

Abstract
Keap1 is a BTB-Kelch substrate adaptor protein that regulates steady-state levels of Nrf2, a bZIP transcription factor, in response to oxidative stress. We have determined the structure of the Kelch domain of Keap1 bound to a 16-mer peptide from Nrf2 containing a highly conserved DxETGE motif. The Nrf2 peptide contains two short antiparallel β-strands connected by two overlapping type I β-turns stabilized by the aspartate and threonine residues. The β-turn region fits into a binding pocket on the top face of the Kelch domain and the glutamate residues form multiple hydrogen bonds with highly conserved residues in Keap1. Mutagenesis experiments confirmed the role of individual amino acids for binding of Nrf2 to Keap1 and for Keap1-mediated repression of Nrf2-dependent gene expression. Our results provide a detailed picture of how a BTB-Kelch substrate adaptor protein binds to its cognate substrate and will enable the rational design of novel chemopreventive agents.
Keywords: BTB-Kelch proteins, chemoprevention, cullin-dependent ubiquitin ligases, oxidative stress, substrate adaptor proteins
Introduction
Eukaryote cells are constantly exposed to reactive molecules from endogenous and exogenous sources (Finkel and Holbrook, 2000; Jackson and Loeb, 2001). These molecules, including reactive oxygen species, electrophilic chemicals, and heavy metals, damage biological macromolecules and impair normal cellular functions (Davies, 2000; Imlay, 2003). Oxidative damage has been implicated in diverse pathophysiological processes, including cancer, cardiovascular disease, diabetes and neurodegeneration (Ames and Shigenaga, 1993; Jackson and Loeb, 2001; Bonnefont-Rousselot, 2002; Ceconi et al, 2003; Ghanbari et al, 2004; Mhatre et al, 2004). Therapeutic approaches to protect against damage caused by reactive molecules would have broad ramifications for improving human health (Cash et al, 2002; Hamilton et al, 2004).
Eukaryote cells have evolved multiple protective mechanisms against reactive molecules. A major mechanism is the coordinated induction of enzymes that neutralize reactive molecules, eliminate damaged macromolecules and restore cellular redox homeostasis. In metazoan organisms, this cytoprotective response is controlled, in large part, by the Cap N' Collar (CNC) transcription factors, which constitute a unique subset within the bZIP transcription factor family (Mathers et al, 2004). In mammals, the CNC transcription factors include Nrf1, Nrf2, Nrf3, Bach1 and Bach2 (Motohashi et al, 2002). Phenotypic analysis of mice containing targeted deletions of individual or multiple CNC family members has revealed that Nrf1 and Nrf2 are the major regulators of cytoprotective gene expression (Leung et al, 2003; Yu and Kensler, 2005). Nrf1 and Nrf2 share a common domain structure, including a conserved N-terminal regulatory domain termed Neh2, a central transactivation domain, and a C-terminal bZIP domain required for heterodimer formation with members of the small Maf protein family, for DNA binding and nuclear import and export (Chan et al, 1993; Moi et al, 1994; Katoh et al, 2001; Bloom et al, 2002; Motohashi et al, 2002; Li et al, 2005).
The Keap1 protein is the major negative regulator of cytoprotective gene expression (Itoh et al, 1999; Dhakshinamoorthy and Jaiswal, 2001). Keap1, a BTB-Kelch protein, is a substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex (Cullinan et al, 2004; Kobayashi et al, 2004; Zhang et al, 2004). Under basal conditions, Keap1 targets Nrf2 for ubiquitin-dependent degradation and represses Nrf2-dependent gene expression (Wakabayashi et al, 2003; Zhang and Hannink, 2003). In cells exposed to reactive chemicals or oxidative stress, Nrf2 is no longer targeted for ubiquitin-dependent degradation (Zhang and Hannink, 2003; Zhang et al, 2004; Kobayashi et al, 2006). Instead, steady-state levels of Nrf2 increase, resulting in activation of Nrf2-dependent gene expression.
