

Abstract
OmpG, a monomeric pore-forming protein from Escherichia coli outer membranes, was refolded from inclusion bodies and crystallized in two different conformations. The OmpG channel is a 14-stranded β-barrel, with short periplasmic turns and seven extracellular loops. Crystals grown at neutral pH show the channel in the open state at 2.3 Å resolution. In the 2.7 Å structure of crystals grown at pH 5.6, the pore is blocked by loop 6, which folds across the channel. The rearrangement of loop 6 appears to be triggered by a pair of histidine residues, which repel one another at acidic pH, resulting in the breakage of neighbouring H-bonds and a lengthening of loop 6 from 10 to 17 residues. A total of 151 ordered LDAO detergent molecules were found in the 2.3 Å structure, mostly on the hydrophobic outer surface of OmpG, mimicking the outer membrane lipid bilayer, with three LDAO molecules in the open pore. In the 2.7 Å structure, OmpG binds one OG and one glucose molecule as sugar substrates in the closed pore.
Keywords: 2D crystallization, membrane protein, protein function, X-ray crystallography
Introduction
The outer membrane of Gram-negative bacteria acts as a selective permeability barrier and prevents uncontrolled exchange of solutes and nutrients such as sugars, nucleotides, amino acids and ions (Benz et al, 1978; Nikaido, 2003). The uptake of these substances from the medium to the periplasm occurs through channel-forming integral membrane proteins known as outer-membrane porins, which function as molecular filters. Outer-membrane porins were among the first membrane proteins to be crystallized (Garavito and Rosenbusch, 1980), and high-resolution structures of the pore-forming proteins OmpF (Cowan et al, 1992), PhoE (Cowan et al, 1992), LamB (Schirmer et al, 1995), ScrY (Forst et al, 1998) and others (Weiss et al, 1991) have been determined. So far, all outer-membrane porins involved in solute uptake are membrane-spanning β-barrels of 16 or 18 strands surrounding an aqueous pore. They conform to a common pattern whereby three monomers associate into trimers. This is their oligomeric state in 2D (Jap, 1989; Sass et al, 1989) and 3D crystals, and also their functional state in lipid bilayers (Benz et al, 1978).
Other β-barrel outer-membrane proteins do not conform to this pattern. Tsx, a distorted 12-stranded barrel (Ye and Van den Berg, 2004) and FadL, a 14-stranded barrel (van den Berg et al, 2004) form narrow channels specific for nucleosides or long fatty acids, respectively. Small 8-stranded β-barrels seem to serve primarily as membrane anchors (OmpA), or promote bacterial adhesion to mammalian cells (OmpX), and may be promising candidates for vaccine development (NspA). Others are membrane-bound enzymes, such as the protease OmpT or the phospholipase OmpLA. Yet others are large barrels with a plug domain, and are specifically adapted to the sequestering of essential compounds such as siderophores for iron uptake (e.g. FhuA; Ferguson et al, 1998), or vitamin B12 (BtuB; Kurisu et al, 2003), demonstrating the great versatility of β-barrel outer-membrane proteins. Similar β-barrel-forming membrane proteins are thought to participate in solute exchange and protein translocation in the outer membranes of chloroplasts (Schleiff et al, 2003) and mitochondria (Pfanner and Wiedemann, 2002).
In Escherichia coli the main porins for sugar uptake are LamB (Szmelcman et al, 1976) and ScrY (Schmid et al, 1991). In mutants where LamB is either nonfunctional or deleted, the uptake of sugars is facilitated by OmpG (Misra and Benson, 1989). The gene for OmpG encodes a polypeptide with 301 amino acids, and the mature OmpG protein shows all features of an outer-membrane porin: a signal sequence of 21 amino acids at its N-terminus, which is cleaved during export, absence of long hydrophobic stretches, lack of cystein residues (Fajardo et al, 1998) and a C-terminal phenylalanine, which is important for membrane insertion (Struyve et al, 1991). Circular dichroism spectroscopy of detergent-solubilized OmpG indicated that, like other outer-membrane porins, it consists largely of β-sheet with little, if any, α-helix content. Proteoliposome swelling assays have shown that OmpG is a nonselective channel for mono-, di- and trisaccharides, with an unusually large limiting pore diameter of 2 nm (Fajardo et al, 1998).
Chemical crosslinking and two-dimensional electrophoresis suggested that OmpG is a monomer (Fajardo et al, 1998), unlike the classical outer-membrane porins which are trimers (Nikaido, 2003). Projection maps at 6 Å resolution obtained by electron cryo-microscopy of two different two-dimensional (2D) crystal forms revealed a ring-shaped density indicative of a monomeric β-barrel, with no evidence of oligomer formation. Initial secondary structure predictions of OmpG suggested 16 β-strands (Fajardo et al, 1998), but more recent estimates (Conlan et al, 2000; Behlau et al, 2001) put this number at 14, which agrees better with the diameter of the β-barrel in the projection maps.
To investigate OmpG and its role in nutrient uptake in detail, we expressed OmpG at high level in E. coli, refolded it from inclusion bodies, crystallized the refolded protein and determined its structure in two different states. OmpG is indeed a 14-stranded β-barrel and a pore-forming monomer. A long extracellular loop assumes two distinct, well-defined conformations, apparently in response to the pH of the medium. At neutral pH this loop projects into the extracellular medium, leaving the pore wide open, whereas at low pH it folds across the pore channel and blocks it, suggesting a direct role in pH-dependent pore gating.
Results and discussion
Pore architecture
OmpG forms a β-barrel of 14 antiparallel β-strands (S1–S14, Figure 1). The orientation of OmpG in the membrane is indicated by the location of the N and C termini next to one another, which are on the periplasmic side in all outer-membrane porins. The β-strands are connected by six short turns (T1–T6) of three to five residues on the periplasmic side, and by 7 longer loops (L1–L7) on the extracellular side. L4 contains one full turn of an α-helix, whereas the short helices found so far in loops and turns of other outer-membrane porins are of the 310 variety (Cowan et al, 1992, 1995; Dutzler et al, 1999). The average angle between the β-strands and the membrane plane is ∼60°. Except for the first four N-terminal residues, the main chain was traced completely both in the 2.3 Å map derived from triclinic crystals and in the 2.7 Å map of the orthorhombic crystals (Table I). All side chain densities were well-defined except for the loop residues 24, 26, 58–62 and 224–227, indicating a degree of flexibility in L1, L2 and L6.
