

Abstract
N-Acyl ethanolamines (NAEs) constitute a large and diverse class of signaling lipids that includes the endogenous cannabinoid anandamide. Like other lipid transmitters, NAEs are thought to be biosynthesized and degraded on-demand rather than being stored in vesicles prior to signaling. The identification of enzymes involved in NAE metabolism is therefore imperative to achieve a complete understanding of this lipid signaling system and control it for potential therapeutic gain. Recently, an N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) was identified as a candidate enzyme involved in the biosynthesis of NAEs. Here, we describe the generation and characterization of mice with a targeted disruption in the NAPE-PLD gene [NAPE-PLD(-/-) mice]. Brain tissue from NAPE-PLD- (-/-) mice showed more than a 5-fold reduction in the calcium-dependent conversion of NAPEs to NAEs bearing both saturated and polyunsaturated N-acyl chains. However, only the former group of NAEs was decreased in level in NAPE-PLD(-/-) brains, and these reductions were most dramatic for NAEs bearing very long acyl chains (≥C20). Further studies identified a calcium-independent PLD activity in brains from NAPE-PLD(-/-) mice that accepted multiple NAPEs as substrates, including the anandamide precursor C20:4 NAPE. The illumination of distinct enzymatic pathways for the biosynthesis of long chain saturated and polyunsaturated NAEs suggests a strategy to control the activity of specific subsets of these lipids without globally affecting the function of the NAE family as a whole
Unlike classical neurotransmitters, which are stored in membrane-delineated vesicles prior to release, lipid signaling molecules are thought to be produced by neurons at the moment of their intended action (1, 2). This “on-demand” model implicates the participating biosynthetic and degradative enzymes as key regulators of lipid signaling tone. One large and diverse class of lipid transmitters is the N-acyl ethanolamines (NAEs1), which includes the endogenous cannabinoid N-arachidonoyl ethanolamine, or anandamide (3), the antiinflammatory lipid, N-palmitoyl ethanolamine (PEA) (4), and the appetite-suppressing substance N-oleoyl ethanolamine (OEA) (5). Consistent with a primary role for enzymes in controlling NAE signaling in vivo, genetic or chemical disruption of the NAE-degrading enzyme fatty acid amide hydrolase [FAAH (6, 7)] results in highly elevated endogenous levels of NAEs and corresponding reductions in pain sensitivity (8-11), anxiety (12), and inflammation (13,14). These results have established FAAH as a key terminator of NAE activity in vivo. In contrast, the enzymes involved in generating endogenous NAE signals remain more enigmatic.
The biosynthesis of NAEs has been proposed to occur by a two-step enzymatic process wherein, first, a calcium-activated transacylase transfers the sn-1 acyl chain of a phospholipid onto the amine of phosphatidylethanolamine (PE) to generate an N-acyl PE (NAPE). This NAPE intermediate is then converted by a phospholipase D (PLD) into an NAE and phosphatidic acid (15). The identity of the transacylase remains unknown; however, a candidate enzyme responsible for the PLD step of NAE biosynthesis has recently been molecularly characterized (16). This NAPE- PLD enzyme, which belongs to the metallo-lactamase family of phosphodiesterases, is activated by calcium (17) and highly expressed in brain (16), suggesting that it may be a principal enzyme responsible for the generation of NAE signals in the nervous system. Here, we have tested the contribution that NAPE-PLD makes to NAE biosynthesis in vivo by generating and characterizing mice with a targeted disruption of the NAPE-PLD gene.
EXPERIMENTAL PROCEDURES
Generation of NAPE-PLD(-/-) Mice. The NAPE-PLD gene was obtained as part of a commercial BAC clone (Invitrogen). The gene disruption construct was generated using PCR-amplified 5′ and 3′ homologous recombination fragments surrounding exon 4 of the NAPE-PLD gene, which were subcloned into the SalI and XhoI sites, respectively, that reside on opposing sides of a PGK-Neo cassette in the pKO-NTKV vector. Primers for 5′ homologous end: 5′-TCT TCC CTG GGC CAT TCC-3′; 5′-GCC AGC TGG TCT CTG TGC-3′. Primers for 3′ homologous end: 5′-TTC CAA TCC CTG CCC TGC AGC-3′; 5′-CAA GAC GCA CAA GCA GGC AG-3′. Homologous recombinant 129SvJ embryonic stem cell clones were identified by Southern blot analysis, and two such clones were used to generate chimeric mice on a C57BL/6 background. Chimeras from one of the two clones gave germline transmission of the mutated gene. All mice used in this study were second or third generation offspring from intercrosses of 129SvJ-C57BL/6 PLD+/- mice. See Supporting Information for a full genotyping protocol.