Keap1, as a substrate adaptor, bridges both Cul3 and Nrf2 using its N-terminal BTB and central linker domains to bind Cul3 and its C-terminal Kelch domain to bind the Neh2 domain of Nrf2 (Cullinan et al, 2004; Kobayashi et al, 2004; Zhang et al, 2004). Lysine residues within the Neh2 domain of Nrf2 are targeted for ubiquitin transfer mediated by the Cul3-associated Rbx1 protein and a ubiquitin-charged E2 protein (Zhang et al, 2004). Cyclical association and dissociation of this E3 ubiquitin ligase complex, mediated by the opposing actions of CAND1 and Cul3 neddylation, enables efficient ubiquitination of Nrf2 and repression of Nrf2-dependent gene expression (Lo and Hannink, 2006). This ubiquitin ligase complex is perturbed by reactive chemicals and oxidative stress, which modify a number of reactive cysteine residues located in the BTB and linker domains of Keap1 (Dinkova-Kostova et al, 2002; Zhang and Hannink, 2003; Wakabayashi et al, 2004; Eggler et al, 2005; Hong et al, 2005a, 2005b).
We have determined the structure of the Kelch domain of Keap1 bound to a peptide containing a conserved DxETGE motif located in the Neh2 domain of Nrf2. The binding pocket in Keap1 contains multiple charged and hydrophobic residues that contact the side chains of the two glutamate residues and the peptide backbone. The DxETGE motif contains a β-turn region stabilized by the aspartate and threonine residues. Threonine phosphorylation prevents binding of the Nrf2-derived peptide to Keap1 and phosphomimetic mutations enable Nrf2 to escape Keap1-mediated repression. Keap1 dimerizes via its BTB domain and both Kelch domains within the Keap1 dimer are capable of independently binding to Nrf2. Our results provide insight into how Keap1 targets Nrf2 for ubiquitin-dependent degradation and will facilitate the rational design of novel therapeutic compounds that block binding of Nrf2 to Keap1.
Results
The structure of a complex between the Kelch domain of Keap1 and an Nrf2-derived peptide reveals how Nrf2 fits into the substrate binding pocket of Keap1
The Kelch domain of Keap1 is a six-bladed β-propeller in which each blade of the propeller (I–VI) is comprised of four β-strands (A–D) (Figure 1A) (Li et al, 2004). The β-strands are connected by loops of varying lengths that project from the central core of the β-propeller. By convention (Wall et al, 1995), the short loops that connect either β-strands A and B (A–B) or β-strands C and D (C–D) define the bottom face of the β-propeller, whereas the longer loops that connect either β-strands D and A (D–A) or β-strands B and C (B–C) define the top face of the β-propeller.
Figure 1.
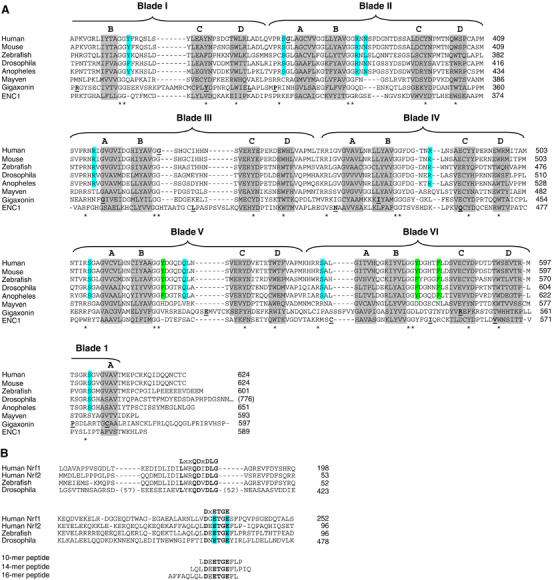
(A) The sequence of the Kelch domains from the Keap1 protein of five different species (human, mouse, zebrafish, Drosophila melanogaster, and Anopheles gambia) is shown, along with the sequence of three human BTB-Kelch proteins, mayven, gigaxonin, and ENC1. The six blades of the β-propeller structure are indicated above the alignment and the four β-strands (A–D) that comprise each blade are highlighted in gray. The A strand for blade I is located at the extreme C-terminus of the protein. Disease-associated mutations in Keap1, gigaxonin, and ENC1 proteins are underlined in bold (Bomont et al, 2000, 2003; Bruno et al, 2004; Kuhlenbaumer et al, 2002; Liang et al, 2004; Padmanabhan et al, 2006). Highly conserved residues that define the Kelch repeat are indicated by asterisks. Amino acids in Keap1 whose side chains contact the Nrf2-derived peptide in the crystal structure are highlighted in blue, whereas those amino acids that only make van der Waals contacts with the Nrf2-derived peptide are highlighted in green. (B) The Neh2 domain from the human Nrf1 and Nrf2 proteins is shown, along with the corresponding regions of Nrf2-related proteins from zebrafish and D. melanogaster. Two conserved motifs that have been implicated in binding to Keap1 are shown. The glutamate residues of the DxETGE motif are highlighted in blue. The sequence of the three Nrf2-derived peptides used in our experiments is shown.
