

Abstract
Metalloproteases are a large, diverse class of enzymes involved in many physiological and disease processes. Metalloproteases are regulated by post-translational mechanisms that diminish the effectiveness of conventional genomic and proteomic methods for their functional characterization. Chemical probes directed at active sites offer a potential way to measure metalloprotease activities in biological systems; however, large variations in structure limit the scope of any single small-molecule probe aimed at profiling this enzyme class. Here, we address this problem by creating a library of metalloprotease-directed probes that show complementary target selectivity. These probes were applied as a ‘cocktail’ to proteomes and their labeling profiles were analyzed collectively using an advanced liquid chromatography-mass spectrometry platform. More than 20 metalloproteases were identified, including members from nearly all of the major branches of this enzyme class. These findings suggest that chemical proteomic methods can serve as a universal strategy to profile the activity of the metalloprotease superfamily in complex biological systems.
Enzyme superfamilies present several challenges for post-genomic researchers interested in the assignment of protein function1,2. First, enzyme superfamilies possess many members that, despite sharing a common catalytic mechanism, have widely varied biochemical, cellular and physiological activities. In addition, a large fraction of these enzymes currently fall into the category of uncharacterized proteins, meaning that they are defined solely as predicted protein products based on genome sequence analysis. Finally, most enzyme classes are regulated by post-translational mechanisms3 that hinder their functional characterization using conventional ‘expression-based’ genomic and proteomic methods.
Activity-based protein profiling (ABPP) is a chemical proteomic method that uses active site-directed probes to report on the functional state of enzymes in complex biological systems4-6. ABPP probes have been shown to selectively label active enzymes but not their inactive precursor7,8 or inhibitor-bound8-10 forms, and thus they offer a powerful means to measure dynamics in the activity of enzymes that may occur in the absence of changes in the abundance of their corresponding transcript and/or protein. The biological application of ABPP has led to the discovery of enzyme activities elevated in several disease states, including aggressive cancer cells9,11 and tumors12,13, invasive malaria parasites14 and obese livers15.
Certain ABPP probes that exploit conserved mechanistic features to achieve active-site labeling have proven very effective at capturing a large fraction of their target enzyme superfamily in cell and tissue proteomes. Prototype examples of such class-wide ABPP probes include the fluorophosphonates4,16 and acyloxymethyl ketones17 that target serine hydrolases and cysteine proteases, respectively. Both of these enzyme classes use conserved active-site nucleophiles (serine and cysteine, respectively) to form covalent adducts with substrates (acylenzyme intermediates) as part of their catalytic cycle, a mechanism that has cultivated the design of irreversible inhibitors and probes with broad intraclass selectivity and minimal interclass cross-reactivity. However, many enzyme classes do not use a protein-bound nucleophile for catalysis, thus raising an important question: can ABPP probes be systematically developed for enzyme superfamilies that do not engage in covalent catalysis? In general support of this possibility, active site-directed proteomic probes have been generated for a wide range of mechanistically distinct enzymes, including oxidoreductases18, glutathione S-transferases15, glycosidases19,20, kinases21 and metalloproteases8,22. However, in each of these cases, only a single or handful of enzymes from each class was targeted in proteomes, leaving uncertain whether ABPP probes can be generated for the global profiling of enzyme superfamilies of varied catalytic mechanism.
Of the numerous enzyme classes for which a universal strategy for ABPP would be desired, the metalloproteases stand out as particularly important for several reasons. First, the metalloprotease superfamily is exceptionally large and diverse, with more than 100 members encoded by the human genome23. These enzymes play key roles in many physiological and pathological processes, including tissue remodeling24, peptide hormone signaling25 and cancer24,26,27. Metalloproteases are also regulated by post-translational mechanisms in vivo27, including production as inactive precursor enzymes (zymogens) and inhibition by endogenous binding proteins (TIMPs). Initial efforts have succeeded in generating biotin- and fluorophore-tagged photoreactive hydroxamate (Hx) probes that selectively label active metalloproteases8,22 but not their zymogen or TIMP-bound forms8. However, these first-generation probes were found to target only a limited number of metalloproteases in proteomes, most likely owing to substantial variations in their affinity for different subgroups of the metalloprotease superfamily. Here, we present a general solution to this problem by creating a library of structurally diverse photoreactive Hx probes that have complementary metalloprotease selectivity profiles. These probes can be administered to cell and tissue proteomes as a cocktail and their labeling profiles analyzed collectively using an advanced liquid chromatography-mass spectrometry (LC-MS) platform for ABPP. Using this approach, we identified more than 20 metalloproteases as targets of the probe library, including members from most of the major subfamilies of this enzyme class (for example, matrix metalloproteases (MMPs), adamalysins (ADAMs), aminopeptidases and AAA proteases). Some of these metalloprotease activities were markedly elevated in invasive tumor cells, suggesting that they may be involved in cancer pathogenesis.
