

Abstract
The nuclear factor of activated T cell (NFAT) proteins are a family of Ca2+/calcineurin-responsive transcription factors primarily recognized for their central roles in T lymphocyte activation and cardiac valve development. We demonstrate that NFATc1 is commonly overexpressed in pancreatic carcinomas and enhances the malignant potential of tumor cells through transcriptional activation of the c-myc oncogene. Activated NFATc1 directly binds to a specific element within the proximal c-myc promoter and upregulates c-myc transcription, ultimately resulting in increased cell proliferation and enhanced anchorage-independent growth. Conversely, c-myc transcription and anchorage-dependent and -independent cell growth is significantly attenuated by inhibition of Ca2+/calcineurin signaling or siRNA-mediated knock down of NFATc1 expression. Together, these results demonstrate that ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway is an important mechanism of oncogenic c-myc activation in pancreatic cancer.
Keywords: c-myc, NFATc1/NFAT2, pancreatic cancer, proliferation
Introduction
The nuclear factor of activated T cells (NFAT) family of transcription factors consists of four closely related proteins, NFATc1 (NFATc, NFAT2), NFATc2 (NFATp, NFAT1), NFATc3 (NFATx, NFAT4), and NFATc4 (NFAT3), as well as a more distant relative, NFAT5 (TonEBP: tonicity element-binding protein). The DNA-binding domain, which shows extensive homologies to the Rel DNA-binding domain of Rel/NF-κB transcription factors, is conserved to approximately 70% among the prototypic family members NFATc1 through NFATc4, with NFAT5 showing ca. 40% sequence similarity (reviewed by Feske et al, 2003). The NFAT proteins c1–c4 are primarily regulated at the level of their subcellular localization through the actions of the Ca2+/calmodulin-dependent serine/threonine phosphatase calcineurin (Loh et al, 1996b). In resting cells, NFAT factors are located in the cytoplasm, where they exist in a heavily phosphorylated inactive state. Activation of NFAT factors is achieved through initiation of signaling pathways that produce a rise in intracellular free calcium levels. The increase in intracellular calcium concentrations leads to activation of calcineurin, which dephosphorylates multiple phosphoserines on NFAT proteins and thereby induces their nuclear translocation (Luo et al, 1996; Shibasaki et al, 1996). The process of calcium mobilization and calcineurin activation has primarily been studied in different cells of the immune system and has been shown to be triggered by engagement of surface receptors, such as the antigen receptor in T and B cells, or activation of receptor tyrosine kinases (Yellaturu et al, 2002; Liu et al, 2004). Once in the nucleus, NFAT proteins bind to specific promoter elements and, depending on the promoter context, regulate the transcription of their target genes either alone or in cooperation with nuclear partner proteins. (Loh et al, 1996a; Chen et al, 1998; Garcia-Cozar et al, 1998).
NFAT proteins preferentially bind to a GGAAA consensus sequence which is found in the promoter/enhancer regions of many immune response genes, including the cytokines IL-2, IL-4, IFNγ, TNFα, and several cell surface proteins such as CD40L and FasL. Because of the presence of constitutively active intracellular kinases, which rephosphorylate and inactivate NFAT factors, calcineurin is continuously required to maintain the active state of NFAT proteins (Neal and Clipstone, 2001; Sheridan et al, 2002; Hogan et al, 2003). Both the nuclear translocation of NFAT as well as the NFAT-dependent activation of gene transcription are reversed by either a decrease in intracellular calcium concentrations or treatment of cells with the calcineurin inhibitors FK506 and cyclosporin A (CsA) (Park et al, 1995; Rao et al, 1997; Horsley and Pavlath, 2002). Thus, calcineurin-mediated NFAT activation provides a direct and important link between intracellular Ca2+ signaling and the transcriptional regulation of genes that are integral to the initiation and maintenance of a productive immune response (Hogan et al, 2003).
In recent years, it has become increasingly evident that in addition to their central role in the immune response, NFAT proteins participate in the regulation of genes influencing the development and differentiation of numerous mammalian cell types. It has been shown, for instance, that NFAT members control multiple steps in myogenesis, chondrocyte differentiation and the development of the cardiovascular system (Friday et al, 2000; Graef et al, 2001b; Crabtree and Olson, 2002; Graef et al, 2003; Chang et al, 2004; Hirotani et al, 2004; Mammucari et al, 2005). In addition, recent evidence suggests that NFAT proteins, and in particular NFATc1, regulate important cellular processes such as proliferation and apoptosis in different cell types including epithelial cells, fibroblasts, and preadipocytes (Hogan et al, 2003; Neal and Clipstone, 2003; Viola et al, 2005). In contrast, only little information is available on expression and activation of the calcineurin–NFAT transcription pathway and its function in transformed cells. Here, we have identified NFATc1 as ectopically expressed and highly activated in pancreatic cancer cells in vivo and in vitro. Inhibition of Ca2+/calcineurin/NFAT signaling in cultured pancreatic cancer cells by treatment with CsA or FK506 or siRNA-mediated knock down of NFATc1 expression led to a marked decrease in anchorage-dependent and -independent growth rates owing to the induction of a cell cycle arrest. We show that the pro-proliferative effect of NFAT signaling is mediated by direct binding of activated NFATc1 to the promoter of the c-myc oncogene, resulting in upregulation of c-myc transcription and accelerated G1/S phase transition. These results provide important novel insights into the mechanisms of oncogenic c-myc activation in pancreatic cancer, especially in tumors that lack genomic amplifications of the c-myc gene locus.
Results
Expression of NFATc1 and calcineurin in pancreatic cancer
In a microarray analysis of pancreatic tissues (M Buchholz, unpublished data), we have identified NFATc1 as significantly overexpressed in pancreatic cancer tissues (n=37) as compared to chronic pancreatitis and healthy pancreas control samples (n=25) (Figure 1A). Differential expression was confirmed by RT–PCR analysis (Figure 1B). In order to assess whether NFATc1 was expressed by the tumor cells themselves or by infiltrating immune cells, we performed immunostaining on a series of pancreatic cancer tissue sections (n=23) using an anti-NFATc1 antibody. Immune cells staining positive for NFATc1 were observed in some of the specimens, but independent of the presence of immune cells, strong nuclear staining of tumor cells was observed in 16 of the 23 samples (69.5%) (Figure 1C, upper panel). Moreover, NFATc1-positive tumor cells consistently also stained positive for the NFAT-activating phosphatase calcineurin (Figure 1C, lower panel). In fact, calcineurin staining was observed both in the cytoplasm and the nuclei of tumor cells, confirming previous findings in primary keratinocytes and indicating that NFAT nuclear localization is accompanied by some relocalization of calcineurin from the cytoplasm into the nucleus (Al Daraji et al, 2002). Conversely, staining for both proteins was consistently weak or absent in normal ducts and acini or in tissue sections obtained from patients with chronic pancreatitis (n=7; data not shown). In analogy to the in vivo situation, concurrent expression of NFATc1 and calcineurin, although at varying levels, was observed in all seven pancreatic cancer cell lines analyzed by RT–PCR and Western blot analysis (Figure 1D).
Figure 1.