Western Blotting. Tissue homogenates were centrifuged at 100000g for 1 h to generate membrane and soluble fractions. Membrane protein (75 μg) from each tissue was analyzed by standard SDS-PAGE (10% NuPAGE Bis-Tris gel, Invitrogen) and Western blotting procedures using anti-NAPE-PLD polyclonal antibodies (1:500 dilution) generated against an NAPE-PLD-GST fusion protein (antibodies kindly provided by Dr. Ken Mackie).
Reverse Transcription-PCR Analysis of NAPE-PLD mRNA Expression. NAPE-PLD primers: 5′-CAGCGGCGTTC-CAGGTTCC-3′ and 5′-GCTCCGATGGGAATGGCCGC-3′. GAPDH primers: 5′-TGTCTTCACCACCATGGAGAAG-GC-3′ and 5′-TGGCAGTGATGGCATGGAACTGTGG-3′. Two primers spanning exons 3 and 4 (deleted exon) were used in the SuperScript III one-step RT-PCR system with Platinum Taq DNA polymerase (Invitrogen) following the manufacturer’s recommended protocol. cDNA synthesis was performed at 55 °C for 30 min followed by initial denaturation at 94 °C for 2 min. PCR amplification was performed at a denaturing temperature of 94 °C for 15 s followed by annealing at 60 °C for 30 s and extension at 68 °C for 30 s (20-30 cycles).
NAPE-PLD Activity Assay. Tissue homogenates (100 μg total protein) were incubated with 100 μM 1,2-dioleoyl-N-[14C]-acyl PE (synthesized as described in Supporting Information) in 100 μL of 50 mM Tris-HCl pH 8.0, with or without 10 mM CaCl2 at 37 °C for 1.5 h. Except where indicated, assays were conducted with an N-[14C]-palmitoyl PE substrate. Reactions were stopped by the addition of a mixture of chloroform/methanol (2:1 v/v, 1.5 mL) and water (400 μL). After extraction, the organic layer was concentrated under a stream of nitrogen and the mixture was resuspended in 20 μL of chloroform/methanol and spotted on a silica gel thin layer plate (5 cm) and developed in methanol, chloroform, ammonium hydroxide (10:90:1 v/v). Distribution of radioactivity on the plate was quantified by a phosphorimager (Packard), where NAE and NAPE signals were identified by comparison to 14 C-lipid standards. The relative [14C]-NAE concentration was calculated from percentage values of the total radioactivity (NAE + NAPE) on the TLC plates.
Measurement of Brain Levels of NAEs and NAPEs. Mice were anesthetized by CO2/O2 and killed by decapitation. Brains were removed and immediately snap frozen using liquid nitrogen. Each tissue was weighed and subsequently dounce homogenized in 8 mL of a chloroform:methanol: Tris pH 8.0 (2:1:1) solution containing standards for NAE or NAPE measurements (0.02 and 0.2 nmol of d4-anandmide and d4-OEA, respectively, for NAE measurements; 10 nmol of N-C15:0 PE for NAPE measurements). NAE measurements were performed by LC-electrospray MS using an Agilent 1100-MSD SL instrument as described previously (18,19). For NAPE measurements, each tissue homogenate was poured into an 8 mL glass vial and centrifuged for 10 min at 1400g to separate phases. The bottom organic layer was isolated and concentrated under a stream of nitrogen. Hydrolysis was carried out by vigorously stirring the concentrated lipid fraction using 2 mL of a chloroform: methanol:2 N LiOH (2:1:1) solution. After 6 h, the solution was quenched with 0.5 mL of 3 N HCl. The organic layer was isolated and concentrated under a stream of nitrogen. This layer was then dissolved in chloroform and directly analyzed by LC-MS. Hydrolyzed lipids were normalized to the N-C15:0 NAPE standard. See Supporting Information for details on experiments performed with the FAAH inhibitor URB597.