Figure 1.
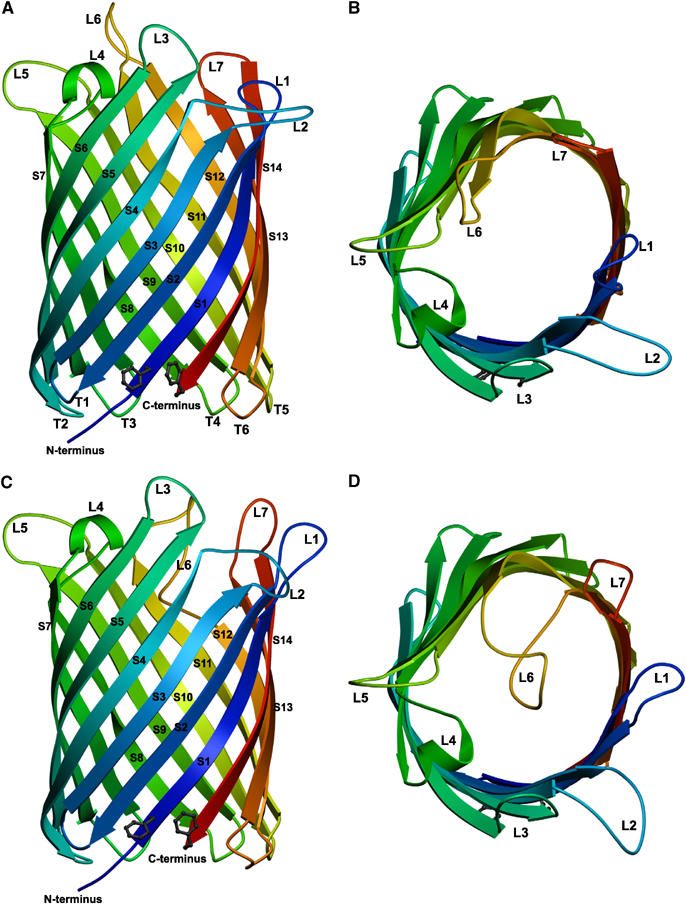
Overall structure of OmpG, showing the 14-stranded, antiparallel β-barrel in the open (A, B) and closed (C, D) conformation viewed along the membrane (A, C) and from the extracellular side (B, D). The β-strands S1–S14 are rainbow-coloured starting from the N terminus (blue) to the C terminus (red) on the periplasmic side, where adjacent strands are connected by short turns T1–T6. Loops L1–L7 extend outward into the extracellular space in the open conformation, but L6 folds across the barrel entrance in the closed conformation.
Table 1.
Data collection and refinement statistics
| SeMet | |||
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | ||
| Cell dimensions (Å) | a=56.3 b=71.4 c=122.6 | ||
| α=β=γ=90° | |||
| Resolution (Å) | 20–2.9 (3.0–2.9) | ||
| Wavelength | λpeak=0.9791 | λinfl=0.9797 | λremote=0.9000 |
| Robs | 16.1 (43.5) | 12.1 (29.2) | 12.9 (34.6) |
| I/σI | 7.94 (3.22) | 6.79 (2.84) | 6.15 (2.59) |
| Completeness (%) | 99.4 (99.0) | 98.2 (97.4) | 98.1 (98.4) |
| Redundancy | 4.7 | 2.4 | 2.6 |
| Native 1 | Native 2 | |
|---|---|---|
| Data collection | ||
| Space group | P1 | P212121 |
| Cell dimensions (Å) | a=70.6 b=77.0 c=103.9 | a=70.4 b=71.1 c=191.6 |
| α=79.3° β=73.4° γ=74.3° | α=β=γ=90° | |
| Wavelength | 1.176 | 0.9764 |
| Resolution (Å) | 15–2.3 (2.4–2.3) | 20–2.7 (2.8–2.7) |
| Robs | 9.8 (24.6) | 7.5 (22.8) |
| I/σI | 7.2 (3.8) | 10.2 (4.9) |
| Completeness (%) | 94.5 (88.2) | 98.6 (99.7) |
| Redundancy | 2.95 | 5.8 |
| Refinement | ||
| Resolution (Å) | 15–2.3 | 19.8–2.7 |
| No. of unique reflections | 79 984 | 25 487 |
| Rwork/Rfree | 22.37/26.94 | 24.4/30.4 |
| No. of atoms | 12 129 | 5006 |
| Protein | 9192 | 4602 |
| B-factors | 56.6 | 48.7 |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.02 | 0.02 |
| Bond angles (deg) | 2.5 | 2.2 |
The β-strands and turns are indistinguishable in both crystal forms (Figure 1A and C), with an r.m.s.d. of 0.74 Å. The OmpG pore is elliptical in cross-section, with one side slightly flattened at strands S6–S10 (Figure 1B and D). In the centre of the membrane, the pore diameter measured between Cα atoms of opposite β-strands is 29 by 25, and 15 Å × 12 Å when taking sidechains into account. The OmpG channel is lined predominantly by charged and polar residues, mainly Glu and Arg, arranged in clusters, as well as strings of aromatic residues (Figure 2A). Interestingly, clusters of the same charge are positioned opposite one another in the barrel (Figure 2B and C). The purpose of this clustering is not clear, but it might create an electrostatic barrier that prevents ions and charged compounds from entering the channel.
Figure 2.

Stereo views of OmpG. (A) Lines of aromatic residues (‘aromatic slides') leading from the extracellular channel entrance to the pore interior, perhaps serving as guide rails for substrate molecules. Top (B) and side view (C) of charged sidechains in the pore, with clusters of positively (mostly Arg, blue) and negatively charged residues (mostly Glu, red) on opposite sides of the barrel wall. (D) Loops L6 and L7 in the open (green) and closed (purple) conformation. His231 in S12 and His261 in S13 together act as a pH-sensitive switch, with the protonated sidechains repelling one another at low pH. In the open conformation, L6 is stabilized by hydrogen bonds from His231 to Ser218 and His 261.
