

Abstract
The structures of T7 RNA polymerase (T7 RNAP) captured in the initiation and elongation phases of transcription, that of φ29 DNA polymerase bound to a primer protein and those of the multisubunit RNAPs bound to initiating factors provide insights into how these proteins can initiate RNA synthesis and synthesize 6–10 nucleotides while remaining bound to the site of initiation. Structural insight into the translocation of the product transcript and the separation of the downstream duplex DNA is provided by the structures of the four states of nucleotide incorporation. Single molecule and biochemical studies show a distribution of primer terminus positions that is altered by the binding of NTP and PPi ligands. This article reviews the insights that imaging the structure of polynucleotide polymerases at different steps of the polymerization reaction has provided on the mechanisms of the polymerization reaction. Movies are shown that allow the direct visualization of the conformational changes that the polymerases undergo during the different steps of polymerization.
Keywords: DNA polymerase, polymerase translocation, RNA polymerase
Introduction
Many enzymes not only catalyze chemical reactions, but also act as mechanical machines in the process of executing their functions. Numerous obvious enzymes such as muscle myosin come to mind, but the polynucleotide polymerases also utilize the energy of NTP (or dNTP) to do work. DNA-dependent RNA polymerases (RNAPs), as well as certain DNA polymerases (DNAPs) that initiate at the 3′ end of a template strand, are able to recognize and remain bound to their initiation site throughout the initiation phase of primer extension and then transition to an elongation phase in which the initiation site is released. A question to be addressed here is how these enzymes are able to accommodate the elongating duplex product while remaining attached to their initiation site; furthermore, what is the nature of the structural transition to the elongation phase and how is it achieved? A second mechanical property of polymerases to be addressed here is how and when the incorporation of nucleotides into a growing primer chain results in translocation of the product duplex and a concomitant separation of the strands of the downstream duplex DNA; additionally, in the case of RNAPs, the RNA transcript must also be peeled off the template by the enzyme as it translocates.
While the most complete description of the structural basis of the transition from the initiation phase of transcription to the elongation phase exists for T7 RNAP (Cheetham and Steitz, 1999; Tahirov et al, 2002; Yin and Steitz, 2002), significant structural insights are known for both the multisubunit bacterial and eukaryotic yeast RNAPs (Cramer et al, 2001; Gnatt et al, 2001; Murakami et al, 2002; Vassylyev et al, 2002; Bushnell et al, 2004). Clues into the nature of the initiation to elongation process in a protein primed DNAP have been obtained from the structure of phage φ29 DNAP complexed with terminal protein (TP), the protein primer (Kamtekar et al, 2006). A common feature of these initiating polymerases appears to be the accumulation of the duplex product in the active site that in various ways gradually displaces the protein or the part of the polymerase that provides the polymerase's specificity for the initiation site.
Both RNAPs and DNAPs translocate along the DNA template as the duplex product is synthesized, and RNAP as well as some DNAPs are able to open the downstream DNA duplex without requiring a separate helicase protein. Structural data on both A family and B family DNAPs as well as T7 RNAP are consistent with a mechanism of translocation that is promoted by the dissociation of PPi (Yin and Steitz, 2004; Berman and Steitz unpublished), whereas DNA footprinting as well as single molecule studies of bacterial RNAP are consistent with a mechanism in which translocation is proposed to be associated with the binding of the incoming NTP (Abbondanzieri et al, 2005; Tuske et al, 2005). The term ‘power stroke' has often been used in the literature to describe a mechanism in which pyrophosphate dissociation results in an enzyme conformational change that is in turn associated with or perhaps even ‘pushes' translocation of the product duplex. The term ‘Brownian ratchet' has been used to describe a mechanism in which the binding of the incoming NTP captures and stabilizes the translocated state of the product among a number of states that are in equilibrium in its absence (Guajardo and Sousa, 1997). These terms convey two extreme polls of the possibilities. However, as there is an equilibrium among states of duplex translocation whose most stable state is altered by the protein conformational changes associated with the binding or dissociation of the ligands PPi or NTP, the distinction between these terms is vague. To know whether the product is being ‘pushed' by the conformational change or the change is capturing an equilibrium state of translocation would require knowing both the rates of ligand-induced conformational change and of one-dimensional diffusion of the DNA, which are generally unknown. Hence, the goal here is to describe the structural, biochemical and kinetic bases for understanding the mechanisms of translocation and strand separation without using the terms ‘power stroke' or ‘Brownian ratchet'.
The transition from the initiation to the elongation phase
All DNA-dependent RNAPs initiate RNA synthesis after binding to a specific promoter DNA sequence and opening the DNA duplex. With both multisubunit RNAPs and the single subunit T7 RNAP the enzyme remains bound to the promoter sequence throughout the initiation phase, which involves the synthesis of an 8- to 12-nucleotide transcript. This phase, often referred to as the abortive synthesis phase, results in the synthesis and release of numerous short transcripts and only after the transition to the elongation phase does transcription proceed with complete processively until completion of the entire RNA transcript followed by termination. Various models have been previously proposed to account for the enzyme's ability to remain bound to the promoter while synthesizing RNA. These included the inchworm model whereby the enzyme's active site catalyzing nucleotide incorporation progresses down the template while the promoter binding site remains fixed on the DNA, or the scrunching model where the product is accumulated in the polymerase active site.
T7 RNA polymerase
The T7 RNAP is a 98 000 Da molecular weight, single subunit whose polymerase domain is homologous to that of the DNA polymerase I (Pol I) family of polymerases and is completely unrelated to the multisubunit RNAPs. It binds to a 17-base-pair (bp) promoter DNA that it recognizes through base-specific interactions made by side chains emanating from a β hairpin called the specificity loop that binds in a DNA major groove and a loop that interacts within the minor groove (Cheetham and Steitz, 1999; Cheetham et al, 1999). The promoter binding site is formed in part by an N-terminal domain of 300 amino-acid residues whose structure is unique to this polymerase, as well as the β hairpin specificity loop, which is an insertion into the fingers domain of the polymerase. Comparison of the structure of the T7 RNAP bound to a 17-bp promoter DNA with that of its complex to a promoter containing a template overhang and a 3-nucleotide transcript shows that the promoter recognition is identical, but the template strand has accumulated in the active site after the synthesis of 3 nucleotides in order to position the +4 template base appropriately for nucleotide insertion; thus, a small amount of scrunching has occurred (Cheetham et al, 1999). It was notable, however, that the cleft in which the 3-bp heteroduplex resides in the initiation complex is not sufficiently large to accommodate even a fourth base pair in the post-translocation state. While these data suggested the possibility that the transcript peeled off the DNA after only 3 bp, biochemical data, such as the crosslinking of template and transcript strands, strongly implied that the heteroduplex would be as long as 8 bp (Temiakov et al, 2000). The apparent incompatibility of these results led to a review entitled ‘T7 RNA polymerase transcription complex: what you see is not what you get', which suggested that perhaps the co-crystal structure was ‘captured in an act of unproductive synthesis' as a consequence of the template sequence used (Severinov, 2001). Clearly, there was more to this interesting story.