RESULTS
Targeted Disruption of the NAPE-PLD Gene. To generate mice lacking NAPE-PLD [NAPE-PLD(-/-) mice], exon 4 of the NAPE-PLD gene was removed by homologous recombination. This exon encodes the majority of the protein sequence (aa 98-313; total length of the protein is 396 amino acids), including the conserved HXHXDH catalytic residues responsible for zinc-binding and catalysis in the metallolactamase family (20) (Figure 1A). Two homologously recombinant 129S6/SvEv embryonic stem cell clones were identified by Southern blotting (Figure 1B) and used to generate chimeric mice on a C57Bl/6 background. One of these clones gave germline transfer of the mutated gene (Figure 1C) and was used to create NAPE-PLD(-/-) mice on an outbred background.
Figure 1.
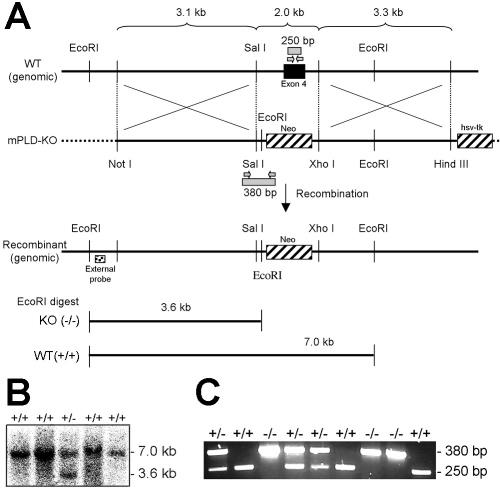
Generation of NAPE-PLD(-/-) mice. (A) The genomic structure surrounding the deleted NAPE-PLD exon 4. Only relevant restriction sites are designated. The deleted exon encodes amino acids 98-313, including the conserved HXHXDH catalytic motif.(B) Southern blot analysis of EcoRI digested embryonic stem cell genomic DNA using the indicated external probe, where the 3.6 and 7.0 kb bands correspond to NAPE-PLD(-/-) and -(+/+) genotypes, respectively. (C) PCR analysis of mouse genomic DNA, where the 250 and 380 bp bands correspond to NAPE-PLD(+/+) and -(-/-) genotypes, respectively.
NAPE-PLD(-/-) mice were born at the expected Mendelian frequency, were viable and healthy, and showed no overt differences in their cage behavior compared to wild type littermates. Reverse-transcription PCR (Figure 2A) and Western blotting (Figure 2B) with anti-NAPE-PLD antibodies confirmed the absence of NAPE-PLD mRNA and protein in tissues from NAPE-PLD(-/-) mice. Tissues from NAPE- PLD(-/-) mice showed significantly lower NAPE-PLD catalytic activity with an N-palmitoyl (C16:0) PE substrate that ranged from as much as a 10-fold (spinal cord) to 2-fold (testis) reduction compared to wild type tissues (Figure 3A). Brain tissue from NAPE-PLD(-/-) mice also displayed significantly lower activity with the polyunsaturated substrate N-arachidonoyl (C20:4) NAPE, while testis tissue from NAPE-PLD(+/+) and -(-/-) animals did not differ significantly in activity with this lipid (Figure 3B). These data confirm that NAPE-PLD is a principal enzyme responsible for the calcium-dependent conversion of NAPEs to NAEs observed in mouse tissue homogenates.
Figure 2.

Absence of NAPE-PLD mRNA and protein in tissues from NAPE-PLD(-/-) mice. (A) Reverse-transcription PCR analysis of tissues from NAPE-PLD(+/+) and -(-/-) mice. Amplification of the mRNA for the housekeeping enzyme GAPDH was used as a control for mRNA integrity. (B) Western blot of nervous system tissues from NAPE-PLD(+/+) and -(-/-) mice using anti-NAPE-PLD polyclonal antibodies (generously donated by K. Mackie).
Figure 3.