The Neh2 domain contains two regions that are highly conserved from humans to flies (Figure 1B). One region, amino acids 17–32 in the human Nrf2 protein, contains an LxxQDxDLG motif (McMahon et al, 2004; Katoh et al, 2005). A second region, amino acids 77–82 in the human Nrf2 protein, contains a DxETGE motif (Kobayashi et al, 2002). Both of these motifs within Nrf2 bind to the same site in Keap1 but the affinity of Keap1 for the DxETGE motif is approximately 100-fold higher than for the LxxQDxDLG motif (Tong et al, 2006).
To determine if the high-affinity DxETGE motif is sufficient for binding to Keap1, the ability of several different DxETGE-containing peptides to disrupt the Keap1–Nrf2 complex was determined. A complex between a Keap1 protein containing a C-terminal chitin binding domain (Keap1-CBD) and an HA-tagged Nrf2 protein was isolated by affinity purification on chitin beads. The beads were incubated with increasing amounts of the peptides and levels of bound HA-Nrf2 determined. A marked reduction in the amount of HA-Nrf2 that remained bound to the Keap1-CBD protein was observed following incubation with a 16-mer Nrf2-derived peptide containing amino acids 69–84 (AFFAQLQLDEETGEFL) (Figure 2A, lanes 11–14). A 14-mer peptide, containing amino acids 74–87 of Nrf2 (LQLDEETGEFLPIQ), was able to displace Nrf2 from Keap1 as effectively as the 16-mer (Figure 2A, lanes 7–10). A 10-mer Nrf2-derived peptide, containing amino acids 76–85 (LDEETGEFLP), was also able to displace Nrf2 from Keap1, although not quite as effectively as the two longer peptides (Figure 2A, lanes 3–6). This 10-mer peptide lacks one or more backbone interactions that stabilize the longer peptides (see Figure 4C). Isothermal titration calorimetry revealed that the 16-mer peptide bound to the Kelch domain of Keap1 with a Kd value of 20 nM (Figure 2B), in good agreement with the published Kd values ranging from 5 to 9 nM for binding of the Neh2 domain to the full-length Keap1 protein (Eggler et al, 2005; Tong et al, 2006).
Figure 2.
