cysteine mutation in COL2A1: evidence for three distinct founders. Hum Mutat. 1998;12:172–176. doi: 10.1002/(SICI)1098-1004(1998)12:3<172::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Williams CJ, Considine EL, Knowlton RG, Reginato A, Neumann G, Harrison D, Buxton P, Jimenez S, Prockop DJ. Spondyloepiphyseal dysplasia and precocious osteoarthritis in a family with an Arg75→Cys mutation in the procollagen type II gene (COL2A1). Hum Genet. 1993;92:499–505. doi: 10.1007/BF00216458. [DOI] [PubMed] [Google Scholar]
- Bleasel JF, Bisagni-Faure A, Holderbaum D, Vacher-Lavenu MC, Haqqi TM, Moskowitz RW, Menkes CJ. Type II procollagen gene (COL2A1) mutation in exon 11 associated with spondyloepiphyseal dysplasia, tall stature and precocious osteoarthritis. J Rheumatol. 1995;22:255–261. [PubMed] [Google Scholar]
- Reginato AJ, Passano GM, Neumann G, Falasca GF, Diaz-Valdez M, Jimenez SA, Williams CJ. Familial spondyloepiphyseal dysplasia tarda, brachydactyly, and precocious osteoarthritis associated with an arginine 75→cysteine mutation in the procollagen type II gene in a kindred of Chiloe Islanders. I. Clinical, radiographic, and pathologic findings. Arthritis Rheum. 1994;37:1078–1086. doi: 10.1002/art.1780370714. [DOI] [PubMed] [Google Scholar]
- Bjelle AO. Morphological study of articular cartilage in pyrophosphate arthropathy. (Chondrocalcinosis articularis or calcium pyrophosphate dihydrate crystal deposition diseases). Ann Rheum Dis. 1972;6:449–456. doi: 10.1136/ard.31.6.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelle A. Cartilage matrix in hereditary pyrophosphate arthropathy. J Rheumatol. 1981;8:959–964. [PubMed] [Google Scholar]
- Williams CJ, McCarron S, Considine E. A point mutation in one allele of the type II procollagengene produces a Gly976-Ser substitution of the gene in a family with severe degenerative arthropathy of the associated with probable epiphyseal dysplasia. Am J Hum Genet. 1993;53:A1252. [Google Scholar]
- Karzentein PL, Campbell DF, Machado MA, Horton WA, Lee B, Ramirez P. A type II collagen defect in a new family with SED tarda and early-onset osteoarthritis (OA) [abstract]. Arthritis Rheum. 1992;35:S41. [Google Scholar]
- Meulenbelt I, Bijkerk C, Breedveld FC, Slagboom PE. Genetic linkage analysis of 14 candidate gene loci in a family with autosomal dominant osteoarthritis without dysplasia. J Med Genet. 1997;34:1024–1027. doi: 10.1136/jmg.34.12.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritvaniemi P, Korkko J, Bonaventure J, Vikkula M, Hyland J, Paassilta P, Kaitila I, Kaariainen H, Sokolov BP, Hakala M, Pertti M, Meerson EM, Klemola T, Williams C, Peltonen L, Kivirikko KI, Prockop DJ, Ala-Kokko LA. Identification of COL2A1 gene mutations in patients with chondrodysplasias and familial osteoarthritis. Arthritis Rheum. 1995;38:999–1004. doi: 10.1002/art.1780380717. [DOI] [PubMed] [Google Scholar]
- Vikkula M, Nissila M, Hirvensalo E, Nuotio P, Palotie A, Aho K, Peltonen L. Multiallelic polymorphism of the cartilage collagen gene: no association with osteoarthrosis. Ann Rheum Dis. 1993;52:762–764. doi: 10.1136/ard.52.10.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin J, Irven C, Fergusson C, Sykes B. Sibling pair analysis shows no linkage of generalized osteoarthritis to the loci encoding type II collagen, cartilage link protein or cartilage matrix protein. Br J Rheumatol. 1994;33:1103–1106. doi: 10.1093/rheumatology/33.12.1103. [DOI] [PubMed] [Google Scholar]