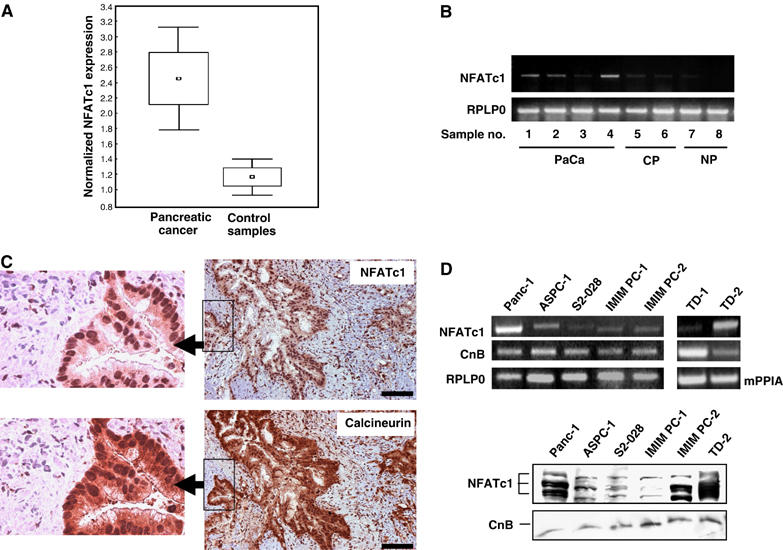
NFATc1 and calcineurin are ectopically expressed in pancreatic cancer. (A) Box-and-whisker plot illustrating normalized NFATc1 expression levels in pancreatic cancer (n=37) and normal/inflammatory control tissues (n=25) as determined in microarray experiments. (B) RT–PCR analyses using NFATc1-specific primers to validate the microarray data. Depicted are representative results for eight individual tissue samples. The human ribosomal protein, large, P0 gene (RPLP0) was used as housekeeping gene control. PaCa=pancreatic carcinoma; CP=chronic pancreatitis; NP=normal pancreas. (C) Serial sections of pancreatic cancer tissue stained for NFATc1 (upper panel) and calcineurin B (lower panel). Strong nuclear staining for NFATc1 in cancer cells was observed in 16 of 23 specimens. NFATc1-positive tumor cells consistently also stained positive for cytoplasmic and nuclear calcineurin B. Scale bars: 50 μm. (D) Expression levels of NFATc1 and calcineurin in pancreatic cancer cell lines as determined by RT–PCR (upper panel) and Western blot (lower panel) analyses. The human RPLP0 gene was used as housekeeping gene control for the human cell lines and the murine cyclophilin A gene (mPPIA) for the murine cell lines in the RT–PCR experiments. The Western blot for NFATc1 shows three major bands corresponding to different phosphorylation states of the protein. CnB=calcineurin B.
NFATc1 is constitutively activated in pancreatic cancer cells
Both the nuclear localization of NFATc1 in the tumor cells as well as the coexpression with calcineurin strongly suggested that the calcineurin–NFATc1 signaling and transcription pathway is constitutively active in pancreatic cancer cells in vivo and in vitro. This was confirmed by subcellular fractionation experiments using cultured pancreatic cancer cells, which demonstrated that NFATc1 was predominantly located in the nucleus and was present in the active (hypophosphorylated) form (Figure 2A, lanes 1 and 2). Treatment of the cells with the Ca2+/calcineurin inhibitor CsA rapidly and efficiently inactivated NFATc1, as evidenced by the change of phosphorylation status as well as nuclear export of NFATc1 (Figure 2A, lanes 3–8). Of note, although highly active under normal cell culture conditions (i.e. in the presence of serum), the calcineurin–NFATc1 signaling pathway was subject to additional stimulation by a further increase in intracellular calcium concentrations. Treatment of pancreatic cancer cells with ionomycin, a calcium ionophore, stimulated nuclear import of NFATc1 (Figure 2B) and, most importantly, increased the DNA-binding affinity of NFATc1, as revealed by DNA pulldown experiments using biotinylated double-stranded oligonucleotides containing the GGAAA consensus-binding element (Figure 2C, lanes 1 and 3). Conversely, both the increase in nuclear NFATc1 as well as its binding to the consensus-binding sequence were completely blocked by pretreatment of cells with the calcineurin inhibitor CsA (Figure 2C, lanes 2 and 4). Thus, basal and ionomycin-stimulated activation of NFATc1 were regulated by intracellular levels of calcineurin activity.
Figure 2.
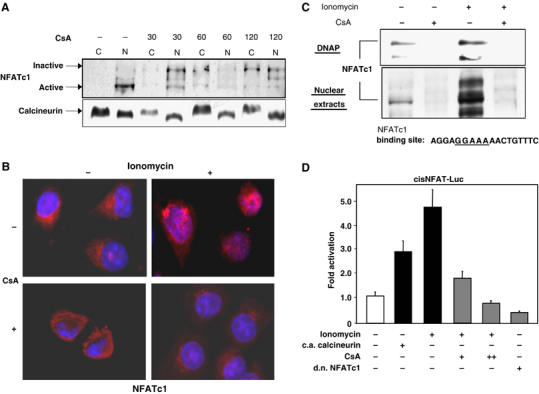
The Ca2+/calcineurin signaling pathway is active in pancreatic cancer cells. (A) Immunoblot analysis of subcellular localization of NFATc1 in pancreatic cancer cells. Panc-1 cells were either left untreated or treated with 1 μM CsA in the presence of serum for the indicated time intervals. Following subcellular fractionation, nuclear and cytoplasmic fractions were analyzed separately for the presence of NFATc1 protein by immunoblotting. (B) Immunocytological detection of NFATc1 localization in Panc-1 cells. Cells were kept in the absence of serum and either left untreated or treated with 1 μM CsA for 60 min, 1 μM ionomycin for 30 min, or a combination of CsA treatment followed by ionomycin treatment. Ionomycin stimulation led to a rapid and efficient translocation of NFATc1 (red signals) from the cytosol to the nucleus, which was completely blocked by CsA pretreatment. (C) DNA pulldown assays using oligonucleotides representing the consensus NFAT-binding sequence. Panc-1 cells were either left untreated or treated with 1 μM CsA for 30 min, 1 μM ionomycin for 15 min, or a combination of CsA treatment followed by ionomycin treatment. DNA–protein complexes from nuclear extracts were collected by precipitation with streptavidin–agarose beads and analyzed by Western blotting. Both, nuclear translocation (lower panel) as well as DNA-binding activity (upper panel) of NFATc1, were significantly enhanced by ionomycin, but completely blocked by CsA treatment. DNAP=DNA pulldown. (D) Activities of an NFAT-responsive reporter gene construct in Panc-1 cells. Cells were cotransfected with constitutively active (c.a.) calcineurin or dominant negative (d.n.) NFATc1 expression constructs and/or treated with 1 μM CsA or 1 μM ionomycin as indicated. Reporter gene activities are expressed as ‘fold activation' relative to untreated controls.
In order to assess the transcriptional activity of NFAT proteins, in particular NFATc1, and its dependency on Ca2+/calcineurin signaling in pancreatic cancer cells, we transiently transfected Panc-1 cells with a luciferase reporter construct (NFAT-Luc) harboring three NFAT-binding sites originally identified in the IL-2 gene promoter. Transactivation of the reporter gene construct was significantly enhanced following activation of calcineurin through either introduction of a constitutively active calcineurin expression construct or treatment with ionomycin, respectively (Figure 2D, bars 1–3). Conversely, as expected from our DNA-binding studies, ionomycin-induced transactivation of the NFAT responsive promoter was inhibited in a dose-dependent manner by pharmacological inhibition of calcineurin by CsA (Figure 2D, bars 4 and 5). Moreover, overexpression of a dominant negative form of NFATc1 in Panc-1 cells reduced the NFAT-Luc reporter activity by 50%, indicating that activated NFATc1 was responsible for at least half of the total NFAT signaling activity in the pancreatic cancer cells (Figure 2D, bar 6). Similar results were obtained in IMIM-PC2 and ASPC-1 cells (data not shown). Together, these results demonstrated that the Ca2+/calcineurin signaling pathway is highly active in pancreatic cancer cells and is sufficient to induce the nuclear translocation and the transcriptional activity of ectopically expressed NFATc1.