The structure of T7 RNAP captured in an elongation complex, however, was able to accommodate all the biochemical data and show that the transition from initiation to elongation was accompanied by a massive conformational change particularly in the N-terminal domain of the polymerase (Tahirov et al, 2002; Yin and Steitz, 2002). This conformational change resulted in the enlargement of the binding cleft for the product enabling it to accommodate up to 8 bp of heteroduplex product, the complete destruction of the promoter binding site and the formation of a tunnel through which the RNA transcript was seen to emerge (Yin and Steitz, 2004). In addition to the 8 bp heteroduplex, 4 nucleotides of single-stranded RNA transcript are seen to be ‘peeled' off the template, consistent with biochemical data (Huang and Sousa, 2000; Temiakov et al, 2000) by a thumb α-helix as well as side-chain interactions with a newly folded H domain and the specificity loop (Figure 1C and D). Furthermore, a 9-nucleotide bubble is formed as predicted from biochemical experiments of Huang and Sousa (2000) in the substrate DNA, stabilized on the one hand by the 8 bp heteroduplex and on the other by extensive interactions of the single-stranded non-template strand with the protein (Figure 1D; Yin and Steitz, 2004). Comparison of the initiation and elongation structures (Figure 1A and B; Supplementary Movies 1 and 2) showed that the N-terminal domain could be most conveniently thought of as consisting of three subdomains. One, an assembly of six α-helices, moved as a rigid body through a rotation of 220° and a translation of 30 Å that both provided space for the elongating product and positioned the subdomain in the location formerly occupied by the promoter DNA. Perhaps most dramatic is a loop of protein that moves from one side of the N-terminal domain to the complete opposite side of the six-helix subdomain and refolds into a two-helix H domain that forms part of the lid of the transcript tunnel through which the RNA product departs.
Figure 1.
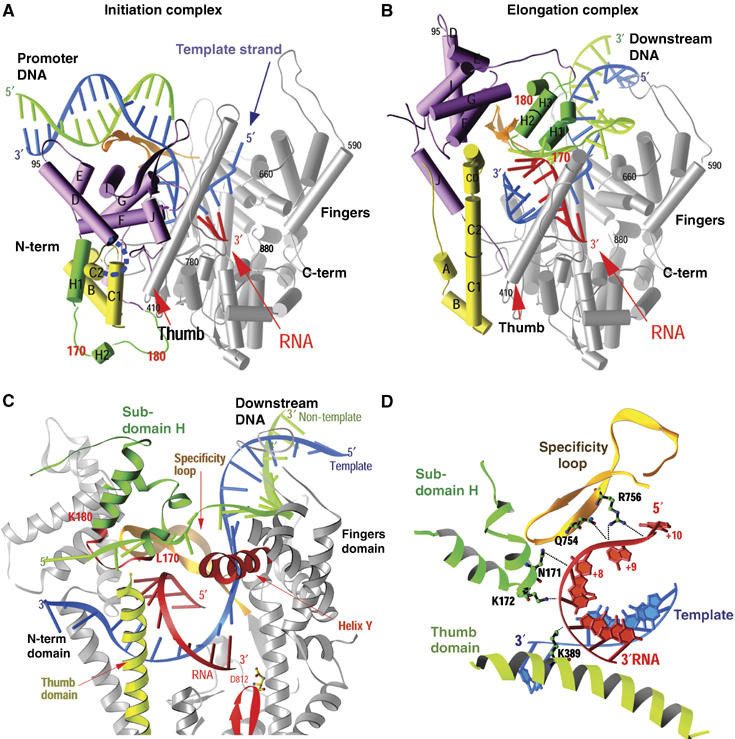
(A–D) Comparison of the structures of the T7 RNAP initiation and elongation complexes (A, B) and views of the transcription bubble (C, D) (Yin and Steitz, 2002). The initiation complex (A) and elongation complex (B) have been orientated equivalently by superimposing their palm domains. Helices are represented by cylinders and beta strands by arrows. The corresponding residues in the NH2-terminal domains of the two complexes that undergo major refolding are colored in yellow, green and purple, and the COOH-terminal domain (residues 300–883) is colored in gray. The template DNA (blue), non-template DNA (green) and RNA (red) are represented with ribbon backbones. The proteolysis-susceptible region (residues 170–180) is a part of subdomain H (green) in the elongation complex and has moved more than 70 Å from its location in the initiation complex. The specificity loop (brown) recognizes the promoter during initiation and contacts the 5′ end of RNA during elongation, whereas the intercalating hairpin (purple) opens the upstream end of the bubble in the initiation phase and is not involved in elongation. The large conformational change in the NH2-terminal region of T7 RNAP facilitates promoter clearance. This figure was made with the program Ribbons. (C) Interactions of the transcription bubble and heteroduplex in the elongation complex with domain H (green and red) and specificity loop (brown). Proteolytic cuts within the red loop in subdomain H reduce elongation synthesis (Ikeda and Richardson, 1987; Muller et al, 1988). Thumb sigma helix (yellow) and sigma helix Y (orange) are analogously involved in strand separation. (D) Side chains from subdomain H (green), the specificity loop (brown) and the thumb that interact with the single-stranded 5′ end of the RNA transcript and facilitate its separation from the template.
The structure of the elongation complex immediately explains both promoter clearance and the processivity of the elongation phase. Promoter clearance is achieved because the promoter binding site is completely destroyed (Figure 1A and B). Not only is the six-helix domain in the position formerly occupied by the promoter, but the anti-parallel β specificity loop that recognizes the promoter in the initiation complex has now moved to form part of the lid of the transcript exit tunnel and is seen to interact with the departing transcript (Yin and Steitz, 2004), consistent with biochemical crosslinking (Temiakov et al, 2000). High processivity is explained by the formation of this transcript exit tunnel that surrounds the transcript and thus functions analogously to the sliding clamp in DNA replication. Once the transcript is threaded through the tunnel, it can no longer dissociate from the enzyme.
The detailed mechanism by which this remarkable conformational change is achieved is at present a matter of speculation. It seems likely that the elongating heteroduplex gradually pushes against the six-helix domain, moving it out of the product site but without destroying the promoter recognition site initially. Formation of a disulfide crosslink between the six-helix domain and the fingers domain that would restrict movement of these domains from the initiation state structure results in synthesis of transcripts of length 5 or 6 nucleotides, only one or two more than the initiation state would appear able to accommodate (Ma et al, 2005). Although an interesting proposal has been put forward (Theis et al, 2004; Supplementary Movie 3), how and when the most major changes are accomplished is at present unknown. Is there a whole series of semi-stable conformational states formed as the primer strand elongates, or is there an equilibrium between a modestly modified initiation state conformation (clearly, the growing heteroduplex has to be accommodated) and the elongation state conformation that gradually favors the elongation state as incorporation proceeds? Attempts to capture the structures of these presumed intermediate states in crystal structures are presently being pursued using various strategies to prevent formation of the aborted complex (Durniak and Steitz, unpublished).