- Chan D, Cole WG, Chow CW, Mundlos S, Bateman JF. A COL2A1 mutation in achondrogenesis type II results in the replacement of type II collagen by type I and III collagens in cartilage. J Biol Chem. 1995;270:1747–1753. [PubMed] [Google Scholar]
- Chan D, Cole WG. Low basal transcription of genes for tissue-specific collagens by fibroblasts and lymphoblastoid cells. Application to the characterization of a glycine 997 to serine substitution in alpha 1(II) collagen chains of a patient with spondyloepiphyseal dysplasia. J Biol Chem. 1991;266:12487–12494. [PubMed] [Google Scholar]
- Chan D, Taylor TK, Cole WG. Characterization of an arginine 789 to cysteine substitution in alpha 1 (II) collagen chains of a patient with spondyloepiphyseal dysplasia. J Biol Chem. 1993;268:15238–15245. [PubMed] [Google Scholar]
- Winterpacht A, Hilbert M, Schwarze U, Mundlos S, Spranger J, Zabel BU. Kniest and Stickler dysplasia phenotypes caused by collagen type II gene (COL2A1) defect. Nat Genet. 1993;3:323–326. doi: 10.1038/ng0493-323. [DOI] [PubMed] [Google Scholar]
- Ahmad NN, Ala-Kokko L, Knowlton RG, Jimenez SA, Weaver EJ, Maguire JI, Tasman W, Prockop DJ. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci USA. 1991;88:6624–6627. doi: 10.1073/pnas.88.15.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg P, Khillan JS, Prockop DJ, Helminen H, Kontusaari S, Ala-Kokko L. Expression of a partially deleted gene of human type II procollagen (COL2A1) in transgenic mice produces a chondrodysplasia. Proc Natl Acad Sci USA. 1991;88:7640–7644. doi: 10.1073/pnas.88.17.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helminen HJ, Kiraly K, Pelttari A, Tammi MI, Vandenberg P, Pereira R, Dhulipala R, Khillan JS, Ala-Kokko L, Hume EL, Sokolov BP, Prockop DJ. An inbred line of transgenic mice expressing an internally deleted gene for type II procollagen (COL2A1). Young mice have a variable phenotype of a chondrodysplasia and older mice have osteoarthritic changes in joints. J Clin Invest. 1993;92:582–595. doi: 10.1172/JCI116625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintala M, Metsaranta M, Garofalo S, de Crombrugghe B, Vuorio E, Ronning O. Abnormal craniofacial morphology and cartilage structure in transgenic mice harboring a Gly → Cys mutation in the cartilage-specific type II collagen gene. J Craniofac Genet Dev Biol. 1993;13:137–146. [PubMed] [Google Scholar]
- Pace JM, Li Y, Seegmiller RE, Teuscher C, Taylor BA, Olsen BR. Disproportionate micromelia (Dmm) in mice caused by a mutation in the C-propeptide coding region of Col2a1. Dev Dyn. 1997;208:25–33. doi: 10.1002/(SICI)1097-0177(199701)208:1<25::AID-AJA3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Briggs MD, Choi H, Warman ML, Loughlin JA, Wordsworth P, Sykes BC, Irven CM, Smith M, Wynne-Davies R, Lipson MH, Biesecker LG, Garber AP, Lachman R, Olsen BR, Rimoin DL, Cohn DH. Genetic mapping of a locus for multiple epiphyseal dysplasia (EDM2) to a region of chromosome 1 containing a type IX collagen gene. Am J Hum Genet. 1994;55:678–684. [PMC free article] [PubMed] [Google Scholar]
- Muragaki Y, Mariman EC, van Beersum SE, Perala M, van Mourik JB, Warman ML, Olsen BR, Hamel BC. A mutation in the gene encoding the alpha 2 chain of the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nat Genet. 1996;12:103–105. doi: 10.1038/ng0196-103. [DOI] [PubMed] [Google Scholar]