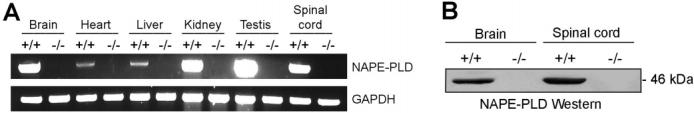
NAPE-PLD activity in tissues from NAPE-PLD(+/+) and -(-/-) mice. (A) Assays were conducted with membrane fractions for NAPE-PLD(+/+) (black bars) and -(-/-) (white bars) tissues as described in Experimental Procedures with N-palmitoyl (C16:0) PE as a substrate. Similar results were obtained with soluble fractions (data not shown). (B) Assays conducted with N-arachidonoyl (C20:4) PE and whole tissue homogenates. **p < 0.01 for NAPE-PLD(-/-) versus -(+/+) tissues (planned comparison). The results are presented as means (standard error (SE). n) 3-4 mice/group.
Endogenous Levels of NAEs and NAPEs in NAPE-PLD-(-/-) Mice. We next measured endogenous brain levels of NAEs and NAPEs in NAPE-PLD(+/+) and -(-/-) mice by liquid chromatography mass spectrometry (LC-MS). Mass measurements were performed by selected ion monitoring and normalized relative to an external non-natural standard (18,19). Significant reductions in the levels of saturated and monounsaturated NAEs were observed in NAPE-PLD(-/-) brains (Figure 4A). These changes were most dramatic for very long chain saturated NAEs (e.g., C20: 0-C24:0), which were 5-10-fold lower in NAPE-PLD-(-/-) brains, while shorter chain NAEs showed a more modest (∼50%) reduction (Figure 4B). In striking contrast to these findings, however, no changes in the levels of the polyunsaturated NAEs, anandamide (C20:4) and C22:6-NAE, were detected in NAPE-PLD(-/-) brains (Figure 4A,B). Complementary profiles of NAPEs were found in NAPE- PLD(+/+) and -(-/-) brains, with the latter samples possessing 5-15-fold higher levels of saturated and monounsaturated N-acyl NAPEs, but little or no (<2-fold) change in polyunsaturated members of this lipid class (Figure 4C,D).
Figure 4.
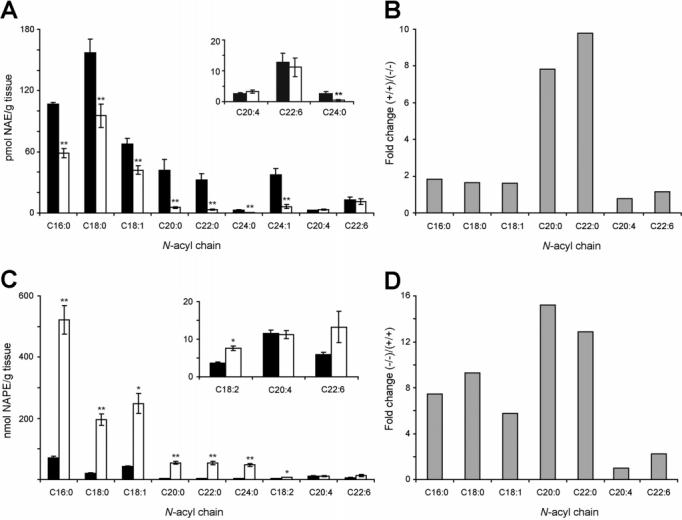
Brain levels of NAEs and NAPEs in NAPE-PLD(+/+) and -(-/-) mice. (A and C) Brain NAE (A) and NAPE (C) levels from NAPE-PLD(+/+) (black bar) and -(-/-) (white bar) mice. Insets: Levels of representative low-abundance NAEs and NAPEs. (B and D) Average fold-change in brain levels of representative NAEs (B) and NAPEs (D) in NAPE-PLD(+/+) and -(-/-) mice. **p < 0.01 for NAPE-PLD(-/-) versus -(+/+) tissues (planned comparison). The results are presented as means (standard error (SE). n) 5-6 mice/group.