Ca2+/calcineurin signaling controls c-myc expression and proliferation in a subset of pancreatic cancer cell lines
Uncontrolled growth is an integral feature of all malignant tumors. This is especially true for pancreatic cancer, whose highly proliferative behavior significantly contributes to the dismal prognosis of this malignancy. In order to elucidate whether the Ca2+/calcineurin/NFATc1 pathway participates in the regulation of pancreatic cancer cell growth, we performed proliferation assays and flow cytometry analyses of cultured cells in the presence or absence of calcineurin inhibitors. Treatment of Panc-1 cells with either CsA or FK506 led to a dramatic reduction in proliferation rates (Figure 3A) in a time- and dose-dependent manner (shown for CsA in Figure 3B). Flow cytometry analysis of CsA-treated and untreated Panc-1 cells revealed that this effect was most likely due to induction of cell cycle arrest, as evidenced by the shift of cells from the S to the G1 phase (Figure 3C, upper left panel). Intriguingly, however, this was not true for all cell lines under investigation. While ASPC-1 cells in similarity to Panc-1 cells reacted with cell cycle arrest to CsA treatment, no significant effect on proliferation and cell cycle progression was observed for IMIM-PC2 and (murine) TD-2 cells (Figure 3C). Incidentally, the nonresponsive cell lines had previously been shown to harbor genomic amplifications of the c-myc gene, whereas the responsive Panc-1 and ASPC-1 cell lines lack genomic amplifications of the c-myc gene, respectively (Schreiner et al, 2003; Holzmann et al, 2004 and unpublished data). As c-myc is well known to promote G1/S phase transition and cell cycle progression, we consequentially hypothesized that the growth-promoting effects of Ca2+/calcineurin signaling might to a great extent be mediated through the regulation of c-myc expression and/or activation. Indeed, c-myc expression was markedly downregulated by CsA treatment in the Panc-1 and ASPC-1 cells, but remained unaffected in IMIM-PC2 and TD-2 cells (Figure 3D).
Figure 3.
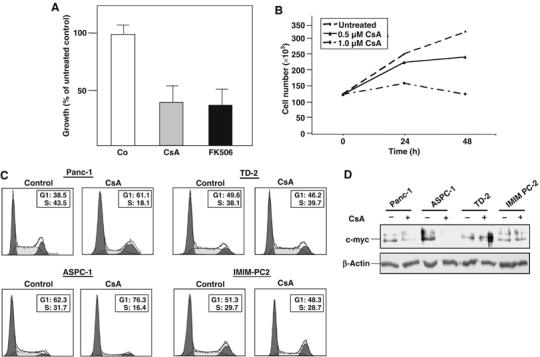
Inhibition of Ca2+/calcineurin signaling attenuates cell cycle progression and c-myc expression in a subset of pancreatic cancer cell lines. (A) Proliferation assays demonstrating reduced growth of Panc-1 cells in response to calcineurin inhibitors. Cells were left untreated or treated with 1 μM CsA or 0.1 μM FK506 for 24 h as indicated. Proliferation was measured by [3H]thymidine incorporation assay. Data are representative of triplicate experiments and are displayed as bars+s.d. (B) Growth inhibition of Panc-1 cells by CsA is time- and dose-dependent. Panc-1 cells were grown for 24 or 48 h in the presence of different amounts of CsA as indicated. Proliferation was measured by [3H]thymidine incorporation assay. (C) Cell cycle analysis of pancreatic cancer cell lines. Cells were left untreated or treated with 1 μM CsA for 24 h and analyzed by propidium iodide staining and flow cytometry. The percentages of cells in the G1 and S phases, respectively, are indicated. CsA treatment resulted in cell cycle arrest, as indicated by a shift from the S to the G1 phase, in Panc-1 and ASPC-1 cells, but not in TD-2 or IMIM-PC2 cells. (D) Western blot analysis of c-myc protein expression in the pancreatic cancer cell lines. Cells were left untreated or treated with 1 μM CsA for 24 h as indicated. Total cell lysates were then analyzed for c-myc protein content using an anti-c-myc antibody. CsA treatment reduced c-myc expression in Panc-1 and ASPC-1 cells, but not in TD-2 or IMIM-PC2 cells.
NFATc1 activation promotes anchorage-dependent and -independent growth via upregulation of c-myc in Ca2+/calcineurin-responsive cell lines
In order to elucidate whether the effects of Ca2+/calcineurin signaling in responsive cell lines were specifically mediated by NFATc1, we transiently knocked down NFATc1 expression in Panc-1 cells by using RNAi technology (Figure 4A) and examined the effects on c-myc expression and cell growth. Loss of NFATc1 expression in siRNA-transfected cells cultured on standard cell culture dishes resulted in a dramatic reduction in proliferation rates as compared to cells transfected with a nonsilencing control siRNA (Figure 4B). Flow cytometry analyses demonstrated that in analogy to the effects of calcineurin–NFAT inhibition by CsA treatment, reduced proliferation was the consequence of a cell cycle arrest induced in the NFATc1 knock-down cells, resulting in a shift of cells from the S to the G1 phase (Figure 4C). Moreover, anchorage-independent growth, which is considered a hallmark of malignant transformation of epithelial cells, was severely impaired by NFATc1 knock-down in Panc-1 cells. The number of colonies formed in soft agar assays was reduced between 70 and 90% for NFATc1 knock-down cells as compared to control cells (Figure 4D).
Figure 4.
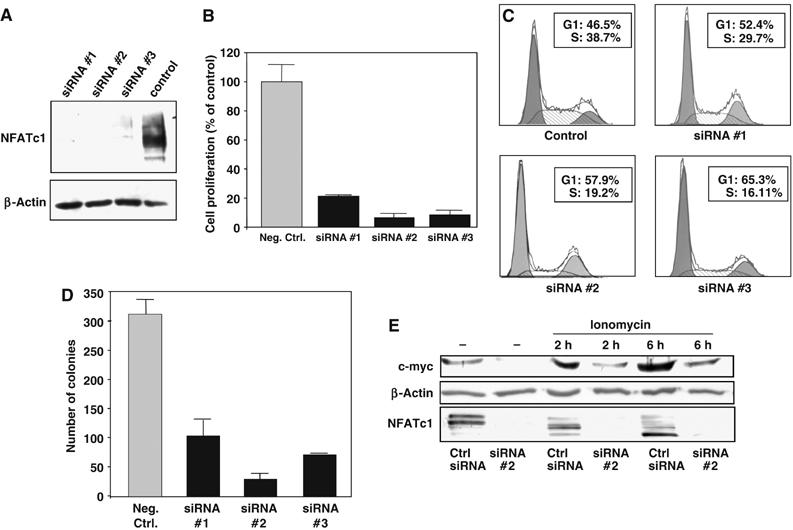
Anchorage-dependent and -independent cell growth and c-myc activation in Ca2+/calcineurin-responsive cell lines is NFATc1-dependent. (A) Western blot analysis of NFATc1 knock down in Panc-1 cells. Cells were transiently transfected with three different NFATc1-specific siRNA sequences or a nonsilencing control siRNA and total cell lysates analyzed 48 h post-transfection using an anti-NFATc1 antibody. (B) Proliferation assays demonstrating dramatically reduced growth of Panc-1 cells after siRNA-mediated knock down of NFATc1. Proliferation was measured by [3H]thymidine incorporation assay. Data are representative of triplicate experiments and are displayed as bars+s.d. (C) Cell cycle analysis of NFATc1 knock-down cells. Cells were transiently transfected with NFATc1-specific siRNA sequences or nonsilencing control siRNA and analyzed 24 h post-transfection by propidium iodide staining and flow cytometry. The percentages of cells in the G1 and S phases, respectively, are indicated. NFATc1 knock down resulted in cell cycle arrest, as indicated by the shift from the S to the G1 phases. (D) Soft agar assays demonstrating significantly impaired anchorage-independent growth of NFATc1 knock-down cells. NFATc1 knock-down and control cells were seeded in soft agar 24 h post-transfection and the number of viable colonies was determined after 10 days. Data are representative of triplicate experiments and are displayed as bars+s.d. (E) Western blot analysis of the effect of NFATc1 knock down on c-myc expression. Panc-1 cells were transiently transfected with the NFATc1-specific siRNA#2 or nonsilencing control siRNA and treated with 1 μM ionomycin for the indicated time periods. NFATc1 and c-myc protein was detected in total cell lysates using specific antibodies. Dephosphorylation, and thus activation, of NFATc1 by ionomycin is evidenced by the shift towards lower molecular weight bands (lower panel).
Importantly, these functional effects were associated with a strong decrease in c-myc expression levels in the NFATc1 knock-down cells (Figure 4E, lanes 1 and 2). Moreover, while induction of Ca2+/calcineurin signaling through ionomycin stimulation led to NFATc1 activation and marked upregulation of c-myc expression in control cells (Figure 4E, lanes 3 and 5), c-myc protein levels remained low in cells lacking NFATc1 expression (Figure 4E, lanes 4 and 6). These data strongly suggested that the activating effect of Ca2+/calcineurin signaling on cell proliferation and anchorage-independent growth were specifically mediated by activation of NFATc1, which in turn upregulated the c-myc proto-oncogene.