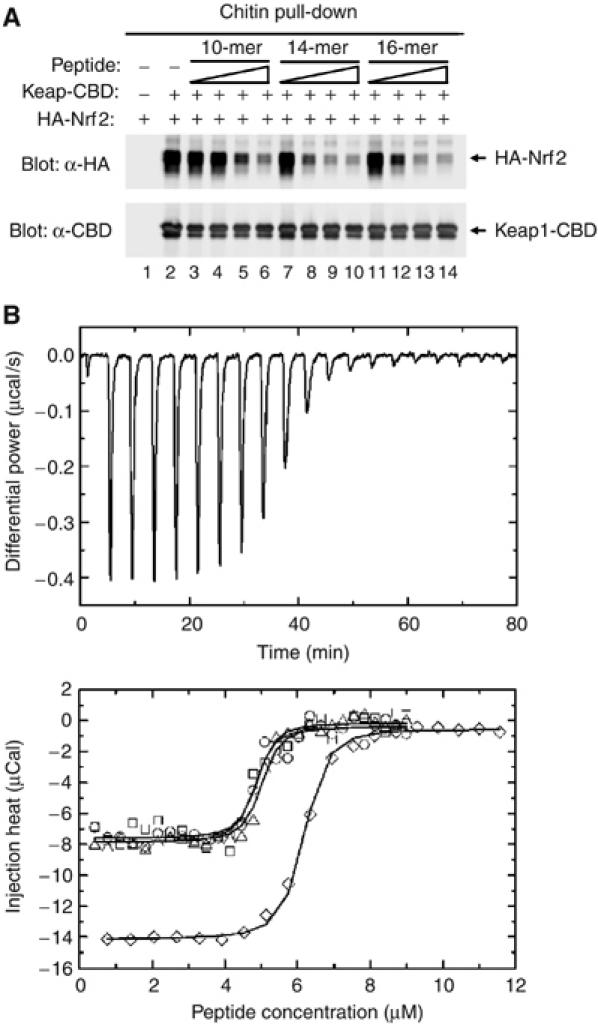
(A) COS1 cells were transfected with expression vectors for Keap1-CBD and HA-Nrf2 and the Keap1:Nrf2 complex isolated on chitin beads. The beads were incubated with the indicated peptides, washed, and proteins that remained bound to the beads were analyzed by immunoblot with anti-HA and anti-CBD antibodies. The amounts of peptides added to each sample were 10 ng (lanes 3, 7, 11), 100 ng (lanes 4, 8, 12), 1 μg (lanes 5, 9, 13), and 10 μg (lanes 6, 10, 14). (B) (Upper panel) Thermographs from a representative isothermal calorimetry experiment are shown, in which a 5 μM solution of the Kelch domain was titrated with 50 μM peptide. (Lower panel) The fitted binding isotherms from four experiments are shown. Three experiments (circles, squares, and triangles) were performed by titrating a 50 μM peptide solution into a 5 μM solution of the Kelch domain. A fourth experiment (diamonds) was performed by titrating an 88 μM solution of the peptide into a 6.25 μM solution of the Kelch domain.
Figure 4.
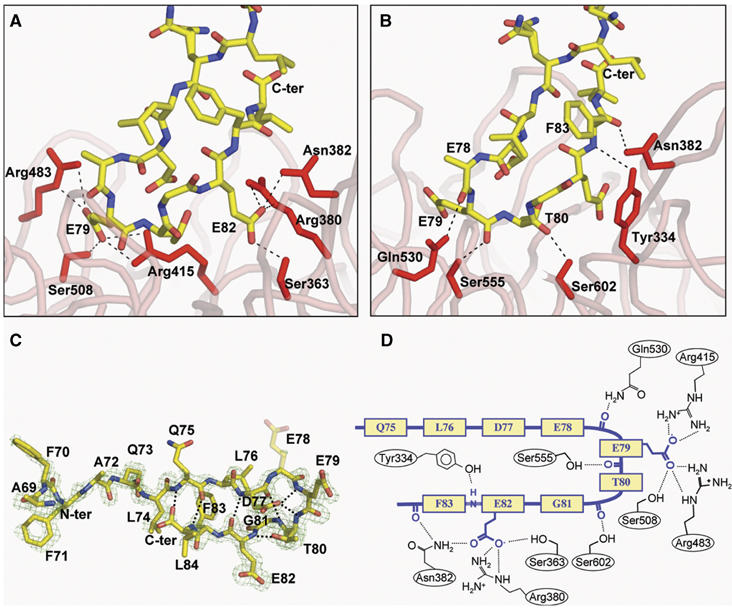
(A) Contacts between the side chain atoms of the Nrf2 peptide and residues in the Kelch domain. (B) Contacts between the backbone atoms of the peptide and residues of the Kelch domain. (C) A stick model of the Nrf2 peptide, with intramolecular hydrogen bonds highlighted. An Fo−Fc electron density omit map contoured at 2.5σ for the vicinity of the peptide is shown in blue. Phases for the map were determined immediately following molecular replacement, before the inclusion of the peptide in the model. The side chain of E78 was not well ordered in the electron density maps and is shown as semi-transparent. (D) A schematic showing both backbone and side-chain contacts to the Nrf2 peptide (yellow/blue) from interacting residues in the Kelch domain.