The Effect of Blocking NAE Degradation in NAPE-PLD- (-/-) Mice. Although NAPE-PLD did not appear to regulate basal levels of anandamide and other polyunsaturated NAEs, it was possible that compensating alterations in the levels of FAAH in NAPE-PLD(-/-) mice might have confounded these measurements. However, no significant changes in FAAH level or activity were observed in NAPE-PLD(-/-) tissues (Supplementary Figure 1, Supporting Information), arguing against a compensatory change in degradation rate for NAEs. We next considered whether NAPE-PLD might selectively participate in the biosynthesis of polyunsaturated NAEs under conditions of elevated signaling tone, as is observed following blockade of FAAH (8,12). To address this question, we crossed NAPE-PLD(-/-) mice with FAAH(-/-) mice (8) and compared the brain levels of NAEs in these double-knockout mice to those observed in FAAH(-/-) mice. As reported previously (8,18), FAAH- (-/-) mice displayed highly elevated levels of NAEs compared to wild type mice (Supplementary Table 1, Supporting Information). The elevated levels of long chain saturated NAEs observed in FAAH(-/-) mice were markedly attenuated in NAPE-PLD(-/-)/FAAH(-/-) mice (Figure 5A,B). In contrast, brains from FAAH(-/-) and NAPE-PLD(-/-)/FAAH(-/-) mice possessed similar levels of polyunsaturated NAEs, including C18:2, C20:4 (anandamide), and C22:6 (Figure 5A,B). Overall, these results were comparable to those observed in NAPE-PLD(+/+) and -(-/-) mice, further supporting a primary role for NAPE- PLD in the biosynthesis of long chain saturated, but not polyunsaturated NAEs.
Figure 5.
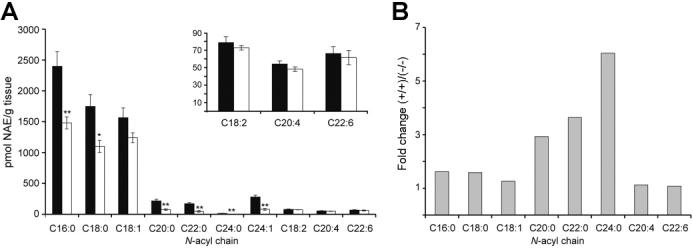
Brain levels of NAEs in NAPE-PLD(-/-)/FAAH(-/-) mice. (A) Brain NAE levels from NAPE-PLD(+/+)/FAAH(-/-) (black bar) and NAPE-PLD(-/-)/FAAH(-/-) (white bar) mice. Inset: Representative polyunsaturated NAE levels. (B) Average fold-change in brain levels of representative NAEs in NAPE-PLD(+/+) and -(-/-) mice on a FAAH(-/-) background. **p < 0.01 for NAPE- PLD(-/-)/FAAH(-/-) versus NAPE-PLD(+/+)/FAAH(-/-) tissues (planned comparison). The results are presented as means (standard error (SE). n) 5-6 mice/group.
FAAH(-/-) mice possess chronically elevated NAEs in their nervous system, and therefore crossing these animals with NAPE-PLD(-/-) mice does not directly test whether NAPE-PLD might contribute to acute changes in anandamide tone. Previous studies have shown that the FAAH inhibitor URB597 causes a significant increase in brain levels of anandamide that peaks at approximately 1-2 h following treatment (21). To test whether NAPE-PLD contributes to the transient increase in anandamide observed following pharmacological inhibition of FAAH, the levels of this NAE were compared in NAPE-PLD(+/+) and -(-/-) mice treated with URB597 (10 mg/kg, ip) or vehicle control. URB597 caused a similar magnitude increase in brain anandamide levels in NAPE-PLD(+/+) and -(-/-) mice at 2 h post-treatment (Supplementary Figure 2, Supporting Information), indicating that the inactivation of NAPE-PLD does not significantly impair the accumulation of anandamide that follows FAAH inhibition.
An Alternative Biochemical Pathway for NAE Production Revealed in NAPE-PLD(-/-) Mice. The simplest interpretation of the results obtained with NAPE-PLD(-/-) mice is that another enzyme or enzymes exist in brain that contribute to the biosynthesis of NAEs, especially those bearing polyunsaturated acyl chains. Consistent with this premise, assays performed in the absence of calcium revealed strong NAPE-PLD activity in brains from NAPE-PLD(-/-) mice that was nearly equivalent in level to the calcium-independent activity detected in NAPE-PLD(+/+) brains (Figure 6A). Interestingly, this activity was partially inhibited by calcium in either brain or testis tissue (Figure 6B), suggesting that standard conditions under which NAPE-PLD assays are performed [i.e., in the presence of 10 mM CaCl2 (16)] may underestimate the contribution that additional PLD enzymes make to NAE biosynthesis. The calcium-independent PLD activity was found in both membrane and soluble fractions of brain (Figure 6C) and accepted both medium-chain saturated and polyunsaturated NAPEs as substrates (Figure 6D). Attempts to measure the activity of this enzyme with longer chain saturated NAPEs (C20:0 or greater) were thwarted by the low solubility of these lipids (data not shown).