Bacteriophage φ29 DNA polymerase
The DNAP of bacteriophage φ29 is able to initiate DNA synthesis at the 3′ end of a template strand by using the hydroxyl group of a serine from a protein called terminal protein to prime replication (Salas, 1991). A complex between the φ29 DNAP and terminal protein initiates DNA synthesis at the second base from the 3′ end of the genome, and then further additions are made subsequent to a sliding back event, which results in base-pairing of the first dAMP with the 3′ terminal base of the genome (Méndez et al, 1997). Terminal protein remains complexed with the polymerase until after the incorporation of between 6 and 10 nucleotides after which it dissociates (Méndez et al, 1997). The crystal structure of a complex between φ29 DNAP and terminal protein suggests a model for the transition from the initiation to elongation phases of DNA synthesis that is consistent with the dissociation of the terminal protein subsequent to incorporation of 6–10 nucleotides (Kamtekar et al, 2006).
φ29 DNAP is a B-family polymerase whose crystal structure is largely similar to that of RB69 DNAP (Wang et al, 1997) except for two sequence insertions, TPR1 and TPR2, that are found only in the polymerase domains of protein primed DNAPs and form discreet subdomains in the structure (Kamtekar et al, 2004). Furthermore, in contrast to other polymerases, the thumb domain is an anti-parallel β ribbon that together with TPR2 forms an arch over the duplex product binding site, thereby accounting, in part, for this enzyme's high processivity (Figure 2A). When it is complexed with the polymerase (Figure 2B), the three domains of terminal protein adopt an extended structure in which its N-terminal domain makes no interactions with the DNAP, its intermediate domain interacts with the TPR1 insertion domain and a C-terminal four-helix domain containing the priming serine residue at its tip binds in the DNA product cleft (Kamtekar et al, 2006; Supplementary Movie 4). The priming domain in some ways mimics the duplex product DNA in both its acidic electrostatic profile and its location on the polymerase in the binding site of the duplex DNA product. The position of this four-helix priming domain explains why the complex between polymerase and terminal protein can initiate exclusively at the ends of linear DNA; the initiation of DNA synthesis by the heterodimer further downstream of the 3′ end of the template strand would lead to steric clashes between the template and terminal protein. This structure suggests that the elongating DNA duplex product must displace the priming domain, which is hypothesized to progressively move out of active site as polymerase elongates the primer strand (Kamtekar et al, 2006). Modeling studies suggest that the priming domain should be able to back out without steric clashes by rotating about a hinge peptide linking the priming domain to the intermediate domain for up to 6 bp synthesized (Figure 3). Subsequently, the terminal protein must dissociate from the polymerase, as, after the incorporation of about 6–7 nucleotides, the serine linked α phosphate would lie closer to the hinge than the approximately 40 Å length of the four-helix bundle. This model is consistent with biochemical data indicating that dissociation occurs after 6–10 nucleotides have been incorporated (Méndez et al, 1997).
Figure 2.
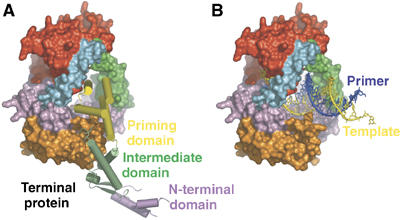
(A, B) The priming domain of terminal protein binds polymerase in the same location that the primer–template DNA binds to polymerase (Kamtekar et al, 2006). (A) A space filling representation of polymerase with a cylinder representation of terminal protein. (B) The primer–template DNA from the structure of an RB69 DNAP ternary complex (Franklin et al, 2001) homology modeled onto the structure of φ29 DNAP. The binding site of the priming domain overlaps that of the DNA.
Figure 3.
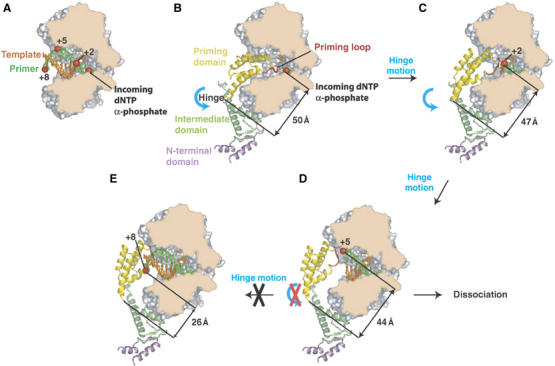
A model for the transition from initiation of replication to elongation (Kamtekar et al, 2006). A space filling representation of the polymerase has been cut through the polymerase to expose the DNA and primer binding cleft. (A) Modeled primer (green) and template (orange) in an elongating complex are represented as sticks. The positions of a phosphate being incorporated, as well as its positions after two, five and eight cycles of incorporation are indicated as red spheres, illustrating the spiral translocation of synthesized DNA. (B) A model for the covalent addition of the first nucleotide onto S232 of terminal protein. Terminal protein is colored by domain, with a plausible path for the loop containing S232 shown in red (this loop is disordered in the current structure). (C) Following the hinge motion indicated in panel B, the interactions between the intermediate domain and polymerase can be maintained. The priming domain has backed away from the polymerase active site enough to allow the incorporation of two more nucleotides. (D) A further hinge motion between the priming domain and the intermediate domain is consistent with the incorporation of 5 nucleotides. (E) A hinge motion alone is insufficient after the incorporation of seven bases. The example shown here is for nucleotide eight; the distance between the α phosphate and the hinge is only 26 Å, which is shorter than the length of the priming domain (approximately 40 Å), suggesting that the intermediate domain must be displaced resulting in the dissociation of polymerase–terminal protein complex.
Multisubunit RNA polymerases
The multisubunit RNAP from bacteria recognizes its promoters using transcription factors that are separate polypeptides. Sigma factor bound to the bacterial RNAP participates in the initiation phase by recognizing the DNA promoter, and it dissociates from the polymerase either during or shortly following the transition to elongation phase. The synthesis of approximately 12 nucleotides of an RNA transcript has been proposed to displace the 3.2 loop of the σ factor, which is lying in the tunnel through which the transcript is proposed to exit (Murakami et al, 2002; Murakami and Darst, 2003). Murakami and Darst (2003) further hypothesize that the displacement of the 3.2 loop by the elongating transcript weakens the interactions between the polymerase and σ, which ultimately leads to promoter escape.
Eukaryotic RNA polymerase II (Pol II) appears to behave in an analogous fashion. It is bound to promoters through general transcription factors that are released by the polymerase when it enters the elongation phase. The binding site on the polymerase for a domain of TFIIB (which together with TATA binding protein recognizes the promoter DNA) overlaps with the location of the heteroduplex in the elongation complex (Bushnell et al, 2004; Chen and Hahn, 2004). Thus, once again, the elongating RNA transcript may progressively displace the TFIIB domain providing a structural basis for the dissociation of the polymerase from promoter DNA.