Open and closed conformation
Major differences were found in the extracellular loops in the two crystal forms. In the triclinic crystals, the loops constrict the outer barrel entrance slightly to an effective diameter of 8 Å × 12 Å, but otherwise they extend into the medium, leaving the pore wide open. In the orthorhombic form, the pore is obstructed on the extracellular side, mainly as a result of a substantial conformational change of L6, which folds across and into the barrel (Figure 1D). This is achieved by an increase in length from 10 to 17 residues (Figure 2D), due to the unzipping of hydrogen bonds between β-strands leading up to L6 and L7. As a result of the same effect, L7 doubles in length from four residues in the open state to eight residues in the closed conformation. L7 and the adjacent L1 move outward to make room for the reoriented L6. To some extent, the conformational changes propagate to L2, while L3, L4 and L5 remain unaffected.
Comparison of projection maps calculated from the atomic coordinates of the open and closed form (Figure 3A and B) with those obtained from 2D crystals of native (Behlau et al, 2001) and refolded OmpG (Figure 3C and D) suggested that OmpG in the 2D crystals was closed. Moreover, the structures of the native (Figure 3C) and refolded protein (Figure 3D) are indistinguishable at 6 Å resolution in projection.
Figure 3.

Projection maps of OmpG monomers calculated from atomic coordinates of the open (A) and closed conformation (B), and determined by electron crystallography of 2D crystals of the native OmpG isolated from E. coli outer membranes (Behlau et al, 2001) (C), or of 2D crystals of refolded recombinant OmpG (D), all at 6 Å resolution. Top (E, F) and side views (G, H) of the noncrystallographic OmpG dimer (E, G) in the triclinic, pH 7.5 crystal form, and the crystallographic dimer (F, H) in the orthorhombic, pH 5.6 crystal form, with the position of a potential lipid bilayer indicated in (G, H).
pH gating
The change in conformation appears to be induced by pH, as the triclinic crystals were grown at pH 7.5, while the orthorhombic crystals formed at pH 5.6. Looking for a pH-sensitive switch that might trigger this rearrangement, we found a pair of solvent-exposed histidine residues (His231 and His261) in adjacent β-strands S12 and S13 (Figure 2D), at the C-terminal end of L6 and the N-terminal end of L7, respectively. At pH 7.5 the centre-to-centre distance of the imidazole rings is 4.9 Å, whereas at pH 5.6, this distance increases to 13.7 Å, consistent with the protonated histidines repelling one another at acidic pH. This, and the refolding of L6 into a more compact conformation, might provide the energy for the unzipping of neighbouring H-bonds. At neutral pH, the two histidines would be uncharged, allowing the H-bonds to reform, and L6 to assume its extended conformation projecting into the extracellular space.
Our findings explain the pH-dependent channel gating that was observed in electrophysiological measurements with recombinant, refolded OmpG reconstituted into lipid bilayers (Conlan and Bayley, 2003). At neutral and basic pH the OmpG channel was open, but it was closed at acidic pH below pH 5. At pH 6, OmpG fluctuated between the open and closed state. These observations are entirely consistent with our two structures, which show the open conformation of OmpG at pH 7.5, and the closed conformation at pH 5.6.
In the triclinic crystals at neutral pH, OmpG binds a number of Ca2+ ions from the crystallization buffer in its hydrophilic surface regions. On the periplasmic side there are one or two per monomer at T6, and 5–8 per monomer in the extracellular loops (not shown). Most likely Gd3+, which closes the pore (Conlan et al, 2000), binds to one of the loop sites, perhaps exerting an effect similar to that of low pH on L6.
There are no reports on the potential physiological role of pH-dependent channel closure of OmpG in the literature. OmpG may be a ‘rescue porin' that is expressed under special circumstances if other outer-membrane porins are insufficient for nutrient uptake, for example, of larger oligosaccharides (Fajardo et al, 1998), or are otherwise unavailable. It is reasonable to suggest that the large, comparatively nonspecific pore of OmpG should need to be closed at ∼pH 6 or below, to prevent a massive influx of protons into the periplasm in an acidic environment, which may be particularly undesirable under conditions of nutrient stress. Previous claims that the size of the channel in other, trimeric porins is reduced at acidic pH have not been substantiated (Saint et al, 1996), and X-ray crystallography of OmpF at 3.2 Å resolution failed to show a corresponding conformational change (Saint et al, 1996). So far, OmpG is thus the only outer-membrane porin for which pH-dependent gating has been demonstrated (Conlan and Bayley, 2003) and validated by X-ray crystallography of the open and closed states.
OmpG is a monomeric outer-membrane porin
Unlike the classical trimeric porins, OmpG seems to function as a monomer (Conlan et al, 2000), and there is no evidence to suggest a physiological oligomer. Although hydrophobic interaction between OmpG monomers gives rise to apparent dimers in the 3D crystal lattice, these are clearly nonphysiological, as indicated by the up/down orientation of the two monomers (Figure 3E–H). The c12 symmetry of one 2D crystal form (Behlau et al, 2001) stipulates an alternating up and down orientation of adjacent molecules, so that OmpG is monomeric also in the lipid environment of membrane crystals. The interactions that give rise to the c12 crystal form seem to be the same as the crystal contacts found in the orthorhombic 3D crystals (Figure 3F and H). Otherwise the hydrophobic interactions between OmpG monomers in all six crystal forms we obtained appear to be different, which in itself is a strong argument against an oligomer. Nor is there any need for OmpG to oligomerize in order to form a functional pore, because its β-strands are all of similar length, and the wall of OmpG is equally high all the way round. This is not the case in the classical outer-membrane porins, which can achieve a pore wall of uniform height only by forming trimers.
The monomeric structure is fully consistent with electrophysiological measurements, which indicate individual, single channels in OmpG reconstituted into lipid bilayers (Conlan et al, 2000), while the trimeric outer-membrane porins show three connected channels (Benz et al, 1978). The pores of these 16- or 18-stranded porins are restricted by loops, which do not undergo large conformational changes (Nikaido, 2003), and are stably integrated into the larger barrels.