RESULTS
Synthesis of a library of ABPP probes for metalloproteases
Initial efforts to create chemical probes for the proteomic profiling of metalloprotease activities have produced two classes of reagents. The first example of a metalloprotease-directed probe was designed based on Hx inhibitors of MMPs (marimastat and ilomastat): the core structure of these reagents was derivatized with a benzophenone (BP) photo-cross-linker and rhodamine (Rh) reporter group for the covalent labeling and detection of metalloprotease activities, respectively8. The resulting HxBP-Rh reagent served as an effective ABPP probe for a select number of metalloproteases. However, metalloproteases have large variations in active-site structure that limit the universal applicability of any single probe directed toward this enzyme class. A second study also described the characterization of photoreactive, reporter-tagged Hx probes, in this case generated in a library format based on a Gly-Gly-X-NHOH peptide scaffold22. Although this approach enabled the straightforward synthesis of numerous candidate metalloprotease probes, the core peptide structure of these probes is not well-suited for achieving high-affinity interactions with most metalloproteases. Indeed, several previous studies aimed at designing metalloprotease inhibitors have shown that these enzymes interact much more strongly (by two to three orders of magnitude) with inhibitors bearing a succinyl linkage separating the Hx and R1 groups as compared to a standard peptide scaffold28-30. To date, the application of peptide Hx probes has mostly been restricted to labeling of metalloproteases in purified form22. Finally, both types of first-generation metalloprotease probes incorporated large reporter tags (fluorophores or biotin) directly into their structures, which might be expected to obstruct interactions with certain metalloproteases.
On the way toward the goal of creating a universal strategy for the proteomic profiling of metalloprotease activities, we envisaged the need for several technical advances. First, a diverse library of probes based on an optimal binding scaffold (for example, succinyl Hx) would be required to accommodate the broad range of active-site structures found in the metalloprotease superfamily. Second, these probes should be created in ‘tag-free’ form such that bulky reporter groups that might otherwise adversely affect probe-metalloprotease interactions are avoided. Finally, an analytical method should be devised to permit the pooled application of multiple metalloprotease probes to individual proteomic samples. We sought to meet the first two objectives by designing and synthesizing three distinct alkyne-tagged libraries of HxBP probes based on the succinyl Hx scaffold (HxBPyne probes; Fig. 1a). In each library, a single position was varied with up to ten amino acid side chains, resulting the generation of approximately 20 distinct HxBPyne probes (see Supplementary Fig. 1 online for additional information on the probe library). The alkyne group was incorporated into each probe as a sterically benign substitute for bulky fluorophore or biotin reporter tags. After proteome labeling, this latent affinity handle could then be exploited to append reporter tags to probe-labeled proteins through the click chemistry (CC) reaction31,32.
Figure 1.

Functional proteomic analysis of metalloprotease activities using active site-directed chemical probes. (a) General structure of the alkyne-tagged hydroxamate-benzophenone (HxBPyne) probe library for the proteome profiling of metalloprotease activities. The structures of the R1 and R2 substituents are shown and named based on the amino acid that they mimic (R1 sublibrary, 15-23; Trp-R2 sublibrary, 11-14; R2 sublibrary, 8-10). (b) Application of HxBPyne probes to proteomes and analysis of results by gel-based (upper) or MudPIT-based (lower) ABPP. In gel-based ABPP, individual HxBPyne-treated proteomes are reacted with an azide-rhodamine (Rh) reporter tag under CC conditions and separated by SDS-PAGE, and labeled metalloproteases are visualized by in-gel fluorescence scanning. In ABPP-MudPIT, proteomes are treated with a cocktail of HxBPyne probes and reacted with an azide-biotin (B) reporter tag under CC conditions. Probe-labeled metalloproteases are then captured on avidin beads, digested with trypsin and analyzed by multidimensional LC-MS.
Proteomic profiling of the HxBPyne probe library
We performed initial screening of the HxBPyne library by gel-based ABPP as follows. Individual probes were added to cell and tissue proteomes and the reactions exposed to UV light for 1 h, treated with an azide-Rh tag under CC conditions31,32 and analyzed by one-dimensional (1D) SDS-PAGE (Fig. 1b). Members of the probe library showed highly distinct proteome reactivity profiles (Supplementary Fig. 2 online), indicating that the variable R1 and R2 groups exerted a strong influence over specific probe-protein interactions. A representative set of probes showing complementary reactivity profiles were compared across a broad concentration range (1-100 nM) to estimate the strength of their interactions with proteins in the proteome (Fig. 2). These experiments revealed clear differences in the relative probe-selectivity profiles of individual proteins. For example, a 102-kDa protein showed extremely robust labeling with the LysR2 probe (9), such that no diminution in signal intensity was observed across the entire concentration range (Fig. 2a,b). Treatment of probe-labeled proteomes with an azide-biotin tag under CC conditions, followed by avidin enrichment and LC-MS analysis, identified this protein as the metalloprotease puromycin-sensitive aminopeptidase (PSAP). Recombinant expression of PSAP in COS7 cells confirmed this metalloprotease’s preferential reactivity with the LysR2 probe (Fig. 2c). Three additional protein targets in the mouse liver proteome, identified as the metalloproteases adipocyte-derived leucine aminopeptidase (ALAP; 105 kDa), dipeptidylpeptidase III (DPPIII, 80 kDa) and leucine aminopeptidase III (LAPIII, 55 kDa), showed preferential labeling with other members of the probe library (PheR2 (10), LeuR2 (8), and PheR1 (18) probes, respectively) (Fig. 2a,b). Finally, a protein target in the cytosolic proteome of the human breast cancer cell line MCF7, identified as the metalloprotease leukotriene A4 hydrolase (LA4H), showed exclusive reactivity with the AspR1 (16) probe (Supplementary Fig. 3 online).
Figure 2.