Although the specific mechanisms of initiation site recognition and transitions to the elongation phase differ among the three RNAPs and φ29 DNAP, there appear to be some analogous structural principles involved (Kettenberger et al, 2004; Kamtekar et al, 2006). In each case, the elongating duplex or heteroduplex product gradually pushes a part of the initiation site recognition element away from the site of nucleotide incorporation eventually resulting in the polymerase dissociating from the initiation site. The high processivity of the three RNAPs in the elongation phase results from the transcripts exiting through a tunnel in the enzyme (Murakami et al, 2002; Murakami and Darst, 2003; Westover et al, 2004a, 2004b; Yin and Steitz, 2004). In the case of φ29 DNAP, the processivity is a consequence of the polymerase surrounding both the downstream template strand and the duplex product (Kamtekar et al, 2004).
Translocation and strand separation
T7 RNA polymerase
The structural basis of heteroduplex product translocation and separation of downstream duplex DNA by the monomeric T7 RNAP can be understood from high-resolution crystal structures of the four states of nucleotide incorporation by T7 RNAP in the elongation phase of DNA synthesis (Yin and Steitz, 2002; Yin and Steitz, 2004). These structures include that of the enzyme in a post-translocated product complex (Yin and Steitz, 2002), a complex with the post-translocated product with the incoming nucleotide bound in a pre-insertion site (Temiakov et al, 2004), the structure of a ternary complex with the incoming NTP positioned for incorporation and finally the product complex in a pre-translocation state including bound pyrophosphate (Yin and Steitz, 2004). Two of these states, the binary complex with primer–template DNA and the ternary complex including the incoming deoxy-NTP, have been determined for several DNA Pol I enzymes as well (Doublié and Ellenberger, 1998; Doublié et al, 1998; Li et al, 1998; Johnson et al, 2003), and the structures of these states are largely identical to the corresponding complexes with the T7 RNAP. Furthermore, the initiation complex of T7 RNAP with a 3-nucleotide transcript, but without PPi, is in the post-translocated state. The conclusion from these studies is that a conformational change in the ‘fingers' domain of the Pol I family polymerases from an ‘open' to a ‘closed' state is driven by the binding of the NTP (or dNTP) to the O helix followed by its positioning in the insertion site, and the change from a ‘closed' to an ‘open' state is associated with the release of the product pyrophosphate. Further, it is the conformational change resulting from the release of the pyrophosphate product that is associated both with translocation of the heteroduplex product and with strand separation of the downstream DNA (Yin and Steitz, 2004).
The structures of the four stages of nucleotide incorporation by T7 RNAP have been determined for the enzyme in its elongation phase of RNA synthesis (Figure 4; Supplementary Movie 5). The structure of the product complex after nucleotide incorporation and pyrophosphate dissociation is in the post-translocation state and the base that is to template the next incoming nucleotide is located in a pre-insertion pocket outside the active site (Kiefer et al, 1998; Li et al, 1998; Yin and Steitz, 2002; Johnson et al, 2003). The substrate NTP binds initially to the open complex at a site located on the O helix, apparently base-paired to the next template base, but not in a position for insertion (Temiakov et al, 2004). A conformational change that can be described as the rotation of a five-helix assembly about a pivot point then occurs that results in the formation of a closed complex in which the incoming nucleotide is properly positioned for insertion and the templating base moves from a pre-insertion pocket to pair with this incoming base (Yin and Steitz, 2004). This conformational change is driven by the ionic links that are able to form in the closed conformation between the three phosphates of the NTP and the two magnesium ions bound to the enzyme on one side and an arginine residue of the O helix on the other. The structure of T7 RNAP in the pre-translocation product complex that forms in the presence of pyrophosphate is identical to the structure of the enzyme in the ternary complex. The enzyme continues to be held in the closed conformation after nucleotide incorporation, once again, by the pyrophosphate product forming an ionic crosslink between a magnesium ion bound to the active site carboxylates and an arginine on the O helix. Dissociation of pyrophosphate completes this cycle and results in the formation of an open complex accompanied by translocation of the product heteroduplex.
Figure 4.
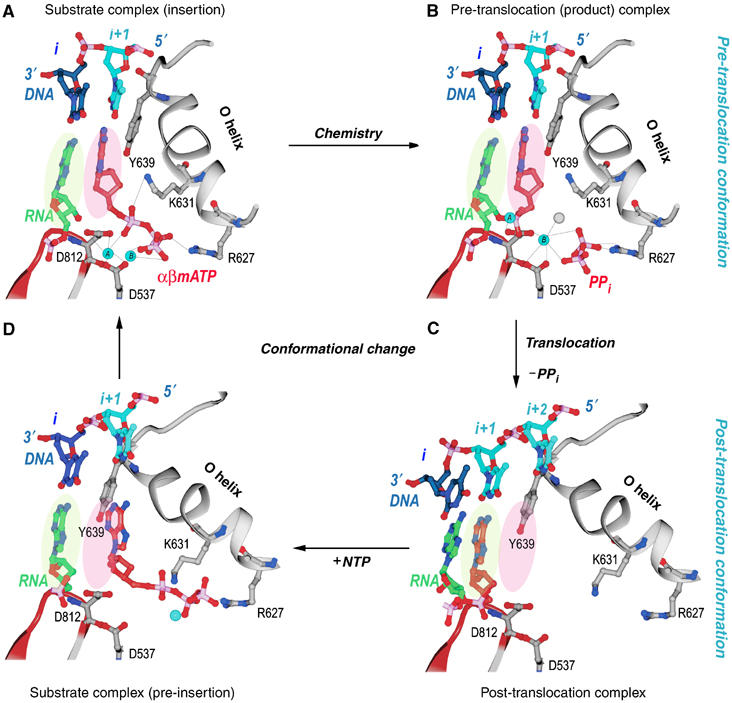
Structural changes at the active site during a single nucleotide addition cycle (Yin and Steitz, 2004). This figure shows the O helix with its phosphate binding K631 and R627, a β turn–β motif bearing the metal binding catalytic D812 and D537, template nucleotides in blue, the RNA primer terminus in green, as well as the P and N sites in green and pink ovals. (A) The NTP (red) is bound to the N site in position to be inserted with its metal-bound triphosphate moiety crosslinking the O helix to the active site aspartic acid residues. Template nucleotide i+1 (light blue) forms a base pair with the correct incoming nucleotide. (B) The product of the phosphoryl transfer reaction shows a Mg ion (blue) bound to PPi (red), which crosslinks D531 to R677, thereby maintaining RNAP in an identical conformation as in the substrate complex. The 3′ end of RNA remains in the N site in a pre-translocation state. (C) Release of Mg-PPi results in the loss of the link between the O helix and D531, which promotes the rotation of the O helix and translocation of the 3′ end of the RNA to the P site. The RNAP conformational change also places Y639 into the N site and positions the i+2 template nucleotide into the flipped-out, pre-insertion position. (D) A modeled NTP pre-insertion complex with NTP bound to the post-translocated RNAP. Although the base binding site is blocked by the side chain of Y639, the triphosphate binding site on the O helix is accessible.