- van Mourik JB, Hamel BC, Mariman EC. A large family with multiple epiphyseal dysplasia linked to COL9A2 gene. Am J Med Genet. 1998;77:234–240. doi: 10.1002/(sici)1096-8628(19980518)77:3<234::aid-ajmg9>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- Paassilta P, Lohiniva J, Annunen S, Bonaventure J, Le Merrer M, Pai L, Ala-Kokko L. A third locus for multiple epiphyseal dysplasia. Am J Hum Genet. 1999;64:1036–1044. doi: 10.1086/302328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spayde EC, Joshi AP, Wilcox WR, Briggs M, Cohn DH, Olsen BR. Exon skipping mutation in the COL9A2 gene in a family with multiple epiphyseal dysplasia. Matrix Biol. 2000;19:121–128. doi: 10.1016/s0945-053x(00)00055-x. [DOI] [PubMed] [Google Scholar]
- Lohiniva J, Paassilta P, Seppanen U, Vierimaa O, Kivirikko S, Ala-Kokko L. Splicing mutations in the COL3 domain of collagen IX cause multiple epiphyseal dysplasia. Am J Med Genet. 2000;90:216–222. [PubMed] [Google Scholar]
- Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perala M, Carter L, Spector TD, Kolodziej L, Seppanen U, Glazar R, Krolewski J, Latos-Bielenska A, Ala-Kokko L. A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am J Hum Genet. 2001;69:969–980. doi: 10.1086/324023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K, Ono K, Miyazaki J, Olsen BR, Muragaki Y, Adachi E, Yamamura K, Kimura T. Osteoarthritis associated with mild chondrodysplasia in transgenic mice expressing alpha 1(IX) collagen chains with a central deletion. Proc Natl Acad Sci USA. 1993;90:2870–2874. doi: 10.1073/pnas.90.7.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler R, Schnegelsberg PN, Dausman J, Shinya T, Muragaki Y, McCarthy MT, Olsen BR, Jaenisch R. Mice lacking alpha 1 (IX) collagen develop noninflammatory degenerative joint disease. Proc Natl Acad Sci USA. 1994;91:5070–5074. doi: 10.1073/pnas.91.11.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendler M, Eich-Bender SG, Vaughan L, Winterhalter KH, Bruckner P. Cartilage contains mixed fibrils of collagen types II, IX, and XI. J Cell Biol. 1989;108:191–197. doi: 10.1083/jcb.108.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AJ, Yates JRW, Williams R, Payne SJ, Pope FM, Scott JD, Snead MP. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha-1(XI) collagen. Hum Molec Genet. 1996;5:1339–1343. doi: 10.1093/hmg/5.9.1339. [DOI] [PubMed] [Google Scholar]
- Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH, Warman ML. Marshall syndrome associated with a splicing defect at the COL11A1 gene. Am J Hum Genet. 1998;62:816–823. doi: 10.1086/301789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Richards AJ, Yates JRW, Scott JD, Pope M, Snead MP. Stickler syndrome: further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur J Hum Genet. 1999;7:807–814. doi: 10.1038/sj.ejhg.5200377. [DOI] [PubMed] [Google Scholar]
- Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, Mulliken JB, Tranebjaerg L, Brooks DG, Cox GF, Cruysberg JR, Curtis MA, Davenport SL, Friedrich CA, Kaitila I, Krawczynski MR, Latos-Bielenska A, Mukai S, Olsen BR, Shinno N, Somer M, Vikkula M, Zlotogora J, Prockop DJ, Ala-Kokko L. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65:974–983. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikkula M, Mariman EC, Lui VC, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SE, de Waal Malefijt MC, van den Hoogen FH, Ropers H-H, Mayne R, Cheah K, Olsen BR, Warman ML, Brunner HG. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 1995;80:431–437. doi: 10.1016/0092-8674(95)90493-x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Buma P, de Waal Malefijt MC, van den Hoogen FH, Brunner HG. Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical description of three patients homozygous for a missense mutation in the COL11A2 gene. Am J Med Genet. 1997;70:315–323. doi: 10.1002/(sici)1096-8628(19970613)70:3<315::aid-ajmg19>3.3.co;2-y. [DOI] [PubMed] [Google Scholar]