Profiling metalloprotease activities in proteomes with the HxBPyne probe library. (a) Concentration-dependent labeling profiles for representative HxBPyne probes added to mouse liver proteome (grayscale image of fluorescent gels showing probe-proteome reactions conjugated by CC with a Rh-azide reporter tag). Highly distinct proteome labeling profiles are seen for individual probes. Metalloprotease targets showing different probe selectivities were isolated and identified in separate reactions using biotin-azide tags and avidin chromatography-LC-MS procedures, as described previously13.(b) Concentration dependence for reactivity of metalloproteases with their corresponding optimal probes. (c) Recombinant expression and HxBPyne labeling profiles of PSAP and ALAP. Both recombinant metalloproteases show probe selectivity profiles similar to those of their native counterparts (a). The LysR2 reactive protein observed at low levels in the mock lane is most likely endogenous PSAP.
Concurrently with these proteome screening experiments, we also analyzed the probe library for reactivity against a set of commercially available MMPs. Human MMPs were added to background mouse tissue proteomes and treated with individual members of the HxBPyne library (1 mM). The MMPs generally showed similar probe selectivity profiles, reacting most strongly with the LeuR2 and PheR2 probes (Supplementary Fig. 4 online), which correlated well with predictions of their substrate preferences observed with peptide substrate libraries33. Notably, most of the MMPs showed a marked preference for labeling by the HxBPyne probes over their Rh-tagged counterparts, with certain enzymes showing nearly exclusive reactivity with the former probes (Fig. 3a). The only exceptions were MMP2 and MMP9, which reacted equivalently with HxBPyne and HxBP-Rh probes (data not shown). These results underscore the significant impact (in this case, negative) that reporter tags can exert on probe-protein interactions, a factor that can be circumvented with CC-based methods. All MMP-probe reactions were completely blocked by the inclusion of the MMP inhibitor GM6001 (10 mM), confirming the active-site specificity of these labeling events (Supplementary Fig. 4). Serial dilution of MMPs into a constant background of tissue proteome provided a sensitivity limit for their detection with the LeuR2 probe of 0.25-2.5 mg MMP per mg proteome (0.03-0.25% of total proteome) (Fig. 3b).
Figure 3.
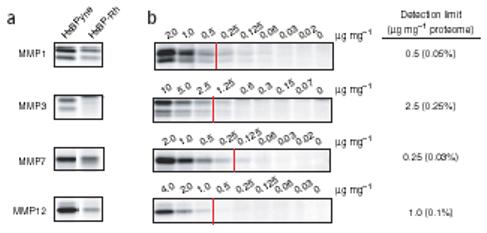
Profiling MMP activities by gel-based ABPP. (a) Comparison of the reactivity of MMPs with the original Rh-tagged HxBP probe8 and its alkyne counterpart (LeuR2 HxBPyne probe). MMPs generally show much stronger labeling with the alkyne probe. (b) Sensitivity of detection of MMPs by LeuR2 HxBPyne probe. Human recombinant MMPs were added at the indicated concentrations into mouse liver proteome and the samples treated with 1 mM of LeuR2 HxBPyne probe. Detection limits was defined as signals that were 200% of the background fluorescence signals observed in lanes lacking added MMPs.
From these initial studies comparing the reactivity profiles of members of the HxBPyne library with both endogenously expressed and recombinant metalloproteases, we identified a subgroup of probes (AspR1, AspR2 (7), LeuR2, PheR2) that collectively targeted the vast majority of enzymes labeled by the library as a whole. We next set out to develop a method whereby this ‘optimal probe set’ could be applied as a cocktail to profile a broad range of metalloprotease activities in individual proteomes with maximal resolution and sensitivity.
A high-resolution platform for profiling metalloprotease activities
As exemplified above, 1D gel-based ABPP offers a rapid means to analyze many probe-proteome reactions in parallel. However, owing to inherent resolution and sensitivity limits, 1D gels are not suitable for the comprehensive characterization of probe targets in proteomes. We recently described an advanced gel-free strategy for ABPP that unites this method with the multidimensional protein identification technology (MudPIT34) (Fig. 1b)13. This ABPP-MudPIT platform exploits the exceptional resolution and sensitivity of multidimensional LC-MS to enable the simultaneous characterization of numerous probe-labeled enzyme activities in individual proteomic samples. Initial ABPP-MudPIT studies applied single probes to proteomes13, but we postulated that the resolving power of this approach should afford probe ‘multiplexing’ opportunities such that, for example, our optimal set of metalloprotease probes could be applied as a mixture to proteomes and their combined protein labeling profiles acquired as a single integrated dataset. First, however, we sought to evaluate the sensitivity of metalloprotease detection by ABPP-MudPIT.
Recombinant MMPs were serially diluted into tissue proteomes over a concentration range of 1,000-10 ng MMP per mg proteome, treated with the LeuR2 HxBPyne probe (100 nM) under UV exposure, and subjected to ABPP-MudPIT analysis (Fig. 1b; see Methods for details of procedure). All MMPs could be detected in both the 1,000 and 100 ng MMP per mg proteome reactions (Fig. 4a). MMP1, but not the other MMPs, could also be detected in the 10 ng MMP per mg proteome reaction. To confirm that the observed signals were due to active-site labeling of MMPs, samples were pretreated with excess of an alkane variant of the LeuR2 probe (24) (LeuR2 HxBPane), which was found to block more than 90% of labeling of all MMPs as judged by spectral counting (Fig. 4a). Thus, ABPP-MudPIT detected MMP activities at levels ranging from 0.001-0.01% of total proteome, which corresponds to an approximately 5-50-fold increase in sensitivity as compared to gel-based methods.