Detergent structure mimics lipid bilayer
In addition to the protein, we found a total of 151 elongated densities of the shape and size of detergent molecules in the triclinic unit cell, which were fitted with LDAO (Figure 4A). Another ∼20 densities that resembled partly disordered detergent molecules were left empty. Well-ordered LDAO molecules were distributed more or less evenly around the hydrophobic outside of the OmpG barrel (Figure 5A–C), except for an ∼250 Å2 area of direct hydrophobic contact between adjacent OmpG molecules (Figure 5B). Each of the four different OmpG molecules in the AU had between 27 and 44 LDAO molecules associated with it. Of these, 15 were found in the same positions in all four OmpGs, while another eight were common to two or three of them.
Figure 4.

Stereo views of structural details with electron density. (A) Five LDAO molecules (green) in the detergent layer on the outer surface of OmpG in triclinic crystals (pH 7.5) at 2.3 Å resolution. (B) Trp118 and Thr155 creating a local polar environment at the hydrophobic interface between adjacent OmpG monomers in the triclinic crystal form, accommodating an LDAO molecule with buried head group. (C) OG and glucose (yellow) with neighbouring residues in the interior of the closed pore in the orthorhombic crystals (pH 5.6) at 2.7 Å resolution (B).
Figure 5.
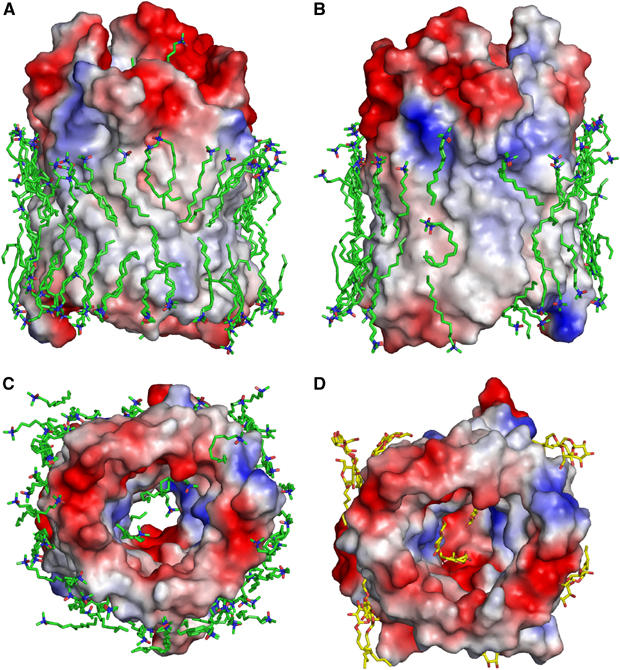
Equipotential surfaces of OmpG with bound detergent molecules. Views of the open, pH 7.5 conformation in two opposite directions along the membrane (A, B) and from the periplasmic side (C). (D) Periplasmic view of the closed pH 5.6 conformation. Two layers of LDAO detergent molecules (green in A–C) form an almost complete belt around OmpG, mimicking the outer membrane lipid on the hydrophobic barrel surface. The ∼250 Å2 hydrophobic crystal contact surface between adjacent OmpG molecules is bare (B). The curved LDAO molecule with buried head group (see Figure 4b) participates in crystal contacts. Three well-defined LDAO molecules are bound within the open pore (C). In the low-pH crystal form, OmpG binds one molecule each of OG and glucose (both yellow) in the closed pore, and 13 OG molecules on its outer hydrophobic surface, in positions that are occupied by LDAO in the other crystal form (D). Surface areas bearing a negative charge at neutral pH are red, positively charged areas are blue. Uncharged, hydrophobic surface areas are colourless.
The hydrocarbon chains of nearly all LDAO molecules around the perimeter are in van der Waals contact with the hydrophobic outer surface of the OmpG barrel over most of their length (Figure 5A and B). Some sit in distinct grooves that would be filled by lipid fatty acid chains in the native membrane. Note that OmpG was refolded without any lipid, so that the detergent can occupy all potential lipid-binding sites on the protein surface. As an ensemble, the LDAO molecules thus mimic the membrane lipid in the immediate surroundings of OmpG, and provide a striking example of an almost complete, ordered bilayer around a membrane protein.
With one exception, the polar detergent head groups of all LDAO molecules are arranged in a ring around the periplasmic and extracellular side of OmpG that would coincide with the periplasmic and exterior membrane surface. One LDAO molecule is sandwiched between two Trp118 of adjacent OmpG molecules (Figure 4B), which, together with the OH groups of Thr155, evidently create a sufficiently polar local environment to accommodate the amine oxide head group.
The hydrophobic region between the detergent head groups forms a continuous band around the outer perimeter of OmpG that is on average 26 Å wide. This correlates closely with the average distance between the belts of aromatic side chains along the periplasmic and extracellular membrane surface (Figure 6). Similar aromatic belts in other membrane proteins demarcate the hydrophobic surface immersed in the lipid bilayer. However, while this zone measures 30–35 Å in α-helical inner membrane proteins (e.g. in the bacterial reaction centre; Deisenhofer and Michel, 1989), its narrower dimension in OmpG and the other outer-membrane porins (Cowan et al, 1992; Forst et al, 1993) indicates that the hydrophobic bilayer core of the outer E. coli membrane is significantly thinner than that of the inner membrane. While inner membrane lipids typically have fatty acid chains of 16 or 18 carbon atoms, the hydrocarbon chains in outer-membrane lipids are on average four carbon atoms shorter. Indeed, five of the six hydrophobic chains of lipid A, the main component of the E. coli outer membrane outer leaflet, are in effect 12 carbon atoms long (Nikaido, 2003), the remaining one having 14 carbon atoms. With its 13 Å chain of 12 carbon atoms, LDAO is thus ideally suited to replace lipids in outer-membrane porins, which may account for the exquisite order in the first detergent shell around OmpG.
Figure 6.

Belts of aromatic residues near the periplasmic and extracellular membrane surface. Note that the conformation of these residues is the same in the open (green) and closed (purple) state of OmpG.