Figure 6.

A calcium-independent NAPE-PLD activity present in tissues from NAPE-PLD(-/-) mice. (A) Significant reductions in brain NAPE-PLD activity in NAPE-PLD(-/-) mice are observed only in the presence of calcium (10 mM CaCl2; left bars, data taken from Figure 3B). In the absence of calcium (right bars), similar NAPE-PLD activity is observed in NAPE-PLD(+/+) and NAPE-PLD(-/-) mice. (B) The NAPE-PLD activity of NAPE-PLD(-/-) tissues is increased in assays conducted without calcium. (C) The calcium-independent NAPE-PLD activity of NAPE-PLD(-/-) brains is enriched in the membrane fraction. (D) Similar levels of calcium-independent NAPE- PLD activity are observed for N-palmitoyl (C16:0) and N-arachidonoyl (C20:4) PE substrates in NAPE-PLD(-/-) brains. **p < 0.01 for NAPE-PLD(-/-)/FAAH(-/-) versus NAPE-PLD(+/+)/FAAH(-/-) tissues (planned comparison). The results are presented as means (standard error (SE). For A, n) 3-4 mice/group. For B-D, n) 3-4 independent experiments/group.
DISCUSSION
The NAE family of lipid transmitters has been implicated in the regulation of numerous physiological and pathological processes, including pain (22), inflammation (4), feeding (5), and emotional/cognitive state (12). Nonetheless, how NAE signaling events are actually controlled in vivo remains enigmatic. NAEs are generally assumed to be biosynthesized at the time of their intended action [often referred to as “on-demand” synthesis (1)], a process that may be stimulated by increases in intracellular calcium (23,24). NAEs are then rapidly degraded by catabolic enzymes such as FAAH (7) to ensure tight temporal and spatial control over their signaling function. This model for NAE signaling is attractive in that it posits a special role for these lipids as “activity-dependent” intercellular messengers in the nervous system (as well as potentially peripheral tissues). Nonetheless, efforts to test this model have been hindered by a lack of understanding of the enzymes involved in NAE biosynthesis.
The recent characterization of a PLD in mammals capable of converting NAPEs to NAEs (16) offered the first potential example of an enzyme that contributes to NAE biosynthesis in vivo. Consistent with a role for this NAPE-PLD in NAE biosynthesis, overexpression of this enzyme in CHO-K1 or HEK-293 cells led to a 1.5-fold increase in the levels of NAEs, including anandamide (25). Also, the levels of NAPE- PLD activity and anandamide in mouse uterus show a strong correlation during the implantation process, suggesting that this enzyme may regulate anandamide production during pregnancy (26). Finally, examination of a Saccharomyces cerevisiae strain in which a yeast homologue of NAPE-PLD was deleted revealed a significant (60%) decrease in saturated and monounsaturated NAEs∼(27). Still, none of these studies have directly tested whether NAPE-PLD is required for NAE biosynthesis and, in particular, anandamide biosynthesis in mammals in vivo. To address this question, we have created NAPE-PLD(-/-) mice and measured NAPE-PLD activity and endogenous levels of NAPEs and NAEs in tissues from these animals. Although a large decrease in calcium-dependent NAPE-PLD activity was observed for both saturated and polyunsaturated NAPE substrates in NAPE-PLD(-/-) brains, only saturated (and monounsaturated) NAEs were decreased in level in these tissues. The magnitude of these reductions escalated with the length of the N-acyl chain, such that the very long chain NAEs (C20:0 or greater) showed a dramatically (>5-fold) lower concentration in NAPE-PLD(-/-) brains. In contrast, polyunsaturated NAEs, including anandamide, were unaltered in brains from NAPE-PLD(-/-) mice. Complementary changes in NAPE levels were observed in NAPE-PLD(-/-) brains, which showed significantly higher levels of N- saturated/monounsaturated NAPEs, but little or no alteration in N-polyunsaturated NAPEs. Similar results were obtained with NAPE-PLD(-/-)/FAAH(-/-) mice or NAPE-PLD- (-/-) mice treated with the FAAH inhibitor URB597, indicating that NAPE-PLD’s selective contribution to the biosynthesis of saturated/monounsaturated NAEs was preserved under conditions of heightened endogenous NAE tone.