Figure 4.

Profiling metalloprotease activities by ABPP-MudPIT. (a) Sensitivity of detection of MMPs by ABPP-MudPIT. Human recombinant MMPs were added at the indicated concentrations to mouse liver proteome and the samples were treated with 100 nM of the LeuR2 HxBPyne probe and analyzed by ABPP-MudPIT as shown in Figure 1b. Relative amounts of MMP activities were estimated by spectral counting13.(b) Endogenous metalloprotease activities identified by ABPP-MudPIT in human cancer cell lines. For a and b, C indicates control reactions run in the presence of 100 × HxBPane probes to compete the specific labeling of metalloproteases by HxBPyne probes. (c) Endogenous metalloprotease activities identified by ABPP-MudPIT that are at significantly different levels in invasive (MUM-2B) and noninvasive (MUM-2C) cancer lines. All data shown were statistically significant (P < 0.05) between comparison groups (for a and b, probe-treated versus C groups; for c, MUM-2B versus MUM-2C groups) as determined by Student’s t-test or Mann-Whitney test. Data represent the average values ± s.e.m. of three independent experiments.
Profiling metalloprotease activities in cancer proteomes
We next investigated whether the optimal HxBPyne probe set, in combination with ABPP-MudPIT, could be used to identify endogenous metalloprotease activities and quantify their relative levels in disease states. For these studies, we compared the metalloprotease activity profiles of human cancer cell lines that differ in key pathogenic traits. Specifically, membrane proteomes from invasive and noninvasive breast carcinoma (MDA-MB-231 and MCF7, respectively) and melanoma (MUM-2B and MUM-2C, respectively) lines9,35 were treated with the optimal probe set (100 nM of each HxBPyne probe; 400 nM total probe) and analyzed by ABPP-MudPIT. Active (that is, probe-labeled) metalloproteases were identified as those enzymes that showed significantly higher spectral counts in HxBPyne-treated proteomes than in control proteomes pretreated with 100 × HxBPane competitor probes (P < 0.05). Several endogenous metalloprotease activities were identified as specific targets of the optimal probe set, including adamalysins, aminopeptidases and AAA proteases (Fig. 4b and Table 1; also see Supplementary Table 1 online). A comparison of the average spectral counts of metalloprotease activities identified a set of enzymes that differed in invasive and noninvasive cancer lines. For example, the invasive MUM-2B line possessed high levels of two metalloprotease activities, alanyl aminopeptidase (AlaAP) and neprilysin, that were absent in its less aggressive sister line MUM-2C (Fig. 4c). The selective expression of these metalloproteases in MUM-2B cells was also observed using antibodies in fluorescence-activated cell sorting and western blotting experiments (Supplementary Fig. 5 online), thus confirming the accuracy of ABPP-MudPIT measurements. The metalloprotease ADAM10 also had greater activity in MUM-2B cells than in MUM-2C cells, whereas the uncharacterized AAA protease AFG3-like protein 2 showed the converse relative activity profile. The metalloprotease activity profiles of MDA-MB-231 and MCF7 cells were generally similar, with the exception of endothelin-converting enzyme, whose activity was three-fold higher in MDA-MB-231 cells (Supplementary Table 1).
The identification of two ADAM proteins as specific targets of the optimal probe set was intriguing, as ABPP probes have not yet been described for this important subclass of integral membrane metalloproteases, which, like the MMPs, have critical roles in cell surface signaling events (for example, ectodomain shedding of growth factors)36,37. Examination of the probe reactivity profiles of recombinant ADAM10 and ADAM17 confirmed that they are specific targets of the probe library and revealed that ADAM17 showed strongest labeling with the AspR1 probe (Fig. 5a,b). This probe selectivity profile contrasted starkly with those shown by MMPs, which had little or no reactivity with the AspR1 probe (Supplementary Fig. 4). The AspR1 probe was also found to be an excellent inhibitor of ADAM17 activity, with an IC50 value of 133 nM as determined using a fluorescent substrate assay (Fig. 5c). These data illuminate potential differences in the active-site structure of MMPs and ADAMs that may be instructive for the design not only of probes but also of inhibitors.
Figure 5.

Characterization of ADAM targets of the HxBPyne library. (a) HxBPyne labeling of recombinant ADAM10 and ADAM17 (4 mg ml-1 in a background of 1 mg ml-1 mouse liver proteome), where red boxes designate the HxBPyne probe with strongest labeling (LeuR2 and AspR1 for ADAM10 and ADAM17, respectively). (b) HxBPyne labeling of ADAM10 and ADAM17 was blocked by 100 × HxBPane probes (C reactions). (c) The AspR1 probe inhibits ADAM17 activity, as determined with a fluorescent substrate assay. Data represent the average values ± s.e.m. of three independent experiments.