By contrast, the detergent surrounding OmpG in the orthorhombic crystals is less well ordered. A total of 13 OG molecules were assigned to characteristically shaped densities (not shown), while several other densities probably correspond to partly ordered OG. The binding sites of all 13 ordered OGs coincide completely or partially with those of LDAO molecules in the other crystal form. The comparatively poor order of OG molecules in the first detergent shell may be related to their shorter carbon chain, which fits the hydrophobic surface of OmpG less well than LDAO.
Crystal packing
The detergent micelles surrounding OmpG clearly extend beyond this shell of frozen LDAO molecules. A rough estimate of the micelle dimensions is obtained from the distance between OmpG molecules in the crystal lattice. Assuming a toroid micelle, and estimating the volume of one LDAO molecule from the ordered first shell as 460 Å3, one obtains a total of 70–100 LDAO per OmpG, in good agreement with the aggregation number. The majority of LDAO molecules in the micelle thus do not occupy defined positions. The triclinic OmpG lattice is characterized by layers of virtually continuous tubes of OmpG cylinders arranged head to tail (Figure 7), held together by extensive hydrophobic protein–protein contacts (Figure 5B), with alternating tubes running in opposite directions. Evidently the LDAO micelles are solid enough to ensure long range order and tight stacking of the layers, even though they are separated by ∼18 Å wide bands of apparently featureless space, containing the bulk of the detergent, plus some solvent. In the resulting, highly unusual crystal packing, there are no protein–protein contacts between layers of OmpG tubes, accounting perhaps for the extreme fragility and poor reproducibility of these crystals.
Figure 7.

Packing of OmpG in the triclinic crystal lattice (pH 7.5), viewed from three mutually perpendicular directions. Head-to-tail interaction of OmpG barrels results in quasi-continuous tubes, which pack into layers through tight interactions of hydrophobic contact surfaces (see Figure 5B). Alternating tubes point in opposite directions. Note that there is no direct protein interaction between layers, which are separated by partly ordered LDAO micelles.
Substrate binding
The open pore of each of the four OmpG molecules in the AU of the pH 7.5 crystals contains three well-ordered LDAO molecules in identical positions (Figure 5C), suggesting that they are firmly bound. The OmpG pore is thus surprisingly amphipathic. In the pH 5.6 crystals grown in the presence of OG, we found two distinct densities in the closed OmpG pore (Figures 4C and 5D), one of which fitted one complete OG molecule. The other density lacked a clear hydrocarbon tail and was fitted with a glucose molecule that either represents the head group of a second, partially disordered detergent molecule or was picked up from the cryoprotectant. The sugar head group of the complete OG molecule is coordinated by hydrogen bonds to residues Arg168, Tyr196, Glu154, Glu253 and Trp131, while the adjacent glucose molecule forms hydrogen bonds with Trp131, Tyr135 and Glu152. Both binding sites coincide with those of LDAO molecules in the other crystal form. The glucose molecule and the OG glucoside head group most likely indicate the positions of sugar binding sites in the OmpG pore, and the side chains involved in sugar binding have the same conformation in both structures.
Nonspecific uptake of oligosaccharides is one of the main roles of OmpG in E. coli (Fajardo et al, 1998). In the classical sugar-uptake porins LamB (Dutzler et al, 1996), ScrY (Forst et al, 1998) and maltoporin from Salmonella typhimurium (Meyer et al, 1997), the entry of disaccharides into the channel is thought to be facilitated by a line of aromatic residues roughly parallel to the barrel axis, sometimes referred to as the ‘greasy slide'. In OmpG we found a strikingly similar arrangement of aromatic residues Tyr98, Tyr96, Tyr94, Phe66 and Tyr50 (Figure 2A), which we prefer to call an ‘aromatic slide', as it is less conspicuous for its hydrophobicity than for its line of tyrosine OH groups. The similarity even extends to Tyr146 in L4, which corresponds exactly to Tyr118 in the restriction loop 3 of LamB, with its hydroxyl group pointing towards the aromatic slide. This similarity is all the more remarkable since it applies to a structural motif that is discontinuous in terms of polypeptide sequence, with contributions from completely different strands and loops in the two porin classes. Its presence in both OmpG and the trimer-forming sugar-uptake porins (Schirmer et al, 1995) suggests that it is an essential feature for solute uptake or pore stability that was either conserved from a most ancient precursor protein, or represents an extraordinary example of convergent evolution.
In OmpG the aromatic slide divides into two branches at Tyr96 and Tyr94. The second branch has no correspondence in the trimeric porins and continues to Phe137, Tyr135 and Trp131, with Tyr146 in L4 pointing towards F137. Interestingly, Trp131 at the end of this second branch participates in coordinating the glucose molecule, suggesting that the OH groups in the aromatic slide may indeed function as a guide rail for incoming substrate. Possibly, the presence of this extra branch makes OmpG, which may be a ‘rescue porin', more effective in the uptake of oligosaccharides.
Biotechnological potential
Because of their ruggedness, pore-forming β-barrel proteins have considerable potential in biotechnology. One candidate is the bacterial toxin α-haemolysin, a heptamer of seven identical 33 kDa monomers, which inserts into the plasma membrane of target cells, forming large (>20 Å) nonspecific and unregulated pores of 14 β-strands (Song and Gouaux, 1998). As a monomer, OmpG is clearly preferable over α-haemolysin or the trimer-forming porins in this regard. OmpG forms large, nonspecific pores, can be produced easily in large quantities, is efficiently reconstituted into artificial lipid bilayers and even forms extensive 2D lattices quite readily. Another attractive feature is its pH-dependent channel gating. Compared to other pore-forming proteins, OmpG thus seems to be not only the most efficient and economical in terms of polypeptide use, but also the most potentially versatile for biosensor development and other biotechnological applications.