We interpret our findings with NAPE-PLD(-/-) mice to indicate that an additional NAE biosynthetic pathway exists in brain that shows overlapping, but distinct substrate selectivity compared to NAPE-PLD (Figure 7). The dramatic impact of NAPE-PLD deletion on the levels of long chain saturated NAEs suggests that this enzyme is a principal regulator of these lipids in the nervous system. In contrast, the biosynthesis of polyunsaturated NAEs, including anandamide, which was unaffected by NAPE-PLD inactivation, appears to be predominately controlled by other enzymes. The shorter chain saturated and monounsaturated NAEs [e.g., PEA (C16:0), OEA (C18:1)], which showed modest, but significant decreases in the brains of NAPE-PLD(-/-) mice, may be partially regulated by both NAPE-PLD-dependent and independent pathways. Whether this NAPE-PLD independent pathway is mediated by another PLD enzyme or reflects a mechanistically distinct route for NAE biosynthesis (28) remains unknown. Consistent with the former possibility, however, significant NAPE-PLD activity was measured in brains from NAPE-PLD(-/-) mice, especially when assays were run in the absence of calcium.
Figure 7.
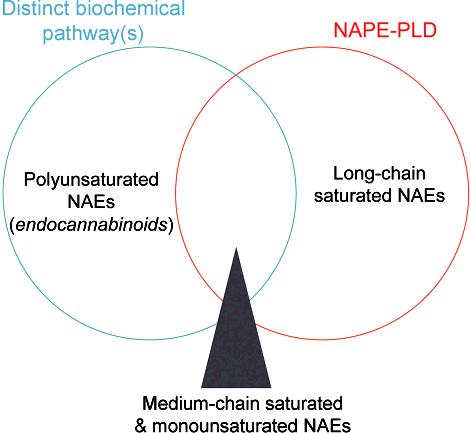
A Venn diagram model highlighting the relative contribution of NAPE-PLD and other biochemical pathways to the biosynthesis of specific subsets of the NAE family.
Finally, it is important to note that our genetic studies do not necessarily exclude a role for NAPE-PLD in anandamide biosynthesis in vivo. For example, compensatory changes may have occurred in NAPE-PLD(-/-) mice that resulted in an underestimation of the role that NAPE-PLD plays in polyunsaturated NAE production in wild type mice. Likewise, in this study, we have focused mostly on evaluating NAPE-PLD function in whole brain, and it is possible that this enzyme could play a significant role in anandamide biosynthesis in specific neural circuits and/or peripheral tissues [e.g., uterus (26)]. Regardless, our initial findings with NAPE-PLD(-/-) mice illuminate a previously unrecognized complexity underlying NAE metabolism, with multiple enzymes making distinct contributions to the biosynthesis of specific subsets of these signaling lipids in vivo. These results have significant implications for our understanding of NAE signaling in vivo. For example, in contrast to the generally accepted model in which all NAEs are produced by a common cellular pathway without particular regard for acyl-chain identity (15,29), we can now entertain the provocative possibility that cells exert significant control over the biosynthesis of specific NAEs by expressing distinct NAPE-PLD enzymes. From a drug discovery perspective, the ability to selectively regulate anandamide biosynthesis without altering the levels of most other NAEs may offer a potentially attractive alternative to cannabinoid receptor antagonists (30) for the treatment of smoking cessation, obesity, and other metabolic disorders.
ACKNOWLEDGMENT
We thank K. Mackie for providing us with the rabbit polyclonal antibodies specific for NAPE-PLD, H. Hoover for technical assistance, and the Cravatt lab for helpful discussions.
Footnotes
This work was supported by the National Institutes of Health (DA015197 and DA017259), the Helen L. Dorris Child and Adolescent Neuro-Psychiatric Disorder Institute, and the Skaggs Institute for Chemical Biology. D.L. is supported by a NARSAD Young Investigator Fellowship. A.S. is supported by a Merck Fellowship of the Life Science Research Foundation and a Burroughs Wellcome Fund Career Award.