The conformational change in the enzyme that is associated with translocation consists largely of a rotation of a five-helix bundle that includes the O helix about a pivot axis. Rotation of the bundle to the ‘open' conformation results in a 3.5 Å movement of a tyrosine residue (Y639) that becomes stacked on the primer–template bases and is responsible for stabilizing the translocated position of the product duplex (Figure 5; Supplementary Movie 5). The position of this tyrosine in the open state not only sterically precludes the return of the heteroduplex to its pre-translocation position, but also blocks the insertion site for the next incoming NTP. Only after the NTP bound to the pre-insertion site on the O helix in the open state promotes the formation of the closed state does the tyrosine side chain move out of the way, thereby allowing the incoming NTP base and the template base to move in to the insertion site and replace it. As this tyrosine is conserved in all Pol I family polymerases, a similar mechanism of translocation is suggested. The translocated state is a result of the open complex being more stable than the closed complex after the dissociation of the electrostatically crosslinking pyrophosphate. In the open state, the rotated five-helix bundle buries 130 Å2 of hydrophobic surface area, which would be expected to contribute about 3 kcal/mol (Chothia, 1974) towards the stabilization of the open conformation states. This modest hydrophobic interaction presumably provides the ‘spring' energy to stabilize formation of the open form and the associated translocation upon PPi dissociation. However, the estimated 3 kcal does not directly yield the equilibrium constant between pre- and post-translocation states of the transcribing complex, as translocation also melts a base pair. Interestingly, the single molecule studies of Thomen et al (2005) estimate a free energy difference of 0.7 kcal between pre- and post-translocation upon PPi release, yielding a 3:1 preference for the post-translocated state, largely consistent with the structural studies. Chemical cleavage studies show a distribution of transcript states that favor pre-translocation in the presence of PPi and post-translocation in the presence of NTP (Guo and Sousa, 2006).
Figure 5.
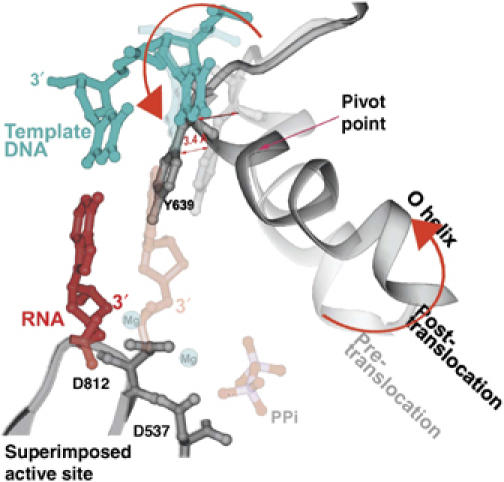
A superposition of the pre- and post-translocation structures at the active site showing the pivoted rotation undergone by the O helix that is associated with translocation (Yin and Steitz, 2004). In the pre-translocation complex (lighter colors), the O helix (light gray) is positioned in the closed conformation by PPi (light red), which is bound to the catalytic carboxyls through Mg. In this conformation, Y639 allows formation of the new base pair (light red and blue). After PPi release, the O helix rotates around Val634, which results in the positive end of the helix moving away from the active site, whereas the other end of the helix moves Y639 by 3.4 Å into the position of the newly formed primer terminus, resulting in translocation.
The B family DNAPs also undergo a transition from an ‘open' form to a ‘closed' form upon binding the primer–template DNA and incoming NTP (Franklin et al, 2001). This change once again involves a rotation of the fingers domain (Supplementary Movie 7), which in this family consists of a two-helix bundle for both RB69 and φ29 DNAPs. Emerging structures of the φ29 DNAP binary and ternary structures convey a related but different detailed mechanism for coupling this fingers conformational change to translocation of the product heteroduplex, as well as insertion of the incoming NTP (Berman, Goodman, Kamtekar and Steitz, unpublished).
Although, of course, an equilibrium must exist between the pre-translocated, ‘closed' product state in the absence of PPi and the translocated, ‘open' state, it is clear that in the absence of PPi, the translocated product state is the most stable in three separate structures of T7 RNAP (Cheetham and Steitz, 1999; Tahirov et al, 2002; Yin and Steitz, 2002), the structures of three binary substrate complexes with A family DNAPs (Doublié et al, 1998; Kiefer et al, 1998; Li et al, 1998) and the B family φ29 DNAP (Berman, Goodman, Kamtekar and Steitz, unpublished). Also, the structures of all A and B family apo-DNAPs, as well as three structures of T7 RNAP without substrates are in the open conformation that is associated with the translocated state. Although one certainly cannot determine the equilibrium constant between the pre- and post-translocated states by X-ray crystallography, the exclusive observation of the translocated state in all A-family polymerase product complexes in the absence of PPi suggests that it is significantly energetically favored. These structures appear to describe a plausible pathway for translocation and delivery of the NTP to the incorporation site (Supplementary Movie 5). The binding of the incoming nucleotide to the translocated open state and converting it to a closed translocation state will stabilize the complex and prevent the primer terminus from entering the pre-translocation position, unless the NTP dissociates. Thus, the binding of NTP can be described as stabilizing the translocated state.
Strand separation
DNA-dependent RNAPs and some DNAPs function as their own helicases, opening downstream duplex DNA as their primer strand is elongated and the enzyme translocates. The general mechanistic principle appears to be that the energy of nucleotide incorporation and associated translocation ‘pulls' the downstream template strand through a cleft or a tunnel that is only large enough to accommodate single-stranded DNA (Kamtekar et al, 2004; Yin and Steitz, 2004). In the case of T7 RNAP, and the homologous DNA Pol I family enzymes that are able to displace the non-template strand, the template strand passes through a narrow cleft before entering the active site, and the template and non-template strands separate at an α-helix containing a tyrosine that stacks on successive base pairs to be separated (Supplementary Movie 6). The RNA transcript is peeled off analogously by a thumb α-helix (Yin and Steitz, 2002). Again, it is posited that the energy for melting 1 bp in an incorporation cycle is contributed by PPi dissociation. In the case of φ29 DNAP, which is completely processive and able to strand displace the entire φ29 genomic DNA, the template strand passes through an upstream tunnel, which similarly excludes the non-template strand (Kamtekar et al, 2004). It seems likely that the hexameric DnaB helicase works in an analogous manner by ‘pulling' one strand of DNA through a tunnel that results in the displacement of the other strand. However, the manner in which the hydrolysis of ATP and the associated protein conformational changes are coupled to single-strand translocational movement is presently unknown and completely unrelated to those exhibited by polymerases.