To confirm this hypothesis, we transiently knocked down c-myc expression and examined the effects on cell cycle regulation by CsA or reconstituted c-myc expression in CsA-treated Panc-1 cells. As demonstrated by Western blot analysis of nuclear extracts, c-myc knock down or re-expression had no effect on the inhibition of NFATc1 activity by CsA (Figure 5A). However, reconstitution of c-myc expression to basal levels nearly abolished the attenuating effect of CsA treatment on cell cycle progression (Figure 5B). Moreover, re-expression of c-myc completely reversed the inhibition of anchorage-independent growth in NFATc1 knock-down cells (Figure 5C and D). Together, these experiments demonstrated that (i) NFATc1 activation is the critical signaling event in the Ca2+/calcineurin responsive cell lines, and that (ii) c-myc is the primary target through which the growth-promoting effects are mediated.
Figure 5.
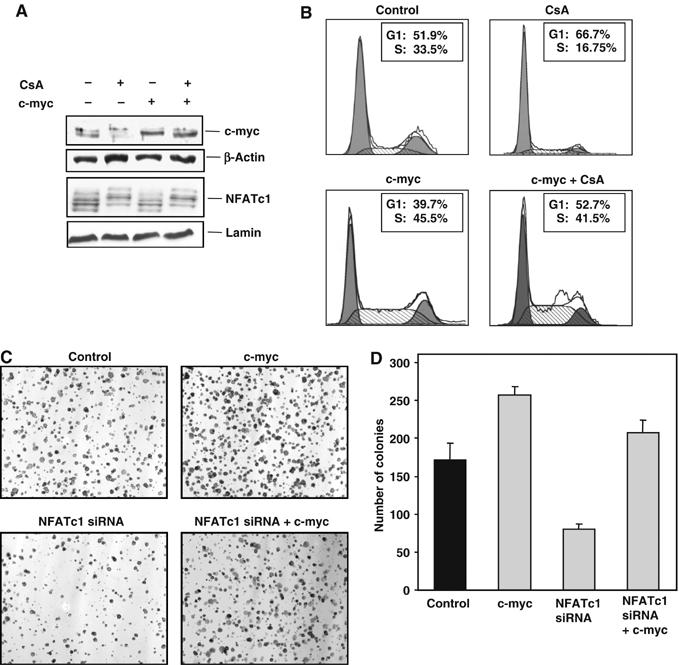
NFATc1-mediated growth promotion requires c-myc activation. (A) Western blot analysis demonstrating c-myc re-expression in CsA-treated Panc-1 cells. Cells were transiently transfected with a tetracycline-inducible c-myc expression construct or an empty vector control. Basal activity of the minimal promoter in the absence of tetracycline or doxycycline was sufficient to reconstitute basal c-myc expression levels in CsA-treated cells (lane 3). Analysis of nuclear extracts revealed that the inhibition of nuclear translocation of NFATc1 by CsA was not influenced by c-myc overexpression (lower panel). (B) Cell cycle analysis of cells re-expressing c-myc. CsA treatment significantly attenuated cell cycle progression in the control-transfected cells (upper panels), but had very little effect on the c-myc-transfected cells (lower panels). (C) Examples of soft agar assays demonstrating strong differences in colony formation after 10 days of incubation. Cells were left untransfected or transfected with the NFATc1-specific siRNA #2, the c-myc expression construct, or both, and the cells were seeded in soft agar 24 h post-transfection. Original magnification: × 100. (D) Statistical evaluation of soft agar assays. Reconstitution of c-myc expression completely abrogated the inhibitory effect of NFATc1 knock down on anchorage-independent growth of Panc-1 cells. Results are representative of triplicate experiments and are displayed as bars+s.d.
NFATc1 directly regulates c-myc promoter activity
To analyze the nature of this regulatory process, we performed a detailed study of the interactions of NFATc1 with the c-myc promoter. Transient transfection of an NFATc1 expression construct into Panc-1 cells expectedly led to a marked increase in c-myc mRNA levels (Figure 6A). Moreover, luciferase reporter gene assays using a c-myc promoter fragment comprising positions −2446 to +334 relative to the P2 transcription start site demonstrated a dose-dependent increase of promoter activity following transfection of increasing amounts of NFATc1 expression constructs (Figure 6B, bars 1–3), which was completely blocked and reduced below baseline levels by incubation of the cells with CsA (Figure 6B, bar 4) or cotransfection with NFATc1 siRNA (Figure 6B, bar 5). Sequence analysis of the promoter revealed a total of 15 putative NFAT-binding sites within the 2778 bp fragment (Figure 6C). Intriguingly, the most proximal putative NFAT-binding site was situated within a previously described regulatory region termed TGFβ inhibitory element (TIE), which has been shown to harbor binding sites for members of several different families of transcription factors and to play a critical role in the regulation of c-myc expression during the G1 phase (Chen et al, 2001; Yagi et al, 2002). In particular, the TIE is responsible for the downregulation of c-myc expression in response to activation of the TGFβ signaling cascade, which in turn is a key feature of efficient growth control in normal epithelial cells.
Figure 6.
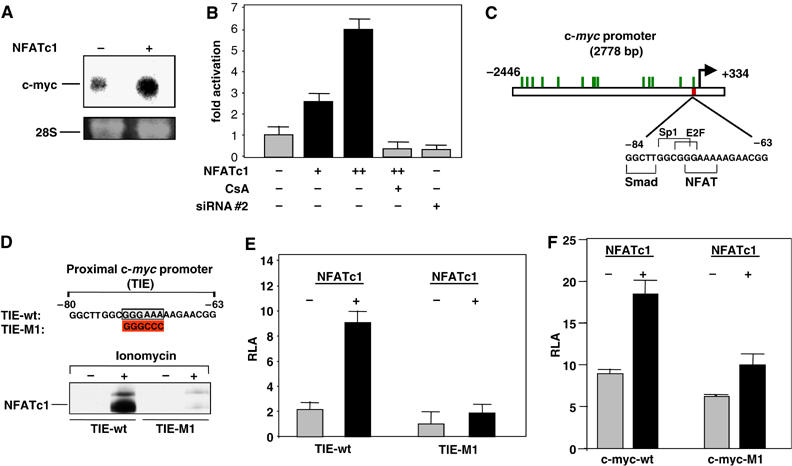
NFATc1 directly regulates c-myc promoter activity. (A) Northern blot analysis demonstrating increased c-myc transcription following transient transfection of Panc-1 cells with an NFATc1 expression construct. Loading control: 28S rRNA. (B) Activities of a reporter gene construct containing the full-length (2778 bp) c-myc promoter sequence. Panc-1 cells were cotransfected with the reporter gene construct and increasing amounts of an NFATc1 expression construct or the NFATc1-specific siRNA#2 and treated with 1 μM CsA where indicated. Firefly luciferase reporter gene activities were normalized to Renilla luciferase activity and expressed as RLA. (C) Location of putative NFAT-binding sites (green bars) within the full-length c-myc promoter fragment. The magnified section shows the sequence of the TGFβ inhibitory element (TIE) with the NFA-T binding site as well as previously described binding sites for the Smad-3, Sp1 and E2F transcription factors. (D) DNA pulldown assays using double-stranded oligonucleotides representing the wild-type (TIE-wt; upper sequence) or mutated (TIE-M1; mutation indicated in lower sequence) TIE. Panc-1 cells were grown in the absence of serum and either left untreated or treated with 1 μM ionomycin for 1 h as indicated. DNA–protein complexes were collected from nuclear extracts by precipitation with streptavidin–agarose beads and analyzed by immunoblotting using an anti-NFATc1 antibody. (E) Activities of reporter gene constructs containing TIE fragments with wild-type (TIE-wt) or mutated (TIE-M1) NFAT-binding sites. Panc-1 cells were cotransfected with the reporter gene constructs and NFATc1 expression vectors as indicated. Firefly luciferase reporter gene activities were normalized to Renilla luciferase activity and expressed as RLA. (F) Activities of reporter gene constructs containing the full-length (2778 bp) c-myc promoter sequence carrying wild-type (c-myc-wt) or mutated (c-myc-M1) NFAT-binding sites within the TIE. Panc-1 cells were cotransfected with the reporter gene constructs and NFATc1 expression vectors as indicated. Firefly luciferase reporter gene activities were normalized to Renilla luciferase activity and expressed as RLA (for colour figure see online).