DISCUSSION
Metalloproteases have fundamental roles in a wide range of mammalian physiological and pathological processes. Given their myriad cellular functions, it is perhaps not surprising that the catalytic activity of metalloproteases is tightly regulated by post-translational mechanisms in vivo. Active site-directed chemical probes can discriminate functional metalloproteases from their inactive precursor or inhibitor-bound forms, thus providing high-content proteomic information that is difficult to obtain by more conventional mRNA or protein expression profiling methods. Still, for chemical proteomics to offer a universal strategy for profiling metalloprotease activities in biological systems, ABPP probes are required that capture a large fraction of this enzyme class. The daunting size and diversity of the metalloprotease superfamily indicates that it might best be tackled through the creation of multiple probes that have complementary target selectivity profiles, along with advanced analytical methods for their consolidated application and characterization. Here we present general solutions for both of these challenges.
Synthesis and proteomic screening of a structurally diverse library of HxBPyne probes enabled the selection of a set of these reagents that showed complementary metalloprotease reactivity profiles. When judging the features of HxBPyne probes that promoted specific interactions with metalloproteases, some general conclusions can be drawn. First, the identity of the R1 and R2 groups, as well as the position of the BP cross-linker, are important in determining an individual probe’s proteome reactivity profile. The contribution that these groups made to enzyme labeling is perhaps best illustrated by noting that none of the metalloproteases identified in this study showed universal reactivity with the probe library. On the contrary, metalloproteases showed notable differences in their respective probe selectivity profiles, with examples of enzymes preferring hydrophobic (for example, MMPs), positively charged (for example, PSAP) and negatively charged (for example, L4AH, ADAM17) probes all being observed. These results emphasize that, even though the Hx group, by coordinating the active-site zinc in a bidentate manner38, most likely has the potential to target most if not all metalloproteases, the strength of these interactions is strongly influenced by additional elements in the probe structure. We also note that the introduction of an alkyne in place of large reporter tags proved beneficial for many probe-metalloprotease interactions, a finding that underscores the versatility afforded by CC methods for probe design.
To capitalize on the complementary metalloprotease reactivity profiles of individual HxBPyne probes, we developed a gel-free strategy for their consolidated application to individual proteomic samples. Key to this approach was the analysis of probe-metalloprotease reactions using a recently introduced ABPP-MudPIT platform13, which provides superior sensitivity and resolution compared to gel-based methods. ABPP-MudPIT analysis of cell and tissue proteomes treated with an optimal set of HxBPyne probes resulted in the identification of more than 20 metalloproteases specifically labeled by these reagents, including several enzyme activities that were elevated in invasive cancer cells. Some of these metalloprotease activities—in particular AlaAP and neprilysin, which were selectively present in invasive, but not noninvasive, melanoma cells—warrant further investigation as potential contributors to cancer pathogenesis. Indeed, AlaAP has recently been identified by gene expression profiling as a potential marker for transdifferentiation of melanocytes into metastatic melanoma39. The inclusion of control experiments containing excess HxBPane competitor probes proved critical for discriminating specifically (that is, active-site) labeled metalloproteases from unlabeled background proteins that bled through the avidin enrichment procedure. For example, although all of the metalloproteases listed in Table 1 showed significantly reduced signals (that is, spectral counts; P < 0.05) in reactions with excess alkane competitor, certain other metalloproteases, such as MMP14, were found at similar levels in both sets of samples (Supplementary Fig. 6 online). Because equivalent signals for MMP14 were also observed in mock proteome reactions lacking HxBPyne probes, we conclude that this MMP corresponds to an unlabeled (probably inactive) background protein present in cancer cell proteomes.
Table 1.
List of metalloprotease targets identified with HxBPyne probe library
| Metalloprotease | Mode of identification | Tissue or cell line | Average spectral counts |
|---|---|---|---|
| ADAM10 (IPI00013897) | LC-MS | MUM-2B | 9 |
| Gel | Recombinant | N/A | |
| ADAM17 (IPI00288894) | LC-MS | MDA-MB-231 | 4 |
| Gel | Recombinant | N/A | |
| ADAMTS4 (IPI00307276) | Gel | Recombinant | N/A |
| Adipocyte-derived leucine aminopeptidase | LC-MS | Mouse brain | 37 |
| (ALAP) (IPI00477831) | Gel | Mouse liver (cyto) | N/A |
| Gel | Recombinant COS7 | N/A | |
| AFG3-like protein 2 (IPI00001091) | LC-MS | MUM-2C | 13 |
| Alanyl aminopeptidase (AlaAP) (IPI00221224) | LC-MS | MUM-2B | 376 |
| Aspartyl aminopeptidase (IPI00072257) | LC-MS | MCF7 | 3 |
| Cystinyl aminopeptidase (IPI00221240) | LC-MS | MDA-MB-231 | 2 |
| Dipeptidylpeptidase 3 (DPPIII) (IPI00020672) | LC-MS | Mouse brain | 18 |
| Gel | Mouse liver (cyto) | N/A | |
| Endothelin-converting enzyme (IPI00002478) | LC-MS | MDA-MB-231 | 3 |
| Eupitrilysin (IPI00219613) | LC-MS | MUM-2B | 11 |
| Leucine aminopeptidase 3 (LAPIII) (IPI00331436) | Gel | Mouse liver (cyto) | N/A |
| Leukotriene A4 hydrolase (LA4H) (IPI00229527) | Gel | MDA-MB-231 | N/A |
| Gel | Recombinant COS7 | N/A | |
| MMP1 (IPI00110075) | LC-MS | Recombinant | 120a |
| MMP2 (IPI00112904) | LC-MS | Recombinant | 10a |
| MMP3 (IPI00123511) | Gel | Recombinant | N/A |
| MMP7(IPI00275232) | LC-MS | Recombinant | 3a |
| MMP9 (IPI00319200) | LC-MS | Recombinant | 8a |
| MMP12 (IPI00321648) | LC-MS | Recombinant | 8a |
| Neprilysin (IPI00247063) | LC-MS | MUM-2B | 27 |
| Paraplegin (IPI00299010) | LC-MS | MUM-2B | 2 |
| Puromycin-sensitive aminopeptidase | LC-MS | MDA-MB-231 | 31 |
| (PSAP) (IPI00026216) | Gel | Mouse liver (cyto) | N/A |
| Gel | Recombinant COS7 | N/A |
Spectral counts of 100 ng recombinant metalloprotease in 1 mg proteome.