Yeast RNA polymerase II
The structure of the yeast RNA Pol II complexed with various bound RNA and DNA substrates that represent states of nucleotide incorporation in the elongation phase has been determined (Gnatt et al, 2001; Kettenberger et al, 2004; Westover et al, 2004a, 2004b). It is not, however, transparently obvious what aspect of the complex determines whether the primer terminus of the heteroduplex resides in the pre- or post-translocation position. The structure of the first complex determined (Gnatt et al, 2001) showed electron density for about 20 bp of downstream duplex DNA and an 8 bp heteroduplex product of RNA transcription synthesized by the enzyme and halted by withholding the next NTP. About 13 nucleotides of a displaced upstream non-template DNA strand as well as some upstream template and product were not ordered in the complex. The primer terminus of the RNA transcript was observed in the pre-translocation position in this complex (Figure 6A and B).
Figure 6.
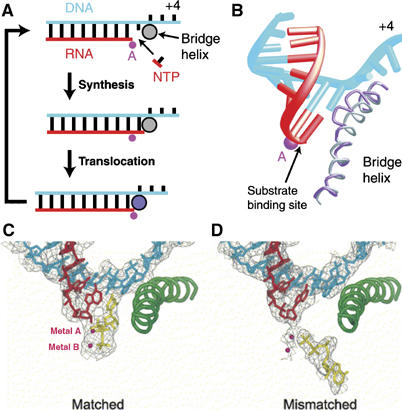
(A, B) Proposed transcription cycle and translocation mechanism (Gnatt et al, 2001). (A) Schematic representation of the nucleotide addition cycle. The NTP fills the open substrate site (top) and forms a phosphodiester bond at the active site (‘Synthesis'). This results in the state of the transcribing complex seen in the crystal structure (middle). Gnatt et al (2001) speculate that ‘Translocation' of the nucleic acids with respect to the active site (marked by a pink dot for metal A) involves a change of the bridge helix from a straight (silver circle) to a bent conformation (violet circle, bottom). Relaxation of the bridge helix back to a straight conformation without movement of the nucleic acids would result in an open substrate site 1 nucleotide downstream and would complete the cycle. (B) Different conformations of the bridge helix in Pol II and bacterial RNAP structures. The bacterial RNAP structure (Zhang et al, 1999) was superimposed on the Pol II transcribing complex by fitting residues around the active site. The resulting fit of the bridge helices of Pol II (silver) and the bacterial polymerase (violet) is shown. The bend in the bridge helix in the bacterial polymerase structure causes a clash of amino-acid side chains (extending from the backbone shown here) with the hybrid base pair at position +1. (C, D) Downstream end of the DNA–RNA hybrid in transcribing complex structures, showing occupancy of the A and E sites (Westover et al, 2004b). (C) Transcribing complex with matched NTP (UTP) in the A site. (D) Transcribing complex with mismatched NTP (ATP) in the E site. DNA is blue, RNA is red and NTPs are in yellow. Mg ions are shown as magenta spheres.
In contrast, the structure of a complex prepared using chemically synthesized RNA and DNA oligonucleotides including a 10-residue RNA transcript, a 28-residue DNA template spanning −10 to +18 and a 14-nucleotide downstream non-template DNA showed the primer terminus located in a post-translocated position (Westover et al, 2004b). Furthermore, the site opposite the next template base (n+1) was empty, but could be occupied by an incoming NTP that was correctly positioned along with two bound metal ions for nucleotide incorporation (Figure 6C). Similarly, Kettenberger et al (2004) determined the structure of a Pol II complex with a DNA duplex containing a mismatched ‘bubble' bound to an RNA transcript. This complex showed density for about 9 downstream base-pairs and a template-RNA product whose 3′ terminus was also in the post-translocated position. The cognate NTP could bind to a pre-insertion site, close to but not identical to the positioning of the insertion site. It is not clear, however, why the earlier complex containing the RNA made by polymerase transcription was in the pre-translocation position, whereas these two complexes containing chemically synthesized transcripts were found in the post-translation position. A key clue to the structural basis of translocation by the multisubunit polymerases may lie in the reason for the difference between these two types of experiments. For example, was pyrophosphate is still present after purification of the transcribed complex? Does the stability of the duplex formed by the specific DNA and RNA sequences used influence the equilibrium between pre- and post-translocated states?
What stabilizes the heteroduplex product in the pre- and post-translocation positions of the multisubunit RNAPs; what conformational changes in the enzyme associated with either ligand binding or dissociation are responsible for the stabilization of each state? Kornberg and co-workers (Gnatt et al, 2001) hypothesized that a conformational change in the bridge helix, which is packed against the primer terminal base pair, from straight to bulged could be responsible (Figure 6B). The bulged form has been observed in the apo-bacterial enzyme and the straight form in all of the published Pol II structures (Zhang et al, 1999; Gnatt et al, 2001; Vassylyev et al, 2002; Kettenberger et al, 2004; Westover et al, 2004a, 2004b). As the translocated state is stable in the absence of a bulged bridge helix, direct evidence for the participation, if any, of a bulged helix in translocation remains to be established.
The first observation, however, that the bridge helix in the bacterial polymerase can change its structure from a bulged to a straight conformation was obtained in the structure of a complex of the enzyme with the antibiotic streptolydigin (Tuske et al, 2005). Although the binding site of this antibiotic does not overlap with that of the substrates, it eliminates polymerase activity, leading the authors to propose that the ability of this bridge helix to cycle between straight and bulged is important for activity and perhaps translocation. Other models for the mechanism of allosteric inhibition are perhaps possible.
When crystals of the transcribing complex in the post-translocated state with a chain-terminating residue at the 3′ end of the RNA were soaked in solution containing cognate NTP, the nucleotide bound in the insertion site, base-paired with the templating base and with its phosphate bound to two Mg2+ ions on one side and an Arg residue on the other side (Westover et al, 2004b), analogously to the case with T7 RNAP (Yin and Steitz, 2004), and indeed all other polymerases (Steitz, 1998, 1999). When, however, these crystals were soaked in 15 mM of a mismatched NTP, the nucleotide bound with its three phosphates in an orientation opposite to that of the correct cognate NTP, but interacting through two divalent metal ions and with its sugar-base directed away from the template strand (Figure 6D). Kornberg and co-workers (Westover et al, 2004b) propose that the NTP bound in this opposite orientation site corresponds to an entrance site, or E site. An alternative explanation could be that at this high concentration, the mismatched nucleotide binds adventitiously largely through the three phosphates that are interacting with the magnesiums and arginines but in an opposite direction, as the base is unable to base-pair with the template base. It would be of some interest to know the binding constants for nucleotide binding to the E site as well as the insertion site. It has been proposed that a correct nucleotide binds first to the E site, in the wrong orientation, and then rotates to bind to the insertion site (Westover et al, 2004b). As the interactions of the phosphates and Mg2+ ions differ significantly in the two orientations, the NTP must first dissociate and then finally rebind in the productive insertion orientation.