- Pihlajamaa T, Prockop DJ, Faber J, Winterpacht A, Zabel B, Giedion A, Wiesbauer P, Spranger J, Ala-Kokko L. Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher-Zweymuller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome). Am J Med Genet. 1998;80:115–120. doi: 10.1002/(sici)1096-8628(19981102)80:2<115::aid-ajmg5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Melkoniemi M, Brunner HG, Manouvrier S, Hennekam R, Superti-Furga A, Kaariainen H, Pauli RM, van Essen T, Warman ML, Bonaventure J, Miny P, Ala-Kokko L. Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am J Hum Genet. 2000;66:368–377. doi: 10.1086/302750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirt WT, Prasad SD, Griffith AJ, Kunst HP, Green GE, Shpargel KB, Runge C, Huybrechts C, Mueller RF, Lynch E, King MC, Brunner HG, Cremers CW, Takanosu M, Li SW, Arita M, Mayne R, Prockop DJ, Van Camp G, Smith RJ. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat Genet. 1999;23:413–419. doi: 10.1038/70516. [DOI] [PubMed] [Google Scholar]
- Mayne R, Brewton RG, Mayne PM, Baker JR. Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. J Biol Chem. 1993;268:9381–9386. [PubMed] [Google Scholar]
- Li Y, Lacerda DA, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, Oxford JT, Morris NP, Andrikopoulos K, Ramirez F, Wardell BB, Lifferth GD, Teuscher C, Woodward SR, Taylor BA, Seegmiller RE, Olsen BR. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- Li SW, Takanosu M, Arita M, Bao Y, Ren ZX, Maier A, Prockop DJ, Mayne R. Targeted disruption of Col11a2 produces a mild cartilage phenotype in transgenic mice: Comparison with the human disorder otospondylomegaepiphyseal dysplasia (OSMED). Dev Dyn. 2001;222:141–152. doi: 10.1002/dvdy.1178. [DOI] [PubMed] [Google Scholar]
- Superti-Furga A, Neumann L, Riebel T, Eich G, Steinmann B, Spranger J, Kunze J. Recessively inherited multiple epiphyseal dysplasia with normal stature, club foot, and double layered patella caused by a DTDST mutation. J Med Genet. 1999;36:621–624. [PMC free article] [PubMed] [Google Scholar]
- Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, Cekleniak JA, Knowlton RG, Cohn DH. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10:330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- Deere M, Sanford T, Francomano CA, Daniels K, Hecht JT. Identification of nine novel mutations in cartilage oligomeric matrix protein in patients with pseudoachondroplasia and multiple epiphyseal dysplasia. Am J Med Genet. 1999;85:486–490. doi: 10.1002/(sici)1096-8628(19990827)85:5<486::aid-ajmg10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Thur J, Rosenberg K, Nitsche DP, Pihlajamaa T, Ala-Kokko L, Heinegard D, Paulsson M, Maurer P. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J Biol Chem. 2001;276:6083–6092. doi: 10.1074/jbc.M009512200. [DOI] [PubMed] [Google Scholar]
- Chapman KL, Mortier GR, Chapman K, Loughlin J, Grant ME, Briggs MD. Mutations in the region encoding the von Wille-brand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat Genet. 2001;28:393–396. doi: 10.1038/ng573. [DOI] [PubMed] [Google Scholar]
- Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem. 2001;276:6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- Marcelino J, Carpten JD, Suwairi WM, Gutierrez OM, Schwartz S, Robbins C, Sood R, Makalowska I, Baxevanis A, Johnstone B, Laxer RM, Zemel L, Kim CA, Herd JK, Ihle J, Williams C, Johnson M, Raman V, Alonso LG, Brunoni D, Gerstein A, Papadopoulos N, Bahabri SA, Trent JM, Warman ML. CACP, encoding a secreted proteoglycan, is mutated in camptodactyly-arthropathy-coxa vara-pericarditis syndrome. Nat Genet. 1999;23:319–322. doi: 10.1038/15496. [DOI] [PubMed] [Google Scholar]