In order to investigate whether NFATc1 was able to directly bind to the TIE, we performed DNA pulldown assays using 30 bp TIE fragments (−63 to −92 relative to the P2 transcription start site) with wild-type or mutated NFAT-binding sites (Figure 6D, upper panel). Strong binding of NFATc1 to the wild-type TIE was observed following ionomycin stimulation of serum-starved cells, whereas no binding was observed for the TIE fragment containing the mutated NFAT-binding site (Figure 6D, lower panel). These observations strongly suggested direct binding of NFATc1 to the TIE, although chromatin immunoprecipitation (ChIP) experiments, due to lack of specificity of the commercially available antibodies, failed to demonstrate NFATc1 binding in vivo.
In addition to the DNA pulldown experiments, reporter gene assays using constructs consisting of the wild-type or mutated TIE fragments fused to the core P2 promoter (−40 to +16) of the c-myc gene showed a five-fold induction of transcriptional activity of the wild-type TIE by overexpression of NFATc1 in Panc-1 cells, while inducibility was completely abolished and basal promoter activity substantially reduced for the mutated construct (Figure 6E). These results thus clearly demonstrated binding to, and transcriptional activation of, the TIE element by NFATc1. To evaluate the relevance of this NFATc1/TIE interaction for the transcriptional regulation of the complete c-myc promoter, we went on to mutate the NFAT-binding site of the TIE within the context of the full-length (2778 bp) c-myc promoter fragment. Reporter gene assays revealed that mutation of this single binding site resulted both in notably diminished basal promoter activity as well as significantly reduced inducibility by NFATc1 (Figure 6F). Taken together, these studies indicated that NFATc1-mediated transcription from the TIE element plays a significant role in Ca2+/calcineurin signaling-induced activation of the c-myc promoter.
Discussion
The present study shows that ectopic NFATc1 expression in conjunction with constitutive activation of the Ca2+/calcineurin signaling pathway is an important novel mechanism of aberrant c-myc activation in pancreatic cancer. Our data thus add to the growing body of evidence suggesting that NFAT transcription factors, in addition to their well-defined roles as transcriptional regulators during an effective immune response, have the potential to control central aspects of cell growth, cell differentiation, and cell death in a wide variety of nonimmune cells. For instance, growth-stimulatory effects of NFATc1 and NFATc2 have been demonstrated in skeletal muscle (Musaro et al, 1999; Horsley and Pavlath, 2003; Pavlath and Horsley, 2003) and heart valve development (Ranger et al, 1998; de la Pompa et al, 1998). Furthermore, NFAT transcription factors have been shown to control peripheral vascular development during angiogenesis (Hernandez et al, 2001; Graef et al, 2001a; Zaichuk et al, 2004), and to play roles in apoptosis regulation both in immune and nonimmune cells (Chuvpilo et al, 2002; Iwai-Kanai and Hasegawa, 2004; Kawamura et al, 2004; Benedito et al, 2005).
As deregulation of the above-mentioned cellular processes is commonly observed in cancer cells and indeed represents some of the hallmark features of malignant transformation, a distinct oncogenic potential of NFAT transcription factors has long been suspected. Of special interest in this context is the role of Ca2+/calcineurin/NFAT signaling in cell cycle control, as calcium signaling has been shown to promote cell cycle progression and G1/S phase transition in a variety of normal cells (Terada et al, 1991; Tomono et al, 1998; Lipskaia and Lompre, 2004) as well as transformed cells (Mosieniak et al, 1998). Moreover, it has been demonstrated that overexpression of a constitutively active NFATc1 mutant is sufficient to induce a transformed phenotype in pre-adipocyte 3T3-L1 fibroblasts, associated with altered expression of cell-cycle-related genes such as cyclin D1, cyclin D2, pRB, and, most notably, c-myc (Neal and Clipstone, 2003).
Our own data demonstrate for the first time that activated NFATc1 is able to transcriptionally upregulate the c-myc proto-oncogene through direct interaction with specific sequence elements within the c-myc promoter. C-myc is frequently overexpressed in many human malignancies (Nesbit et al, 1999), and the prominent role of c-myc in malignant cell transformation is well established. Nonetheless, the details of c-myc's mode of action have proven surprisingly difficult to unravel (Levens, 2003; Patel et al, 2004). Undisputed is c-myc's central role in cell cycle regulation. As a transcription factor, c-myc controls expression of a large number of cell-cycle-associated genes (Amati et al, 1998). Activation of c-myc leads to the upregulation of G1-specific cyclins and cyclin-dependent kinases, while simultaneously inhibiting negative regulators of cell cycle progression. As a result, cells are able to passage through the restriction point and progress from the G1- to the S phase of the cell cycle (Amati et al, 1998). The proliferation-inhibitory effects of FK506 and CsA on pancreatic cancer cells are thus very well explained by the transcriptional downregulation of c-myc in response to the loss of Ca2+/calcineurin signaling activity.
In recent years, however, it has become increasingly evident that the functions of c-myc in normal and transformed cells extend far beyond its role in cell cycle control. C-myc has been implicated in the regulation of growth, differentiation, apoptosis, angiogenesis, DNA repair, and even basic metabolic processes such as protein synthesis and ribosome assembly. It is thus not surprising that deregulation of c-myc activity is a central feature of malignant transformation in many human cancers (for an overview, see Adhikary and Eilers, 2005). Mechanisms influencing the transcriptional activity of c-myc can thus be expected to have significant influence on the malignant phenotype of cancer cells. This also holds true for the transcriptional upregulation of c-myc by NFATc1 in pancreatic cancer cells. Using anchorage-independent growth, that is, the ability to grow in the absence of a solid support matrix, as a well-established indicator of the malignant potential of epithelial cancer cells, we show that upregulation of c-myc via constitutive activation of Ca2+/calcineurin/NFATc1 signaling significantly contributes to the oncogenic potential of a subset of pancreatic cancer cell lines.
In responsive pancreatic cancer cells, c-myc upregulation is mediated through direct interaction of ectopically expressed NFATc1 with the c-myc promoter, as shown in this paper for the most proximal of several putative NFAT-binding sites within the promoter sequence. As mentioned previously, this site is of special interest, as it is situated in the TIE regulatory element which is tightly controlled by mitogenic as well as antiproliferative stimuli. Binding of Smad tumor suppressor proteins to the TIE, for instance, is a key step in TGFβ-mediated downregulation of c-myc expression in normal epithelial cells, which in turn is a prerequisite for efficient growth inhibition in untransformed epithelia (Chen et al, 2002). It has been demonstrated that Smad protein binding to the TIE is eliminated in breast cancer cells, resulting in enhanced cell proliferation (Chen et al, 2001), but binding of signaling-regulated transcriptional activators to the TIE has not been described to date. The cardinal importance of the NFATc1-binding site within the TIE for the regulation of c-myc expression in pancreatic cancer cells is illustrated by the dramatic reduction in both basal and NFATc1-induced activity of the full-length c-myc promoter upon mutation of this particular binding site.