Single molecule studies as well as biochemical results on transcribing bacterial RNAP are consistent with a model of translocation in which the binding of the incoming NTP is responsible for stabilizing the translocated state (Abbondanzieri et al, 2005). Using an ultra-stable optical trapping system, Abbondanzieri et al (2005) showed discreet steps of 3.7±0.6 Å and concluded that RNAP translocates along DNA by a single base pair per nucleotide addition. More significantly, they determined force–velocity relationships for transcription at both saturating and sub-saturating nucleotide concentrations. Global fits of models to the data were inconsistent with a model for movement that is tightly coupled to pyrophosphate release, but consistent with a model that included a secondary NTP binding site in addition to the insertion site.
The structural data are less conclusive in the case of the multisubunit RNAPs, as three of the complexes are in the post-translocated state and one is in the pre-translocated state (Gnatt et al, 2001; Kettenberger et al, 2004; Westover et al, 2004a, 2004b). Solution studies point to an important role for NTP binding in translocation.
Additional similarities between single and multisubunit polymerases
Although T7 RNAP and the multisubunit polymerases differ in several fundamental aspects, most significantly with the complete lack of a structural relatedness, there are aspects that are remarkably similar (Cramer, 2002). Westover et al (2004b) opined that the involvement of two Mg ions in the mechanism of catalysis by the single subunit and multisubunit polymerases is ‘only incidental' and ‘a consequence of the association of Mg2+ with nucleotides in their various forms, and especially with NTPs'. On the contrary, all DNAPs and RNAPs utilize the same two metal ion mechanism of catalysis (Steitz, 1998, 1999). The orientation of the two metal ions relative to the attacking 3′ OH, which metal ion A activates, and to the phosphates of NTP, which they contact, identically is the same in all polymerases; likewise, metal ion B plays the same initial catalytic role. Indeed, one might surmise that the presumed RNA-based enzyme predecessors of the present protein polymerases also utilized the same two metal ion mechanism orienting the Mg2+ ions using backbone phosphates rather than carboxylates, as was recently demonstrated for the group I intron (Stahley and Strobel, 2005). All polymerases have converged on the same chemical basis of catalysis promoted by two metal ions.
Both families of RNAPs achieve nearly complete processivity in the elongation phase by having the transcript pass through an exit tunnel. Perhaps surprisingly the length of the heteroduplex product appears to be the same in both T7 RNAP (Yin and Steitz, 2002) and in yeast Pol II (Gnatt et al, 2001; Kettenberger et al, 2004; Westover et al, 2004b). The multisubunit polymerases have a pore or tunnel through which the incoming NTPs must pass to reach the active site as do all of the A and B family monomeric polymerases, although it is less deep and approaches the heteroduplex along its axis rather than perpendicular to it, as occurs with the multisubunit polymerases (Westover et al, 2004b). Finally, the product heteroduplex and the downstream duplex are at right angles to each other, but with differing relative rotational orientation, in both T7 RNAP and yeast Pol II (Yin and Steitz, 2002; Westover et al, 2004b). The bend between the upstream duplex DNA and the downstream duplex, along with a change in relative twist, is probably associated with assisting the formation of the open ‘bubble' in the duplex, as suggested for T7 RNAP by Yin and Steitz (2002).
In order to visualize the structural basis for the mechanism by which a macromolecular machine achieves its biological function, it is necessary to establish the structures of the complete assembly that executes the function, captured at each step of the process it facilitates. Just as the first movie of a horse running consisted of a series of snapshots capturing images of the many states of process, the structures of macromolecular assemblies must likewise be established for each step. Then, using an approach such as Gerstein's program MORPH (Krebs and Gerstein, 2000), a movie of the process can be constructed. Not only does the structure of an unliganded protein or assembly provide few if any insights into its function, the structure of single state of a reaction pathway is often insufficient; for example, the structure of the T7 RNAP initiation complex provides no explanation of the properties of the elongation phase of the process.
Future attempts to image the processes carried out by macromolecular assemblies will likewise require numerous structures of each stage, which often utilize a cast of characters that may vary at each step of the process. Full understanding of the initiation of protein synthesis by the ribosome, its elongation cycle as well as its termination step will require numerous structures, with each of the various factors involved in executing their functions. Many of these stages have been wonderfully imaged at low resolution by cryo-electron microscopy and provide insights into the conformational changes undergone by this machine during these processes (e.g., Frank and Agrawal, 2000; Spahn et al, 2004). Understanding at the chemical level, however, will require high-resolution crystal structures of these various assemblies.
Many biological processes are executed by assemblies that are less stable or more transitory than the ribosome is during the course of protein synthesis. Replication of DNA at the replication fork involves several different proteins working together, each carrying out a different function. Currently, only the structures of the individual proteins (except for the clamp loader and clamp; Jeruzalmi et al, 2001) are known. Perhaps among the most complex machines known at present is the spliceosome, whose role in pre-mRNA splicing involves several ribonuclear protein complexes that appear and depart at various stages of the process. For the spliceosome and other large assemblies like the nuclear pore, it is likely that only a collaboration between cryo-electron microscopy studies of whole assemblies and crystal structures of individual components will allow visualization of the structural bases of these processes.
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Movie 7
Acknowledgments
I thank Yong Xiong, Baocheng Pan and Satwik Kamtekar for providing unpublished Supplementary Movies 1, 2, 3 and 4. Funding for research on polymerases in the lab was provided by NIH grant GM57510.