- Flannery CR, Hughes CE, Schumacher BL, Tudor D, Aydelotte MB, Kuettner KE, Caterson B. Articular cartilage superficial zone protein (SZP) is homologous to megakaryocyte stimulating factor precursor and Is a multifunctional proteoglycan with potential growth-promoting, cytoprotective, and lubricating properties in cartilage metabolism. Biochem Biophys Res Commun. 1999;254:535–541. doi: 10.1006/bbrc.1998.0104. [DOI] [PubMed] [Google Scholar]
- Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270. doi: 10.1126/science.289.5477.265. [DOI] [PubMed] [Google Scholar]
- Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, Goldblatt J, Borochowitz ZU, Kotzot D, Westermann F, Mundlos S, Braun HS, Laing N, Tinschert S. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28:37–41. doi: 10.1038/ng0501-37. [DOI] [PubMed] [Google Scholar]
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, Baur ST, Shiang R, Grange DK, Beighton P, Gardner J, Hamersma H, Sellars S, Ramesar R, Lidral AC, Sommer A, Raposo do Amaral CM, Gorlin RJ, Mulliken JB, Olsen BR. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68:1321–1326. doi: 10.1086/320612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton A, Johnston M, Ho A, Gurley K, Kingsley D, Wright GD, Dixey J, Doherty M, Hughes AE. A heterozygous mutation in the human ANK gene in a British family with adult onset chondrocalcinosis due to calcium pyrophosphate crystal deposition [abstract]. Arthritis Rheum. 2001;44:S101. [Google Scholar]
- Johnson M, Ho A, McGrath R, Netter P, Loeuille D, Jonveaux P, Gaucher A, Reginato A, Gurley K, Kingsley D, Williams CJ. Analysis of ANK gene reveals heterozygous missense mutation in a French family with calcium pyrophosphate deposition disease (CPPDD). Arthritis Rheum. 2001;9:S161. [Google Scholar]
- Baldwin CT, Farrer LA, Adair R, Dharmavaram R, Jimenez S, Anderson L. Linkage of early-onset osteoarthritis and chondrocalcinosis to human chromosome 8q. Am J Hum Genet. 1995;56:692–697. [PMC free article] [PubMed] [Google Scholar]
- Sowers M. Epidemiology of risk factors for osteoarthritis: systemic factors. Curr Opin Rheumatol. 2001;13:447–451. doi: 10.1097/00002281-200109000-00018. [DOI] [PubMed] [Google Scholar]
- Sharma L. Local factors in osteoarthritis. Curr Opin Rheumatol. 2001;13:441–446. doi: 10.1097/00002281-200109000-00017. [DOI] [PubMed] [Google Scholar]
- Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107–113. doi: 10.1186/ar148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertala A, Ala-Kokko L, Wiaderkiewicz R, Prockop DJ. Collagen II containing a Cys substitution for arg-alpha1-519. Homotrimeric monomers containing the mutation do not assemble into fibrils but alter the self-assembly of the normal protein. J Biol Chem. 1997;272:6457–6464. doi: 10.1074/jbc.272.10.6457. [DOI] [PubMed] [Google Scholar]
- Fertala A, Sieron AL, Adachi E, Jimenez SA. Collagen II containing a Cys substitution for Arg-alpha1-519: abnormal interactions of the mutated molecules with collagen IX. Biochemistry. 2001;40:14422–14428. doi: 10.1021/bi0109109. [DOI] [PubMed] [Google Scholar]
- Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem. 2001;276:6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- Wu JZ, Herzog W, Epstein M. Joint contact mechanics in the early stages of osteoarthritis. Med Eng Phys. 2000;22:1–12. doi: 10.1016/s1350-4533(00)00012-6. [DOI] [PubMed] [Google Scholar]