Conclusion
We determined the structure of OmpG, an unusual outer-membrane porin from E. coli. Unlike the classical outer-membrane porins, which are trimers, OmpG is a structural and functional monomer. Its β-barrel has only 14 strands, whereas those of the classical outer membrane porins have 16 or 18. As another unique feature, OmpG is gated by pH, whereby its uncommonly large channel is open at neutral pH, and closed at acidic pH below 6, presumably to protect the cell against acidification of the periplasm if the protein is expressed under conditions of nutrient stress. By using crystals grown at pH 7.5 or at pH 5.6, we were able to determine the structure of OmpG both in the open and in the closed state at 2.3 and 2.7 Å resolution, respectively. The structures show that pore closure results from the rearrangement of an extracellular loop, L6, which is triggered by the repulsion of two histidine residues in neighbouring β-strands that would be protonated at acidic pH, forming a simple pH-sensitive switch. Like other porins involved in oligosaccharide uptake, OmpG has lines of aromatic residues on its inner pore surface, which may guide incoming sugar molecules into the periplasm. The remarkable correspondence of this discontinuous motif in two classes of porins that share no recognizable sequence homology appears to be a rare example of convergent evolution. OmpG was refolded from inclusion bodies in the absence of membrane lipids, which in the crystals are replaced by tightly bound detergent molecules. An unprecedented feature of the higher-resolution, open structure is an almost complete double ring of well-resolved detergent molecules, representing the first shell of the detergent micelle immobilized by tight contact with the hydrophobic outer surface of the OmpG cylinder. These detergent molecules effectively mimic the lipid bilayer of the outer membrane, which is thus seen to be only 26 Å thick. As a particularly rugged, monomeric membrane porin that is easily refolded from inclusion bodies, OmpG is predestined for biotechnological applications and biosensor development.
Materials and methods
Protein expression and purification
For 3D crystallization, the OmpG gene fragment without its signal sequence (amino acids 22–301) was cloned into the pET26b plasmid vector, resulting in an additional methionine at the N-terminus, and expressed in E. coli strain BL21(DE3)-C41 grown in TB medium. The protein formed inclusion bodies, which were collected by low-speed centrifugation after breaking the cells in a cell disruptor (Constant Systems). The pellet was washed in buffer (25 mM Tris–HCl, pH 8) containing 1 M urea and 1% Triton-X 100, and the inclusion bodies were dissolved in 8 M urea in the same buffer. The solubilized protein was loaded onto an anion exchange column, and unfolded OmpG was eluted by a NaCl step gradient. Refolding of OmpG was achieved by dilution in 1% (wt/vol) n-octy-β-D-glucopyranoside (OG), at a final urea concentration of 3 M. Refolding was monitored by SDS–PAGE, taking advantage of the difference in apparent masses of refolded (28 kDa) and unfolded (36 kDa) OmpG (Conlan et al, 2000). Subsequent ion-exchange chromatography removed small amounts of remaining, unfolded or partially refolded OmpG and served to exchange the buffer. An additional gel filtration step on a Superdex-200 16/60 column was not critical for crystallization. The protein was concentrated to ∼50 mg/ml by ultrafiltration (Centricon) or dialysis against buffer containing 20% PEG-35000 in dialysis tubes with a 12 000 Da cutoff. Buffer was exchanged either by ultrafiltration or dialysis. The desired final protein concentration was adjusted by adding an appropriate amount of buffer to the concentrated protein stock. The final yield was 20–30 mg of refolded OmpG per litre of culture. Protein purity was greater than 95%, as estimated by SDS–PAGE and Coomassie staining.
Selenomethionine-substituted OmpG was expressed similarly in BL21(DE3)-C41 cells in M9 minimal medium, suppressing methionine biosynthesis by a five-fold increase in the concentration of the amino acids Leu, Ile, Lys, Phe, Thr and Val. Precultures were grown overnight in LB medium, which was removed by gentle centrifugation prior to inoculation. Expression and purification was performed as described for the underivatized protein.
2D crystallization
2D crystals of refolded OmpG were obtained by detergent dialysis (Kühlbrandt, 1992) in the presence of E. coli polar lipids (Avanti), essentially as described (Behlau et al, 2001; Hiller et al, 2005). At lipid/protein ratios (LPR) between 0.25 to 1.5 (wt/wt), reconstituted OmpG formed 2D lattices in tubular vesicles that were ∼150 nm wide and up to 1 μm long. At lower LPR 2D crystals were extensive membrane sheets measuring several μm across. Electron micrographs of tubular 2D crystals washed with 4% unbuffered trehalose as a cryo-protectant were recorded at a specimen temperature of ∼4 K in a JEOL 3000 SFF electron microscope. Crystallographic image processing (Crowther et al, 1996) yielded projection amplitudes and phases to ∼8 Å resolution, from which projection maps were calculated.
3D crystallization
OmpG was dialyzed against buffers with different detergents including OG, n-decyl-β-D-maltopyranoside (DM), lauryldimethylamin-N-oxide (LDAO), polyoxyethylene (C8E4) and mixtures of these at final protein concentrations of 2.5, 5, 7.5, 10 and 15 mg/ml. Initial crystallisation conditions were found in Greiner 96 three-well sitting-drop plates using commercial crystallization screens (Hampton, Nextal, Jena) and a Cartesian pipetting robot. Several conditions yielded small crystals, which were optimized in Greiner one-well sitting-drop plates or in hanging drops. Two different crystal forms of data-collection quality were obtained. One crystal form grew over 3 days in hanging drops in 1 μl of 10 mg/ml OmpG in 25 mM Tris at pH 8.0 with 5 mM LDAO mixed with 1 μl of 100 mM HEPES pH=7.5, 30% PEG 4000 and 200 mM CaCl2 at 18°C. The characteristic habit of these very fragile crystals resembled a flattened rice grain of dimensions 700 × 400 × 40 μm3. They diffracted anisotropically to 1.9 Å, and belonged to the triclinic space group P1 (a=71 Å, b=107 Å, c=127 Å, α=90°, β=89.3°, γ=89.9°), with 66% solvent content and four molecules in the asymmetric unit (AU). Another crystal form grew in sitting drops for 4–5 days at 18°C in 0.4 μl of 7.5 mg OmpG in 25 mM Tris at pH 8.0 and 20 mM OG mixed with 0.4 μl of 100 mM Na-Citrate at pH 5.6, 150 mM NaCl and 12% PEG-3350. These low-pH crystals were bipyramidal and belonged to the orthorhombic space group P212121 (a=70.4 Å, b=71.1 Å, c=191.61 Å, α=β=γ=90°), with two molecules in the AU. Soaks with various heavy-metal compounds were not successful, but crystallization in the presence of GdCl3, an inhibitor of channel activity (Conlan et al, 2000), improved the low-pH crystals slightly. However, the reproducibility was low and SAD and MAD data collected on the Gd edge were not usable for phasing.