However, as mentioned above, this mode of regulation is not effective in all pancreatic cancer cells. Human IMIM-PC2 and murine TD2 cells were refractory to the growth-inhibitory effects of FK506 and CsA, and c-myc was not downregulated in these cells in response to calcineurin inhibitor treatment. Intriguingly, in both cell lines, the genomic loci harboring the human and murine c-myc genes, respectively, have been shown to be amplified (Schreiner et al, 2003; Holzmann et al, 2004), whereas Panc-1 and ASPC-1 cells, although expressing similar levels of c-myc protein, carry no genomic amplifications. This is in fact a good representation of the situation in vivo, where overexpression of the c-myc protein is observed in up to 70% of human pancreatic tumors (Li et al, 2005), whereas c-myc gene amplifications only occur in ca. 30% of cases (Schleger et al, 2002). In a significant proportion of cases, mechanisms of transcriptional deregulation must therefore be responsible for the overexpression of c-myc. In view of the high incidence of detection of nuclear (activated) NFATc1 in pancreatic tumors in vivo, we conclude that ectopic expression of NFATc1 in conjunction with aberrant activation of the Ca2+/calcineurin signaling cascade is a major cause of oncogenic c-myc activation in pancreatic cancer cases where genomic amplifications of the c-myc gene locus are absent.
Materials and methods
Material and cell lines
Surgically resected pancreatic adenocarcinoma and chronic pancreatitis tissues were provided by the surgery departments at the Universities of Ulm and Homburg/Saar. Normal pancreas samples were obtained from healthy areas at the borders of chronic pancreatitis resectates. Informed consent was obtained from all patients prior to using tissue or biopsy samples. The study was approved by the local ethics committees at the Universities of Ulm (Germany) and Homburg/Saar (Germany).
The human pancreatic adenocarcinoma cell lines ASPC-1, IMIM-PC1, and IMIM-PC2 (Vila et al, 1995) were provided by FX Real (Insitute Municipale de Investigacion Medica, Barcelona, Spain). S2-028 cells (Taniguchi et al, 1992) were from T Iwamura (Miyazaki Medical College, Miyazaki, Japan). Panc-1 cells were obtained from the American Type Culture Collection (ATCC, RMD, USA). Murine TD-1 and TD-2 cells, which originated from ductal pancreatic tumors arising in transgenic mice overexpressing TGFα under the control of the mouse elastase promoter, were described previously (Schreiner et al, 2003). Panc-1, S2-028, IMIM-PC1, and IMIM-PC2 cells were maintained in Dulbecco's modified minimal essential medium (GIBCO, Invitrogen Corp., NY, USA); ASPC-1, TD-1, and TD-2 cells in RPMI 1640 (Roswell Park Memorial Institute) medium (GIBCO, Invitrogen Corp., NY, USA), both supplemented with 10% FCS (GIBCO, Invitrogen Corp., NY, USA) and 100 μg/ml NormovinTM (Amaxa Inc., Gaithersburg, MD, USA).
Plasmids and siRNA
A full-length human NFATc1 expression vector was provided from A Rao (Harvard Medical School, Boston, MA). The constitutive active calcineurin expression plasmid was from Dr C-W Chow (Albert Einstein College of Medicine, Bronx, NY) and the dominant-negative NFATc1 from J Northrop (Affymax Research Institute, Santa Clara, CA) , respectively. The c-myc-wt (−2446 to +334) reporter construct was a kind gift from J Massague (Memorial Sloan-Kettering Cancer Center, New York, NY). The c-myc expression vector was constructed by cloning the PCR-amplified c-myc open reading frame into the KpnI and SpeI restriction sites of the Tet-inducible mammalian expression vector pBIG2i (Strathdee et al, 1999). To generate the reporter plasmids TIE-wt and TIE-M1, the following double-stranded oligonucleotides were cloned into the pGL3 enhancer vector (Promega, Madison, WI): TIE-wt: 5′-TTCTCAGAGGCTTGGCGGGAAAAAGAACGG-3′ and its complementary strand; TIE-M1: 5′-TTCTCAGAGGCTTGGCGGGCCCAAGAACGG-3′ and its complementary strand. The c-myc-M1 reporter construct was generated from the c-myc-wt (−2446 to +334) reporter construct by using the QuickChange site-directed mutagenesis Kit (Stratagene, La Jolla, CA). Mutagenesis primers were 5′-CTCAGAGGCTTGGCGGGCCCAAGAACGGAG GGAG-3′ and its complementary strand.
Small interfering RNA (siRNA) was transfected into Panc-1 cells using the TransmessengerTM reagent (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The NFATc1-specific siRNA sequences were as follows: siRNA#1: 5′-GGUCAUUUUCGUGGAGAAATT-3′; siRNA NFAT#2: 5′-GAAACUCCGACAUUGAACUTT-3′; siRNA NFAT#3: 5′-GGACUCCAAGGUCAUUUUCTT-3′ (Ambion, Austin, TX, USA). As a negative control, the Silencer Negative Control from Ambion was used. For proliferation assays and flow cytometry analyses, cells were transfected with siRNA twice, with an interval of 24 h.
RT–PCR and Northern blot analysis
RNA was extracted using the RNeasy Midi Kit (Qiagen GmbH, Hilden, Germany) and first-strand cDNA was synthesized from 10 μg total RNA using random primers and Superscript II reverse transcriptase (Invitrogen Life Technologies, Karlsruhe, Germany). Reverse transcription was performed using the Superscript first-strand synthesis kit (Invitrogen) and specific primer pairs which were designed with the PrimerExpress® program (Applied Biosystems, Wellesley, MA, USA). The following primer pairs were used for RT–PCR: mouse calcineurin: forward 5′-CAAGAAGCTTGATTTGGACAA-3′; reverse 5′-CACAGAATTCCTCAAAGGATAT-3′; mouse cyclophilin A: forward 5′-CACCGTGTTCTTCGACATCA-3′; reverse 5′-AGCATTTGCCATGGACAAGAT-3′; mouse NFATc1 forward 5′-CCAGTCATCGGCGGGAAGAAGA-3′; reverse 5′-TATACACCCCCAGACCGCATCAGC-3′; human calcineurin B: forward 5′-TGCCTGCCATCGCTGTTCCTTCAA-3′; reverse 5′-CCCCTCCCTTACCCACCCCCACAC-3′; human NFATc1: forward 5′-GTCCCACCACCGAGCCCACTACG-3′; reverse 5′-GACCATCTTCTTCCCGCCCACGAC-3′; human RPLP0: forward 5′-GCAGCTGATCAAGACTGGA-3′; reverse 5′-CTGGCTAAGTTGGTTGCTTT-3′.
For Northern blot analysis, 20 μg of total RNA were size-fractionated and transferred to Hybond N membranes (Amersham Biosciences, Buckinghamshire, England). Northern blots were hybridized with a 32P-labeled cDNA probe for c-myc, which was generated by random prime labeling with the Megaprime DNA-labeling system (Amersham Biosciences, Buckinghamshire, England). The probe comprised the complete c-myc coding sequence.
Immunohistochemistry and fluorescence microscopy
Panc-1 cells grown on chambered coverslips were left untreated or treated with either CsA (1 μM) for 60 min or ionomycin (1 μM) for 30 min. Cells were then washed, fixed, blocked and probed with anti-NFATc1 antibody (Santa Cruz, Santa Cruz, CA; 1:100). NFATc1 was detected with a fluorochrome-conjugated secondary antibody and nuclei counterstained with DAPI. Coverslips were mounted on glass slides and cells were observed with a fluorescence microscope (Carl Zeiss, Inc., Oberkochen, Germany).
Immunohistochemical analysis was performed as previously described (Wagner et al, 2003). In short, paraffin sections were stained after antigen retrieval with anti-NFATc1 (1:50) or anti-calcineurin (Abcam, Cambridge, UK; 1:1000) antibodies. Antibody binding was visualized using a biotinylated secondary antibody, avidine-conjugated peroxidase (ABC method; Vector Laboratories), and 3,3′diaminobenzidine tetrachloride (DAB) as a substrate, and hematoxylin as counterstain.