- FAAH
- fatty acid amide hydrolase
- NAE
- N-acyl ethanolamine
- NAPE
- N-acyl phosphatidylethanolamine
- PLD
- phospholipase D.
SUPPORTING INFORMATION AVAILABLE
Expanded methods section, a table showing elevated NAE levels in FAAH(-/-) mice, and two figures showing equivalent FAAH expression in NAPE-PLD(+/+) and -(-/-) mice and equivalent anandamide levels in NAPE- PLD(-/-) mice treated with the FAAH inhibitor URB597. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–8. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- 2.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 2004;73:321–54. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 3.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–9. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 4.Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents. Curr. Med. Chem. 2002;9:663–74. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–12. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 6.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–7. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 7.McKinney MK, Cravatt BF. Structure and function of Fatty Acid amide hydrolase. Annu. Rev. Biochem. 2005;74:411–32. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 8.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9371–6. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109:319–27. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–12. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 11.Lichtman AH, Leung D, Shelton C, Saghatelian A, Hardouin C, Boger D, Cravatt BF. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J. Pharmacol. Exp. Ther. 2004;311:441–8. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- 12.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 13.Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10821–6. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br. J. Pharmacol. 2005;146:467–76. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmid HH. Pathways and mechanisms of N-acylethanolamine biosynthesis: can anandamide be generated selectively. Chem. Phys. Lipids. 2000;108:71–87. doi: 10.1016/s0009-3084(00)00188-2. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J. Biol. Chem. 2004;279:5298–305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- 17.Ueda N, Liu Q, Yamanaka K. Marked activation of the N-acylphosphatidylethanolamine-hydrolyzing phosphodiesterase by divalent cations. Biochim. Biophys. Acta. 2001;1532:121–7. doi: 10.1016/s1388-1981(01)00120-2. [DOI] [PubMed] [Google Scholar]
- 18.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased seizure susceptibility and pro-convulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J. Neurosci. 2003;23:3916–23. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry. 2004;43:14332–9. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 20.Daiyasu H, Osaka K, Ishino Y, Toh H. Expansion of the zinc metallohydrolase family of the beta-lactamase fold. FEBS Lett. 2001;503:1–6. doi: 10.1016/s0014-5793(01)02686-2. [DOI] [PubMed] [Google Scholar]
- 21.Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and ole-oylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005;313:352–8. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- 22.Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc. Natl. Acad. Sci. U.S.A. 1999;96:12198–203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–91. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 24.Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J. Neurosci. 1996;16:3934–42. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto Y, Morishita J, Wang J, Schmid PC, Krebsbach RJ, Schmid HH, Ueda N. Mammalian cells stably overexpressing N-acylphosphatidylethanolamine-hydrolysing phospholipase D exhibit significantly decreased levels of N-acylphosphatidylethanolamines. Biochem. J. 2005;389:241–7. doi: 10.1042/BJ20041790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Y, Wang H, Okamoto Y, Ueda N, Kingsley PJ, Marnett LJ, Schmid HH, Das SK, Dey SK. N-acylphosphatidylethanolamine-hydrolyzing phospholipase D is an important determinant of uterine anandamide levels during implantation. J. Biol. Chem. 2005;280:23429–32. doi: 10.1074/jbc.C500168200. [DOI] [PubMed] [Google Scholar]
- 27.Merkel O, Schmid PC, Paltauf F, Schmid HH. Presence and potential signaling function of N-acylethanolamines and their phospholipid precursors in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2005;1734:215–9. doi: 10.1016/j.bbalip.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Liu J, Wang L, Harvey-White J, Kunos G. ICRS 2005 Symposium; 2005. Abstract 119. [Google Scholar]
- 29.Patricelli MP, Cravatt BF. Proteins regulating the biosynthesis and inactivation of neuromodulatory fatty acid amides. Vitam. Horm. 2001;62:95–131. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez JR, Allison DB. Rimonabant Sanofi-Synthelabo. Curr. Opin. InVest. Drugs. 2004;5:430–5. BI060163L. [PubMed] [Google Scholar]