Crystals of selenomethionine-derivatized OmpG were obtained in hanging drops, by mixing 1 μl of 9 mg/ml protein in 25 mM Tris–HCl buffer at pH 8 and 20 mM OG with 1 μl of reservoir solution containing 2.1 M ammonium sulfate and 4% ethylene glycol in 100 mM Tris–HCl pH 7.5. This yielded elongated, rod-shaped orthorhombic crystals containing one molecule in the AU and 66% solvent (space group P212121, a=56.2 Å, b=71.2 Å, c=122.5 Å, α=β=γ=90°).
Data collection, structure determination and refinement
For data collection the crystals were transferred to cryoprotectant solutions and flash-frozen in liquid nitrogen. Best results for the triclinic crystals were obtained by increasing the PEG 3350 concentration to 20–30% by gradual addition of concentrated PEG solution. As the low-pH crystals of underivatized OmpG were stable only in their mother liquor, equilibrated drops with ∼25% glucose were added to the crystals, which were then also flash-frozen in liquid nitrogen. Crystals of selenomethionine-derivatized OmpG were more robust and could be frozen in different cryoprotectants. Best results were obtained with a mixture of paraffin oil and Panatone-N.
SeMet-MAD data to 2.9 Å resolution were collected at three wavelengths from a single crystal at 100 K on beamline I911-3 at the Swedish synchrotron radiation facility MAX-lab at Lund University (Table I). The data sets were processed with XDS (Kabsch, 1993). Four of the expected six selenium sites in the AU were found and used to calculate MAD phases at 3.1 Å with the program SOLVE (Terwilliger, 2004). Electron density map calculation, density modification and phase extension to 2.9 Å were performed with RESOLVE (Terwilliger, 2004), followed by automatic model building. The resulting electron density map was used to build an initial model, which was corrected manually and supplemented with COOT (Emsley and Cowtan, 2004).
Native data from both crystal forms were collected at beamline PXI and PXII at the Swiss Light Source (SLS) and processed with XDS. The initial model obtained from the SeMet-MAD data was used as a search model for the orthorhombic data set in PHASER (McCoy et al, 2005) and was rebuilt and completed in the loop regions. The partially refined model, excluding parts of L1 and L6, then served as a search model for the triclinic data set to find the four molecules in the AU. The model was subjected to iterative rounds of rebuilding into 2Fo−Fc and Fo−Fc electron density maps, and refined using COOT and REFMAC (Murshudov et al, 1997). The two molecules in the orthorhombic unit cell and the four molecules in the triclinic unit cell were averaged by applying noncrystallographic symmetry (NCS) either in REFMAC or in COOT. For structure validation we used COOT and PROCHECK in the CCP4 suite (Collaborative Computational Project, 1994).
Figures were generated using the programs POVSCRIPT (Fenn et al, 2003), PyMOL (Delano, 2004) and POV-Ray (http://www.povray.org). Superpositions were carried out with the SSM Superposition routine (Krissinel and Henrick, 2004) within COOT. For electrostatic surface potential calculation APBS (Baker et al, 2001) and PyMOL were used.
Coordinates of the open (2iwv) and closed form (2iww) of Omph have been deposited in the pdb.
Acknowledgments
We thank Janet Vonck and Deryck Mills for help with electron cryo-microscopy and image processing, Anke Terwisscha van Scheltinga for help and advice on data collection and processing, and beam line staff at MAX-lab at Lund University, Sweden, and the Swiss Light Source, Villingen, for excellent facilities and assistance during data collection.
References
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behlau M, Mills DJ, Quader H, Kühlbrandt W, Vonck J (2001) Projection structure of the monomeric porin OmpG at 6 A resolution. J Mol Biol 305: 71–77 [DOI] [PubMed] [Google Scholar]
- Benz R, Janko K, Boos W, Lauger P (1978) Formation of large, ion-permeable membrane channels by the matrix protein (porin) of Escherichia coli. Biochim Biophys Acta 511: 305–319 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. (1994) The Ccp4 suite—programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Conlan S, Bayley H (2003) Folding of a monomeric porin, OmpG, in detergent solution. Biochemistry 42: 9453–9465 [DOI] [PubMed] [Google Scholar]
- Conlan S, Zhang Y, Cheley S, Bayley H (2000) Biochemical and biophysical characterization of OmpG: a monomeric porin. Biochemistry 39: 11845–11854 [DOI] [PubMed] [Google Scholar]
- Cowan SW, Garavito RM, Jansonius JN, Jenkins JA, Karlsson R, Konig N, Pai EF, Pauptit RA, Rizkallah PJ, Rosenbusch JP, Rummel G, Schirmer T (1995) The structure of Ompf porin in a tetragonal crystal form. Structure 3: 1041–1050 [DOI] [PubMed] [Google Scholar]
- Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit RA, Jansonius JN, Rosenbusch JP (1992) Crystal-structures explain functional-properties of 2 Escherichia-coli porins. Nature 358: 727–733 [DOI] [PubMed] [Google Scholar]
- Crowther RA, Henderson R, Smith JM (1996) MRC image processing programs. J Struct Biol 116: 9–16 [DOI] [PubMed] [Google Scholar]
- Deisenhofer J, Michel H (1989) Nobel lecture. The photosynthetic reaction centre from the purple bacterium Rhodopseudomonas viridis. EMBO J 8: 2149–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano WL (2004) Use of PYMOL as a communications tool for molecular science. Abstr Papers Am Chem Soc 228: U313–U314 [Google Scholar]
- Dutzler R, Rummel G, Alberti S, Hernandez-Alles S, Phale P, Rosenbusch J, Benedi V, Schirmer T (1999) Crystal structure and functional characterization of OmpK36, the osmoporin of Klebsiella pneumoniae. Structure 7: 425–434 [DOI] [PubMed] [Google Scholar]