N/A, not applicable; cyto, cytoplasm. Except where noted, spectral counts shown correspond to the proteome in which the highest level of each metalloprotease was found. For a complete list of ABPP-MudPIT results, see Supplementary Table 1.
A survey of the metalloproteases targeted by the HxBPyne library reveals members of nearly all of the major branches of this enzyme superfamily (Fig. 6). This provocative finding suggests that HxBPyne probes have sufficient versatility to serve as class-wide ABPP probes for metalloproteases. Nonetheless, there remain some segments of the metalloprotease superfamily that were under-represented in the current investigation. What reasons might there be for these apparent holes in metalloprotease coverage? First, some metalloproteases may have evaded detection because they are localized to specific cellular and/or subcellular compartments that were not profiled in this study. For example, the ADAMTS family of metalloproteases is predominantly comprised of secreted enzymes that may not be present at appreciable levels in cell or tissue proteomes. In this regard, we have found that recombinant ADAMTS-4 is specifically labeled by HxBPyne probes (Supplementary Fig. 3), indicating that members of the ADAMTS subfamily of metalloproteases are amenable to ABPP analysis.
Figure 6.

Location of targets of HxBPyne library on a phylogenic tree of the metalloprotease superfamily. With the exception of the carboxypeptidases, members from all major subfamilies of metalloproteases were targeted by the HxBPyne library. The tree was constructed from the peptidase domains of the complete human members of the metalloprotease clan, as designated by the MEROPS database43 (205 total predicted members, 150 putative active members that possess conserved catalytic residues). A pairwise alignment was generated using ClustalW44 and the phylogenic tree constructed using the neighbor-joining method45. Major branches of the tree were designated by different colors, labeled by name, where appropriate, and displayed in hyperbolic space using the Hypertree program46.
Other metalloproteases may exist in active form at levels that are below the current detection limit of gel- or MudPIT-based ABPP. Indeed, the net quantity of active metalloproteases required to perform certain cellular tasks may be quite low and may occur in a background of high concentrations of the inactive (for example, zymogen or inhibitor-bound) forms of these enzymes. For example, MMPs are often found entirely in zymogen and/or TIMP-bound form in cancer cell proteomes40. Further improvements in sensitivity should be attainable by performing ABPP-MudPIT in targeted mode, where the mass spectrometer is focused on measuring the signals of specific metalloprotease-derived tryptic peptides. Additionally, the incorporation into the azido reporter group of a linker that is cleavable chemically20 or through enzyme catalysis41 should facilitate the selective release of active (probe-labeled) metalloproteases from avidin beads, leaving behind any residual inactive (unlabeled) forms of these enzymes that are present as contaminating background proteins.
Finally, there are likely members of the metalloprotease family that are not effectively targeted by any of the ABPP probes in our current library. In this regard, the absence of carboxypeptidases from the list of identified targets of HxBPyne probes is notable. This subclass of metalloproteases may require HxBP probes that more accurately mimic their preferred C-terminal peptide substrates (for example, primary or secondary hydroxamates bearing C-terminal carboxylates42). Other metalloproteases may bind ABPP probes, but show poor photo-cross-linking efficiency, a parameter that can vary from enzyme to enzyme depending on the protein microenvironment surrounding the probe’s photoreactive group. We anticipate that the generation of larger libraries of candidate HxBPyne probes, in combination with expanded (multiplexed) screening of proteomes, should continue to refine the optimal probe set required to profile the majority of the metalloprotease superfamily.
The general process described herein for transforming tight-binding reversible inhibitors into ABPP probes by introducing photoreactive groups and latent affinity handles (for example, alkyne) should prove broadly applicable to many enzyme classes. Notably, our initial results suggest that the converse path of experimental discovery may also prove feasible, namely, proceeding from ABPP probes to high-affinity reversible inhibitors, as was achieved for ADAM17. In this way, ABPP may facilitate the simultaneous discovery of enzyme activities associated with human disease and chemical tools for testing their function in pathological processes.
METHODS
Chemical synthesis. Details concerning the synthesis and characterization of ABPP probes are available in the Supplementary Methods online.