Subcellular fractionation and immunoblotting
Subcellular fractionation was performed as described previously (Schreiber et al, 1989). Briefly, cells were washed twice with ice-cold PBS and collected by centrifugation at 1600 r.p.m. at 4°C. Lysates were then resuspended in buffer A (10 mM Hepes pH 7.9; 10 mM KCl; 0,1 mM EDTA; 0.1 mM EGTA; 1 mM DTT; proteinase inhibitors) for 15 min and subsequently centrifuged for 2 min at 6800 r.p.m. Supernatants were transferred to new cups and centrifuged at 14 000 r.p.m. for additional 20 min. Pellets were resuspended in 30–100 μl buffer C (20 mM Hepes pH 7.9; 0.4 M NaCl; 1 mM EDTA; 1 mM EGTA; 1 mM DTT; proteinase inhibitors) and incubated on ice. A final centrifugation step at 14 000 r.p.m. for 20 min was performed to separate nuclear proteins from cellular debris. For Western blotting, the resulting nuclear protein extracts were electrophoresed through a 7.5 or 12% SDS–polyacrylamide gel and transferred onto PVDF ImmobilonTM-P membranes (Millipore, Billerica, MA, USA) as described previously (Ellenrieder et al, 2004). PVDF membranes were probed with anti-NFATc1 (1:500, abcam, Cambridge, UK), anti-Calcineurin B (1:500, abcam, Cambridge, UK), anti-c-myc (1:250, Sigma-Aldrich, Saint Louis, MI, USA), or anti-β-actin (1:100, Sigma-Aldrich, Saint Louis, MI, USA) antibodies, washed in TBS washing buffer, and incubated with peroxidase-conjugated secondary antibodies. ECLTM Western Blotting Detection Reagent (Amersham Biosciences, UK) was used for visualization.
DNA pulldown assays
Panc-1 cells were treated with ionomycin (1 μM, 30 min), CsA (1 μM, 1 h), or a combination of both before harvesting. In total, 100 μg of nuclear protein per sample were incubated for 3 h with 1 μg of biotinylated double-stranded oligonucleotides containing the GGAAA consensus NFAT-binding sequence of the human interleukin-2 promoter (5′-AGGAGGAAAAACTGTTTC-3′ and its complementary strand), the wild-type TIE element (TIE-wt, −92 to −63 relative to the c-myc P2 transcription start site; 5′-TTCTCAGAGGCTTGGCGGGAAAAAGAACGG-3′ and its complementary strand) or the mutant TIE sequence (TIE-M1; 5′-TTCTCAGAGGCTTGGCGGGCCCAAGAACGG-3′ and its complementary strand). DNA–protein complexes were collected by precipitation with streptavidin–agarose beads (Sigma-Aldrich Corporation, St Louis, MO) for 1 h, washed twice with lysis buffer including proteinase and phosphatase inhibitors and subjected to Western blotting analysis.
Reporter gene assays
For luciferase reporter gene assays, cells were seeded in 24-well tissue culture dishes at 50 000 cells per well and 24 h later transfected with the indicated constructs, along with NFAT-Luc or c-myc reporter plasmids. Treatment with ionomycin (1 μM) (Sigma-Aldrich, Saint Louis, MI, USA) or CsA (1 μM) (Sigma-Aldrich, Saint Louis, MI, USA) was carried out 24 h post transfection. Luciferase assays were performed with a Lumat LB 9501 (Berthold Technologies) luminometer and the Dual-Luciferase®-Reporter Assay System (Promega, Madison, Wis, USA). Firefly luciferase values were normalized to Renilla luciferase activity and were either expressed as relative luciferase activity (RLA) or as mean ‘fold induction' with respect to empty vector control. Mean values are displayed±standard deviations.
Proliferation assays
Cell growth was measured by [3H]thymidine incorporation. Pancreatic cancer cells were seeded in 24-well plates and cultured in medium containing 10% FCS. After 24 h, cells were either transfected with siRNA or treated with CsA or FK506 for 24 h. [3H]thymidine (0.5 μCi/well) was added during the last 5 h of incubation. The cells were then washed with cold 5% TCA and the acid-insoluble fraction was dissolved by incubation with 1 M NaOH for 30 min at 37°C. Radioactivity was evaluated with a scintillation counter. All proliferation assays were performed in triplicates in at least two independent experiments.
Flow cytometry
Flow cytometric analysis was performed as described previously (Michl et al, 2005). For fluorescein diacetate staining, cells were resuspended in 1 ml PBS at 106 cells/ml, and 10 μl of fluorescein diacetate (1 μg/ml) was added to the cells for 10 min. For FITC staining, cells were resuspended in 1 ml PBS containing 5 μl FITC (1 μg/ml), 20 μl propidium iodide (2 mg/ml), and 50 μl RNase (100 μg/ml) for 30 min.
Soft agar assays
Soft agar assays were performed as described previously (Buchholz et al, 2003). In brief, 3 × 104 cells per 3 cm cell culture dish were seeded in DMEM/0.33% bacto-agar onto a bottom layer of DMEM/0.5% bacto-agar. Anchorage-independent growth was measured after 10 days of incubation by counting the number of viable colonies.
Acknowledgments
Grant support: This work was supported by grants of the DFG to MB, TMG, and VE (SFB 518, projects B1 and B16) and the Max Eder program of the German Cancer Research Foundation (Deutsche Krebshilfe) to VE (70-3022-El I).
References
- Adhikary S, Eilers M (2005) Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 6: 635–645 [DOI] [PubMed] [Google Scholar]
- Al Daraji WI, Grant KR, Ryan K, Saxton A, Reynolds NJ (2002) Localization of calcineurin/NFAT in human skin and psoriasis and inhibition of calcineurin/NFAT activation in human keratinocytes by cyclosporin A. J Invest Dermatol 118: 779–788 [DOI] [PubMed] [Google Scholar]
- Amati B, Alevizopoulos K, Vlach J (1998) Myc and the cell cycle. Front Biosci 3: d250–d268 [DOI] [PubMed] [Google Scholar]
- Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, Bonni A (2005) The transcription factor NFAT3 mediates neuronal survival. J Biol Chem 280: 2818–2825 [DOI] [PubMed] [Google Scholar]
- Buchholz M, BieblA, Neesse A, Wagner M, Iwamura T, Leder G, Adler G, Gress TM (2003) SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res 63: 4945–4951 [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR (2004) A field of myocardial–endocardial NFAT signaling underlies heart valve morphogenesis. Cell 118: 649–663 [DOI] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Massague J (2001) Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor beta growth arrest program. Proc Natl Acad Sci USA 98: 992–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Siegel PM, Massague J (2002) E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 110: 19–32 [DOI] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC (1998) Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature 392: 42–48 [DOI] [PubMed] [Google Scholar]
- Chuvpilo S, Jankevics E, Tyrsin D, Akimzhanov A, Moroz D, Jha MK, Schulze-Luehrmann J, Santner-Nanan B, Feoktistova E, Konig T, Avots A, Schmitt E, Berberich-Siebelt F, Schimpl A, Serfling E (2002) Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity 16: 881–895 [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN (2002) NFAT signaling: choreographing the social lives of cells. Cell 109 (Suppl): S67–S79 [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mark TW (1998) Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 392: 182–186 [DOI] [PubMed] [Google Scholar]
- Ellenrieder V, Buck A, Harth A, Jungert K, Buchholz M, Adler G, Urrutia R, Gress TM (2004) KLF11 mediates a critical mechanism in TGF-beta signaling that is inactivated by Erk-MAPK in pancreatic cancer cells. Gastroenterology 127: 607–620 [DOI] [PubMed] [Google Scholar]
- Feske S, Okamura H, Hogan PG, Rao A (2003) Ca2+/calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun 311: 1117–1132 [DOI] [PubMed] [Google Scholar]
- Friday BB, Horsley V, Pavlath GK (2000) Calcineurin activity is required for the initiation of skeletal muscle differentiation. J Cell Biol 149: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cozar FJ, Okamura H, Aramburu JF, Shaw KT, Pelletier L, Showalter R, Villafranca E, Rao A (1998) Two-site interaction of nuclear factor of activated T cells with activated calcineurin. J Biol Chem 273: 23877–23883 [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR (2001a) Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105: 863–875 [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Crabtree GR (2001b) NFAT signaling in vertebrate development. Curr Opin Genet Dev 11: 505–512 [DOI] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR (2003) Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113: 657–670 [DOI] [PubMed] [Google Scholar]
- Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM (2001) Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med 193: 607–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA (2004) The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J Biol Chem 279: 13984–13992 [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17: 2205–2232 [DOI] [PubMed] [Google Scholar]
- Holzmann K, Kohlhammer H, Schwaenen C, Wessendorf S, Kestler HA, Schwoerer A, Rau B, Radlwimmer B, Dohner H, Lichter P, Gress T, Bentz M (2004) Genomic DNA-chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res 64: 4428–4433 [DOI] [PubMed] [Google Scholar]
- Horsley V, Pavlath GK (2002) NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol 156: 771–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley V, Pavlath GK (2003) Prostaglandin F2(alpha) stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J Cell Biol 161: 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai-Kanai E, Hasegawa K (2004) Intracellular signaling pathways for norepinephrine- and endothelin-1-mediated regulation of myocardial cell apoptosis. Mol Cell Biochem 259: 163–168 [DOI] [PubMed] [Google Scholar]
- Kawamura T, Ono K, Morimoto T, Akao M, Iwai-Kanai E, Wada H, Sowa N, Kita T, Hasegawa K (2004) Endothelin-1-dependent nuclear factor of activated T lymphocyte signaling associates with transcriptional coactivator p300 in the activation of the B cell leukemia-2 promoter in cardiac myocytes. Circ Res 94: 1492–1499 [DOI] [PubMed] [Google Scholar]
- Levens DL (2003) Reconstructing MYC. Genes Dev 17: 1071–1077 [DOI] [PubMed] [Google Scholar]
- Li YJ, Wei ZM, Meng YX, Ji XR (2005) Beta-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: relationships with carcinogenesis and metastasis. World J Gastroenterol 11: 2117–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipskaia L, Lompre AM (2004) Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol Cell 96: 55–68 [DOI] [PubMed] [Google Scholar]
- Liu Z, Dronadula N, Rao GN (2004) A novel role for nuclear factor of activated T cells in receptor tyrosine kinase and G protein-coupled receptor agonist-induced vascular smooth muscle cell motility. J Biol Chem 279: 41218–41226 [DOI] [PubMed] [Google Scholar]
- Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A (1996a) Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem 271: 10884–10891 [DOI] [PubMed] [Google Scholar]
- Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A (1996b) Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem 271: 10884–10891 [DOI] [PubMed] [Google Scholar]
- Luo C, Shaw KT, Raghavan A, Aramburu J, Garcia-Cozar F, Perrino BA, Hogan PG, Rao A (1996) Interaction of calcineurin with a domain of the transcription factor NFAT1 that controls nuclear import. Proc Natl Acad Sci USA 93: 8907–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, di Vignano AT, Sharov AA, Neilson J, Havrda MC, Roop DR, Botchkarev VA, Crabtree GR, Dotto GP (2005) Integration of Notch 1 and calcineurin/NFAT signaling pathways in keratinocyte growth and differentiation control. Dev Cell 8: 665–676 [DOI] [PubMed] [Google Scholar]
- Michl P, Ramjaun AR, Pardo OE, Warne PH, Wagner M, Poulsom R, D'Arrigo C, Ryder K, Menke A, Gress T, Downward J (2005) CUTL1 is a target of TGF(beta) signaling that enhances cancer cell motility and invasiveness. Cancer Cell 7: 521–532 [DOI] [PubMed] [Google Scholar]
- Mosieniak G, Pyrzynska B, Kaminska B (1998) Nuclear factor of activated T cells (NFAT) as a new component of the signal transduction pathway in glioma cells. J Neurochem 71: 134–141 [DOI] [PubMed] [Google Scholar]
- Musaro A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N (1999) IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature 400: 581–585 [DOI] [PubMed] [Google Scholar]
- Neal JW, Clipstone NA (2001) Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem 276: 3666–3673 [DOI] [PubMed] [Google Scholar]
- Neal JW, Clipstone NA (2003) A constitutively active NFATc1 mutant induces a transformed phenotype in 3T3-L1 fibroblasts. J Biol Chem 278: 17246–17254 [DOI] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV (1999) MYC oncogenes and human neoplastic disease. Oncogene 18: 3004–3016 [DOI] [PubMed] [Google Scholar]
- Park J, Yaseen NR, Hogan PG, Rao A, Sharma S (1995) Phosphorylation of the transcription factor NFATp inhibits its DNA binding activity in cyclosporin A-treated human B and T cells. J Biol Chem 270: 20653–20659 [DOI] [PubMed] [Google Scholar]
- Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB (2004) Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer 4: 562–568 [DOI] [PubMed] [Google Scholar]
- Pavlath GK, Horsley V (2003) Cell fusion in skeletal muscle—central role of NFATC2 in regulating muscle cell size. Cell Cycle 2: 420–423 [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH (1998) The transcription factor NF-ATc is essential for cardiac valve formation. Nature 392: 186–190 [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG (1997) Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707–747 [DOI] [PubMed] [Google Scholar]
- Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U (2002) c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 15: 462–469 [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W (1989) Rapid detection of octamer binding proteins with ‘mini-extracts', prepared from a small number of cells. Nucleic Acids Res 17: 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner B, Greten FR, Baur DM, Fingerle AA, Zechner U, Bohm C, Schmid M, Hameister H, Schmid RM (2003) Murine pancreatic tumor cell line TD2 bears the characteristic pattern of genetic changes with two independently amplified gene loci. Oncogene 22: 6802–6809 [DOI] [PubMed] [Google Scholar]
- Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P (2002) Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J Biol Chem 277: 48664–48676 [DOI] [PubMed] [Google Scholar]
- Shibasaki F, Price ER, Milan D, McKeon F (1996) Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature 382: 370–373 [DOI] [PubMed] [Google Scholar]
- Strathdee CA, McLeod MR, Hall JR (1999) Efficient control of tetracycline responsive gene expression from an autoregulated bi-directional expression vector. Gene 229: 21–29 [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Iwamura T, Katsuki T (1992) Correlation between spontaneous metastatic potential and type I collagenolytic activity in a human pancreatic cancer cell line (SUIT-2) and sublines. Clin Exp Metast 10: 259–266 [DOI] [PubMed] [Google Scholar]
- Terada N, Lucas JJ, Gelfand EW (1991) Differential regulation of the tumor suppressor molecules, retinoblastoma susceptibility gene product (Rb) and p53, during cell cycle progression of normal human T cells. J Immunol 147: 698–704 [PubMed] [Google Scholar]
- Tomono M, Toyoshima K, Ito M, Amano H, Kiss Z (1998) Inhibitors of calcineurin block expression of cyclins A and E induced by fibroblast growth factor in Swiss 3T3 fibroblasts. Arch Biochem Biophys 353: 374–378 [DOI] [PubMed] [Google Scholar]
- Vila MR, Lloreta J, Schussler MH, Berrozpe G, Welt S, Real FX (1995) New pancreas cancers cell lines that represent distinct stages of ductal differentiation. Lab Invest 72: 395–404 [PubMed] [Google Scholar]
- Viola JP, Carvalho LD, Fonseca BP, Teixeira LK (2005) NFAT transcription factors: from cell cycle to tumor development. Braz J Med Biol Res 38: 335–344 [DOI] [PubMed] [Google Scholar]
- Wagner M, Kunsch S, Duerschmied D, Beil M, Adler G, Mueller F, Gress TM (2003) Transgenic overexpression of the oncofetal RNA binding protein KOC leads to remodeling of the exocrine pancreas. Gastroenterology 124: 1901–1914 [DOI] [PubMed] [Google Scholar]
- Yagi K, Furuhashi M, Aoki H, Goto D, Kuwano H, Sugamura K, Miyazono K, Kato M (2002) c-myc is a downstream target of the Smad pathway. J Biol Chem 277: 854–861 [DOI] [PubMed] [Google Scholar]
- Yellaturu CR, Ghosh SK, Rao RK, Jennings LK, Hassid A, Rao GN (2002) A potential role for nuclear factor of activated T-cells in receptor tyrosine kinase and G-protein-coupled receptor agonist-induced cell proliferation. Biochem J 368: 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaichuk TA, Shroff EH, Emmanuel R, Filleur S, Nelius T, Volpert OV (2004) Nuclear factor of activated T cells balances angiogenesis activation and inhibition. J Exp Med 199: 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]