References
- Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM (2005) Direct observation of base-pair stepping by RNA polymerase. Nature 438: 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell DA, Westover KD, Davis RE, Kornberg RD (2004) Structural basis of transcription and RNA polymerase II–TFIIB cocrystal at 4.5 Å. Science 303: 983–988 [DOI] [PubMed] [Google Scholar]
- Cheetham GMT, Jeruzalmi D, Steitz TA (1999) Structural basis for initiation of transcription from an RNA polymerase–promoter complex. Nature 399: 80–83 [DOI] [PubMed] [Google Scholar]
- Cheetham GMT, Steitz TA (1999) Structure of a transcribing T7 RNA polymerase initiation complex. Science 286: 2305–2309 [DOI] [PubMed] [Google Scholar]
- Chen HT, Hahn S (2004) Mapping the location of TFIIB within the Rna polymerase II transcription preinitiation complex: a model for the structure of the PIC. Cell 119: 169–180 [DOI] [PubMed] [Google Scholar]
- Chothia C (1974) Hydrophobic bonding and accessible surface area in proteins. Nature 248: 338–339 [DOI] [PubMed] [Google Scholar]
- Cramer P (2002) Common structural features of nucleic acid polymerases. Bioessays 24: 724–729 [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell DA, Kornberg RD (2001) Structural basis of transcription: RNA polymerase II at 2.8 Å resolution. Science 292: 1863–1876 [DOI] [PubMed] [Google Scholar]
- Doublié S, Tabor S, Long AJ, Richardson CC, Ellenberger T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391: 251–258 [DOI] [PubMed] [Google Scholar]
- Doublié SD, Ellenberger T (1998) The mechanism of action of T7 DNA polymerase. Curr Opin Struct Biol 8: 704–712 [DOI] [PubMed] [Google Scholar]
- Frank J, Agrawal RK (2000) A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406: 318–322 [DOI] [PubMed] [Google Scholar]
- Franklin MC, Wang J, Steitz TA (2001) Structure of the replicating complex of a pol alpha family DNA polymerase. Cell 105: 657–667 [DOI] [PubMed] [Google Scholar]
- Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD (2001) Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science 292: 1876–1882 [DOI] [PubMed] [Google Scholar]
- Guajardo R, Sousa R (1997) A model for the mechanism of polymerase translocation. J Mol Biol 265: 8–19 [DOI] [PubMed] [Google Scholar]
- Guo Q, Sousa R (2006) Translocation by T7 RNA polymerase: a sensitively poised Brownian ratchet. J Mol Biol (in press) [DOI] [PubMed] [Google Scholar]
- Huang J, Sousa R (2000) T7 RNA polymerase elongation complex structure and movement. J Mol Biol 303: 347–358 [DOI] [PubMed] [Google Scholar]
- Ikeda RA, Richardson CC (1987) Interactions of a proteolytically nicked RNA polymerase of bacteriophage T7 with its promoter. J Biol Chem 262: 3800–3808 [PubMed] [Google Scholar]
- Jeruzalmi D, O'Donnell M, Kuriyan J (2001) Crystal structure of the processivity clamp loader Gamma (γ) complex of E. coli DNA polymerase III. Cell 106: 429–441 [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Taylor JS, Beese LS (2003) Processive DNA synthesis observed in a polymerase crystal structure suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci USA 100: 3895–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamtekar S, Berman AJ, Wang J, Lázaro JM, de Vega M, Blanco L, Salas M, Steitz TA (2004) Insights into strand displacement and processivity from the crystal structure of the protein-primed DNA polymerase of bacteriophage φ29. Mol Cell 16: 609–618 [DOI] [PubMed] [Google Scholar]
- Kamtekar S, Berman AJ, Wang J, Salas M, Steitz TA (2006) Structure of bacteriophage φ29 DNA polymerase bound to its protein primer implications for the transition from initiation to elongation. EMBO J 25: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenberger H, Armache K, Cramer P (2004) Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell 16: 955–965 [DOI] [PubMed] [Google Scholar]
- Kiefer JR, Mao C, Braman JC, Beese LS (1998) Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature 391: 304–407 [DOI] [PubMed] [Google Scholar]
- Krebs W, Gerstein M (2000) The macromolecular morpher, a standardized system for comparing protein conformations in a database framework. Nucl Acids Res 28: 1165–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Korolev S, Waksman G (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J 17: 7514–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Temiakov D, Anikin M, McAllister WT (2005) Probing conformational changes in T7 RNA polymerase during initiation and termination by using engineered disulfide linkages. Proc Natl Acad Sci USA 102: 17612–17617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J, Blanco L, Salas M (1997) Protein-primed DNA replication: a transition between two modes of priming by a unique DNA polymerase. EMBO J 16: 2519–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller DK, Martin CT, Coleman JE (1988) T7 RNA polymerase interacts with its promoter from one side of the DNA Helix. Biochem 27: 5763–5772 [DOI] [PubMed] [Google Scholar]
- Murakami KS, Darst SA (2003) Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol 13: 31–39 [DOI] [PubMed] [Google Scholar]
- Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA (2002) Structural basis of transcription initiation: an RNA polymerase holoenzyme/DNA complex. Science 296: 1285–1290 [DOI] [PubMed] [Google Scholar]
- Salas J (1991) Protein-priming of DNA replication. Annu Rev Biochem 60: 39–71 [DOI] [PubMed] [Google Scholar]
- Severinov K (2001) T7 RNA polymerase transcription complex: what you see is not what you get. Proc Natl Acad Sci USA 98: 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CMT, Gomez-Lorenzo MG, Grassucci RA, Jørgensen R, Andersen GR, Beckmann R, Penczek PA, Ballesta JPG, Frank J (2004) Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J 23: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahley MR, Strobel SA (2005) Structural evidence for a two-metal-ion mechanisms of group I intron splicing. Science 390: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Steitz TA (1998) A mechanism for all polymerases. Nature 391: 231–232 [DOI] [PubMed] [Google Scholar]
- Steitz TA (1999) DNA polymerases: structural diversity and common mechanisms. J Biol Chem 274: 17395–17398 [DOI] [PubMed] [Google Scholar]
- Tahirov TH, Temiakov D, Anikin M, Patlan V, McAllister WT, Vassylyev DG, Yokoyoma S (2002) Structure of a T7 RNA polymerase elongation complex at 2.9 Å resolution. Nature 420: 43–50 [DOI] [PubMed] [Google Scholar]
- Temiakov D, Mentesana PE, Ma K, Mustaev A, Borukhov S, McAllister WT (2000) The specificity loop of T7 RNA polymerase interacts first with the promoter and then with the elongating transcript, suggesting a mechanism for promoter clearance. Proc Natl Acad Sci USA 97: 14109–14114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis K, Gong P, Martin CT (2004) Topological and conformational analysis of the initiation and elongation complex of T7 RNA polymerase suggest a new twist. Biochemistry 43: 12709–12715 [DOI] [PubMed] [Google Scholar]
- Thomen P, Lopez PJ, Heslot F (2005) Unraveling the mechanism of RNA-polymerase forward motion by using mechanical force. Phys Rev Lett 94: 128102–128104 [DOI] [PubMed] [Google Scholar]
- Tuske S, Sarafianos SG, Wang X, Hudson B, Sineva E, Mukhopadhyay J, Birktoft JJ, Leroy O, Ismail S, Clark AD Jr, Dharia C, Napoli A, Laptenko O, Lee J, Borukhov S, Ebright RH, Arnold E (2005) Inhibition of bacterial RNA polymerase by streptolydigin: stabilization of a straight-bridge-helix active-center conformation. Cell 122: 541–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S (2002) Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature 417: 712–719 [DOI] [PubMed] [Google Scholar]
- Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA (1997) Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell 89: 1087–1099 [DOI] [PubMed] [Google Scholar]
- Westover KD, Bushnell DA, Kornberg RD (2004a) Structural basis of transcription: separation of RNA from DNA by RNA polymerase III. Science 303: 1014–1016 [DOI] [PubMed] [Google Scholar]
- Westover KD, Bushnell DA, Kornberg RD (2004b) Structural basis of transcription: nucleotide selection by rotation in the RNA polymerase II active center. Cell 119: 481–489 [DOI] [PubMed] [Google Scholar]
- Yin YW, Steitz TA (2002) Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science 298: 1387–1395 [DOI] [PubMed] [Google Scholar]
- Yin YW, Steitz TA (2004) The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell 116: 393–404 [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA (1999) Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell 98: 811–824 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Movie 7