Labeling of cell and tissue proteomes. Human cell lines including MDA-MB-231, MCF7, MUM-2B and MUM-2C were grown and processed as described previously9,32. Mouse tissue proteomes were prepared as described previously32. Membrane fractions from both cells and tissues were homogenized in PBS and stored at -80 °C until use. Proteome samples were adjusted to a final concentration of 1 mg protein per ml by dilution in PBS before probe labeling. Experiments for visualization by 1D SDS-PAGE were carried out in 43 ml total volume and those for multidimensional protein identification technology (MudPIT) in 946 ml total volume, such that once CC reagents were added, the total reaction volumes were 50 ml and 1 ml, respectively. HxBPyne probes were added at concentrations ranging from 1 nM to 1 mM, as described in the text. HxBPane control probes were added at 100-fold excess concentration over HxBPyne probes. For labeling studies with recombinant enzymes, varying amounts of metalloproteases (MMPs and ADAMs were purchased from Calbiochem and Biomol) were added to a background mouse tissue proteome (liver or kidney, 1 mg total protein per ml). All samples were loaded in an ice-cooled 96-well plate in 50-ml fractions and irradiated at 365 nm using an Spectroline ENF 260C UV lamp for 1 h on ice following previously described procedures8.
CC reactions. After UV cross-linking, reporter-tagged azide reagents (12.5 mM Rh-azide for 1DE analysis or 20 mM biotin-azide for MudPIT analysis31,32) were added, followed by 1 mM TCEP and 100 mM ligand31. Samples were gently vortexed and the cycloaddition initiated by the addition of 1 mM CuSO4. The reactions were incubated at room temperature (25 °C) for 1-1.5 h. For gel-based ABPP, an equal volume of 2× standard SDS loading buffer was added and the samples separated by 1D SDS-PAGE and visualized by in-gel fluorescent scanning using a Hitachi FMBio Ile flatbed scanner (MiriaBio). For ABPP-MudPIT, reactions were quenched by addition of 200 ml PBS and the protein precipitated in a microcentrifuge. The supernatant was removed and the pellet washed twice in 200 ml methanol. The pellet was subsequently dissolved in 1 ml of PBS with 0.2% SDS by sonication. Avidin-coupled agarose beads (50 ml suspension, Sigma) were added and incubated for 1 h at room temperature to bind and enrich biotin-labeled metalloproteases. The beads were then washed twice with 10 mLlPBS with 0.2% SDS, twice with 10 ml 6 M urea, and three times with 10 ml PBS. Beads were next resuspended in 200 μl of 6 M urea and proteins sequentially reduced with TCEP (5 mM, 20 min) and alkylated with iodoacetamide (200 mM, 20 min) in the dark. Beads were then washed and digested with 1 mg trypsin (Promega) in 150 ml 2 M urea overnight. Centrifugation provided the trypsin-digested peptide supernatant, which was acidified by addition of formic acid to a final concentration of 5% formic acid and loaded onto a C18 column for subsequent analysis by 2D-LC in combination with tandem MS using a coupled Agilent 1100 LC-ThermoFinnegan LTQ MS system, as described previously13. Analysis of SEQUEST search results from ABPP-MudPIT runs was carried out as previously described13 and as detailed in Supplementary Methods. A complete list of the metalloprotease targets identified by ABPP-MudPIT, with their spectral counts, is shown in Supplementary Table 1. Certain probe-labeled enzymes were also identified using a trifunctional reporter tag (bearing azide, biotin and Rh groups), avidin-enrichment, separation on and excision from SDS-PAGE gels, and LC-MS analysis, as previously described7,32.
Recombinant expression of metalloproteases. cDNA clones for selected metalloproteases were purchased from commercial sources (Open Biosystems, Invitrogen) and the coding sequence amplified by PCR and cloned into the multiple cloning site of the pcDNA3 eukaryotic expression vector. COS7 cells were transiently transfected with metalloprotease cDNA-pcDNA3 constructs using the FuGENE transfection system (Roche) according to the manufacturer’s protocol. Cells were harvested and processed as described previously18 and treated with probes as described above.
Analysis of the inhibition of ADAM17. The fluorogenic substrate DABCYL-LAQAVRSSSR-EDANS and recombinant ADAM17 protein were purchased from Calbiochem and used for IC50 determination with the AspR1 probe following protocols described previously for MMPs8.
ACKNOWLEDGMENTS
We thank G. Simon for assistance with construction of the metalloprotease family tree diagram, A. Saghatelian for assistance with probe synthesis, M. Madsen for assistance with cell culture, and S. Niessen and B. Wei for assistance with the analysis of MudPIT data. This work was supported by the US National Institutes of Health (CA087660), Activx Biosciences, the Skaggs Institute for Chemical Biology and a DFG Emmy Noether postdoctoral fellowship (S.A.S.).