- Dutzler R, Wang YF, Rizkallah P, Rosenbusch JP, Schirmer T (1996) Crystal structures of various maltooligosaccharides bound to maltoporin reveal a specific sugar translocation pathway. Structure 4: 127–134 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fajardo DA, Cheung J, Ito C, Sugawara E, Nikaido H, Misra R (1998) Biochemistry and regulation of a novel Escherichia coli K-12 porin protein, OmpG, which produces unusually large channels. J Bacteriol 180: 4452–4459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn TD, Ringe D, Petsko GA (2003) POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J Appl Crystallogr 36: 944–947 [Google Scholar]
- Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W (1998) Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 282: 2215–2220 [DOI] [PubMed] [Google Scholar]
- Forst D, Schulein K, Wacker T, Diederichs K, Kreutz W, Benz R, Welte W (1993) Crystallization and preliminary X-ray diffraction analysis of ScrY, a specific bacterial outer membrane porin. J Mol Biol 229: 258–262 [DOI] [PubMed] [Google Scholar]
- Forst D, Welte W, Wacker T, Diederichs K (1998) Structure of the sucrose-specific porin ScrY from Salmonella typhimurium and its complex with sucrose. Nat Struct Biol 5: 37–46 [DOI] [PubMed] [Google Scholar]
- Garavito RM, Rosenbusch JP (1980) Three-dimensional crystals of an integral membrane protein: an initial X-ray analysis. J Cell Biol 86: 327–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Krabben L, Vinothkumar KR, Castellani F, van Rossum BJ, Kühlbrandt W, Oschkinat H (2005) Solid-state magic-angle spinning NMR of outer-membrane protein G from Escherichia coli. Chembiochem 6: 1679–1684 [DOI] [PubMed] [Google Scholar]
- Jap BK (1989) Molecular design of phoe porin and its functional consequences. J Mol Biol 205: 407–419 [DOI] [PubMed] [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D 60: 2256–2268 [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W (1992) 2-Dimensional crystallization of membrane-proteins. Quart Rev Biophys 25: 1–49 [DOI] [PubMed] [Google Scholar]
- Kurisu G, Zakharov SD, Zhalnina MV, Bano S, Eroukova VY, Rokitskaya TI, Antonenko YN, Wiener MC, Cramer WA (2003) The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat Struct Biol 10: 948–954 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr D 61: 458–464 [DOI] [PubMed] [Google Scholar]
- Meyer JE, Hofnung M, Schulz GE (1997) Structure of maltoporin from Salmonella typhimurium ligated with a nitrophenyl-maltotrioside. J Mol Biol 266: 761–775 [DOI] [PubMed] [Google Scholar]
- Misra R, Benson SA (1989) A novel mutation, cog, which results in production of a new porin protein (OmpG) of Escherichia coli K-12. J Bacteriol 171: 4105–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67: 593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N, Wiedemann N (2002) Mitochondrial protein import: two membranes, three translocases. Curr Opin Cell Biol 14: 400–411 [DOI] [PubMed] [Google Scholar]
- Saint N, Prilipov A, Hardmeyer A, Lou KL, Schirmer T, Rosenbusch JP (1996) Replacement of the sole histidinyl residue in OmpF porin from E. coli by threonine (H21T) does not affect channel structure and function. Biochem Biophys Res Commun 223: 118–122 [DOI] [PubMed] [Google Scholar]
- Sass HJ, Buldt G, Beckmann E, Zemlin F, Vanheel M, Zeitler E, Rosenbusch JP, Dorset DL, Massalski A (1989) Densely packed beta-structure at the protein lipid interface of porin is revealed by high-resolution cryo-electron microscopy. J Mol Biol 209: 171–175 [DOI] [PubMed] [Google Scholar]
- Schirmer T, Keller TA, Wang YF, Rosenbusch JP (1995) Structural basis for sugar translocation through maltoporin channels at 3.1-angstrom resolution. Science 267: 512–514 [DOI] [PubMed] [Google Scholar]
- Schleiff E, Soll J, Kuchler M, Kühlbrandt W, Harrer R (2003) Characterization of the translocon of the outer envelope of chloroplasts. J Cell Biol 160: 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid K, Ebner R, Jahreis K, Lengeler JW, Titgemeyer F (1991) A sugar-specific porin, ScrY, is involved in sucrose uptake in enteric bacteria. Mol Microbiol 5: 941–950 [DOI] [PubMed] [Google Scholar]
- Song L, Gouaux E (1998) Crystallization of the alpha-hemolysin heptamer solubilized in decyldimethyl- and decyldiethylphosphine oxide. Acta Crystallogr D 54 (Part 2): 276–278 [DOI] [PubMed] [Google Scholar]
- Struyve M, Moons M, Tommassen J (1991) Carboxy-terminal phenylalanine is essential for the correct assembly of a bacterial outer-membrane protein. J Mol Biol 218: 141–148 [DOI] [PubMed] [Google Scholar]
- Szmelcman S, Schwartz M, Silhavy TJ, Boos W (1976) Maltose transport in Escherichia coli K12. A comparison of transport kinetics in wild-type and lambda-resistant mutants as measured by fluorescence quenching. Eur J Biochem 65: 13–19 [DOI] [PubMed] [Google Scholar]
- Terwilliger T (2004) SOLVE and RESOLVE: automated structure solution, density modification, and model building. J Synchrotron Radiat 11: 49–52 [DOI] [PubMed] [Google Scholar]
- van den Berg B, Black PN, Clemons WM, Rapoport TA (2004) Crystal structure of the long-chain fatty acid transporter FadL. Science 304: 1506–1509 [DOI] [PubMed] [Google Scholar]
- Weiss MS, Abele U, Weckesser J, Welte W, Schiltz E, Schulz GE (1991) Molecular architecture and electrostatic properties of a bacterial porin. Science 254: 1627–1630 [DOI] [PubMed] [Google Scholar]
- Ye J, Van den Berg B (2004) Crystal structure of the bacterial nucleoside transporter Tsx. EMBO J 23: 3187–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]