Footnotes
Note: Supplementary information is available on the Nature Chemical Biology website.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 2.Saghatelian A, Cravatt BF. Assignment of protein function in the postgenomic era. Nat. Chem. Biol. 2005;1:130–142. doi: 10.1038/nchembio0805-130. [DOI] [PubMed] [Google Scholar]
- 3.Kobe B, Kemp BE. Active site-directed protein regulation. Nature. 1999;402:373–376. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jessani N, Cravatt BF. The development and application of methods for activity-based protein profiling. Curr. Opin. Chem. Biol. 2004;8:54–59. doi: 10.1016/j.cbpa.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Speers AE, Cravatt BF. Chemical strategies for activity-based proteomics. ChemBioChem. 2004;5:41–47. doi: 10.1002/cbic.200300721. [DOI] [PubMed] [Google Scholar]
- 7.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 8.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. USA. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jessani N, Liu Y, Humphrey M, Cravatt BF. Enzyme activity profiles of the secreted and membrane proteome that depict cancer invasiveness. Proc. Natl. Acad. Sci. USA. 2002;99:10335–10340. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenbaum DC, et al. Small molecule affinity fingerprinting. A tool for enzyme family subclassification, target identification, and inhibitor design. Chem. Biol. 2002;9:1085–1094. doi: 10.1016/s1074-5521(02)00238-7. [DOI] [PubMed] [Google Scholar]
- 11.Jessani N, et al. Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc. Natl. Acad. Sci. USA. 2004;101:13756–13761. doi: 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joyce JA, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 13.Jessani N, et al. A streamlined platform for high-content functional proteomics of primary human specimens. Nat. Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 14.Greenbaum DC, et al. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science. 2002;298:2002–2006. doi: 10.1126/science.1077426. [DOI] [PubMed] [Google Scholar]
- 15.Barglow KT, Cravatt BF. Discovering disease-associated enzymes by proteome reactivity profiling. Chem. Biol. 2004;11:1523–1531. doi: 10.1016/j.chembiol.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 16.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Direct visualization of serine hydrolase activities in complex proteome using fluorescent active site-directed probes. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Kato D, et al. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005;1:33–38. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- 18.Adam GC, Sorensen EJ, Cravatt BF. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat. Biotechnol. 2002;20:805–809. doi: 10.1038/nbt714. [DOI] [PubMed] [Google Scholar]
- 19.Vocadlo DJ, Bertozzi CR. A strategy for functional proteomic analysis of glycosidase activity from cell lysates. Angew. Chem. Int. Edn. Engl. 2004;43:5338–5342. doi: 10.1002/anie.200454235. [DOI] [PubMed] [Google Scholar]
- 20.Hekmat O, Kim YW, Williams SJ, He S, Withers SG. Active-site peptide “fingerprinting” of glycosidases in complex mixtures by mass spectrometry. Discovery of a novel retaining beta-1,4-glycanase in Cellulomonas fimi. J. Biol. Chem. 2005;280:35126–35135. doi: 10.1074/jbc.M508434200. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, et al. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem. Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Chan EW, Chattopadhaya S, Panicker RC, Huang X, Yao SQ. Developing photoactive affinity probes for proteomic profiling: hydroxamate-based probes for metalloproteases. J. Am. Chem. Soc. 2004;126:14435–14446. doi: 10.1021/ja047044i. [DOI] [PubMed] [Google Scholar]
- 23.Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat. Rev. Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 24.Chang C, Werb Z. The many faces of metalloproteases: cell growth, invasion, angiogenesis, and metastasis. Trends Cell Biol. 2001;11:S37–S43. doi: 10.1016/s0962-8924(01)02122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metallo-endopeptidases: genomics and function. Bioessays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 26.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70:561–573. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- 27.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 28.Johnson WH, Roberts NA, Borkakoti N. Collagenase inhibitors: their design and potential therapeutic use. J. Enzyme Inhib. 1987;2:1–22. doi: 10.3109/14756368709030352. [DOI] [PubMed] [Google Scholar]
- 29.Stoermer D, et al. Synthesis and biological evaluation of hydroxamate-based inhibitors of glutamate carboxypeptidase II. Bioorg. Med. Chem. Lett. 2003;13:2097–2100. doi: 10.1016/s0960-894x(03)00407-4. [DOI] [PubMed] [Google Scholar]
- 30.Jani M, Tordai H, Trexler M, Banyai L, Patthy L. Hydroxamate-based peptide inhibitors of matrix metalloprotease 2. Biochimie. 2005;87:385–392. doi: 10.1016/j.biochi.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 32.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 34.Washburn MP, Wolters D, Yates JR., III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 35.Seftor EA, et al. Molecular determinants of human uveal melanoma invasion and metastasis. Clin. Exp. Metastasis. 2002;19:233–246. doi: 10.1023/a:1015591624171. [DOI] [PubMed] [Google Scholar]
- 36.Moss ML, Bartsch JW. Therapeutic benefits from targeting of ADAM family members. Biochemistry. 2004;43:7227–7235. doi: 10.1021/bi049677f. [DOI] [PubMed] [Google Scholar]
- 37.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 38.Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 39.Seftor EA, et al. Epigenetic transdifferentiation of normal melanocytes by a metastatic melanoma microenvironment. Cancer Res. 2005;65:10164–10169. doi: 10.1158/0008-5472.CAN-05-2497. [DOI] [PubMed] [Google Scholar]
- 40.Ogata Y, Itoh Y, Nagase H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. J. Biol. Chem. 1995;270:18506–18511. doi: 10.1074/jbc.270.31.18506. [DOI] [PubMed] [Google Scholar]
- 41.Speers AE, Cravatt BF. A tandem orthogonal proteolysis strategy for high-content chemical proteomics. J. Am. Chem. Soc. 2005;127:10018–10019. doi: 10.1021/ja0532842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mock WL, Cheng H. Principles of hydroxamate inhibition of metalloproteases: carboxypeptidase A. Biochemistry. 2000;39:13945–13952. doi: 10.1021/bi001497s. [DOI] [PubMed] [Google Scholar]
- 43.Rawlings ND, Tolle DP, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2004;32:D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 46.Bingham J, Sudarsanam S. Visualizing large hierarchical clusters in hyperbolic space. Bioinformatics. 2000;16:660–661. doi: 10.1093/bioinformatics/16.7.660. [DOI] [PubMed] [Google Scholar]