The structure of the human Kelch domain bound to the 16-mer peptide was determined to be 1.5 Å using X-ray crystallography (Figure 3). As predicted by our previous work (Li et al, 2004), and in agreement with the recent structural analysis of the mouse Kelch domain bound to a DxETGE-containing peptide (Padmanabhan et al, 2006), the Nrf2-derived peptide binds in the shallow pocket defined by the D–A and B–C loops on the top face of the Kelch domain (Figure 3). The peptide has two antiparallel β-strands connected by a turn region that has two overlapping type I β-turns (residues 77–80 and 78–81; Figure 4C). The turn region is stabilized by hydrogen bonds involving the side chains of D77 and T80 and the peptide backbone (Figure 4C). Complex formation buries 420 Å2 of the surface area on the Kelch β-propeller.
Figure 3.
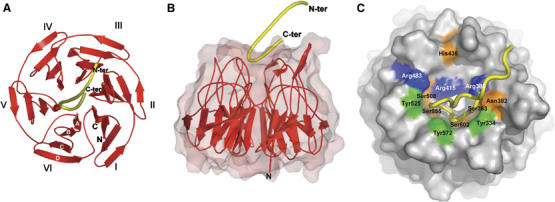
(A) A ribbon diagram of the Kelch β-propeller (red) and bound Nrf2 peptide (yellow tube). The termini of the peptide are labeled N-ter and C-ter; those of the Kelch domain are labeled N and C. The six blades of the β-propeller are labeled I–VI and the four β-strands found in each blade are labeled A–D (white font) on blade VI. (B) Side view of the Kelch domain, with a surface representation of the β-propeller. (C) A surface representation of the Kelch propeller (gray) and peptide (yellow tube). Selected residues are shown in blue (basic), orange (polar), and green (apolar).
Glutamate residues E79 and E82 are the only peptide residues whose side chains make specific interactions with the Kelch domain (Figure 4A and Table I). The carboxylate oxygen atoms of E79 contact the side chains of Arg415, Arg483, and Ser508, whereas the carboxylate oxygen atoms of E82 make hydrogen bonds with the side chains of Ser363, Asn382, and Arg380. The peptide backbone makes five contacts with the Kelch domain, four from the carbonyl oxygen atoms of E78, E79, T80, and F83, and one from a backbone amide group of F83 (Figure 4B).
Table 1.
Mutational analysis of Keap1
| Bladea | Loopb | Amino acidc | Mutant phenotyped | Nrf2 contacte | |
|---|---|---|---|---|---|
| VI/I | D–A | S | 602 | + | T80 bb |
| I | B–C (N) | YFR | 334–336 | − | |
| I | B–C (C) | QSL | 337–339 | − | |
| I | B–C | Y | 334 | − | F83 bb |
| I | B–C | F | 335 | + | |
| I | B–C | R | 336 | + | |
| I | B–C | Q | 337 | + | |
| I/II | D–A | S | 363 | + | E82 sc |
| II | B–C (N) | NNSP | 381–384 | − | |
| II | B–C (C) | DGNT | 385–388 | + | |
| II | B–C | R | 380 | − | E82 sc |
| II | B–C | N | 382 | − | F83 bb; E82 sc |
| II | B–C | P | 384 | + | |
| II | B–C | N | 387 | + | |
| II/III | D–A | R | 415 | − | E79 sc; E79 vw; T80 vw |
| III | B–C (N) | SHG | 431–433 | + | |
| III | B–C (C) | CIH | 434–436 | − | |
| III | B–C | C | 434 | + | |
| III | B–C | H | 436 | − | |
| III/IV | D–A | R | 459 | + | |
| IV | B–C (N) | FDG | 478–480 | − | |
| IV | B–C (C) | TNR | 481–483 | − | |
| IV | B–C | F | 478 | − | |
| IV | B–C | R | 483 | − | E79 sc |
| IV/V | D–A | S | 508 | + | E79 sc |
| V | B–C (N) | YDG | 525–527 | − | |
| V | B–C (C) | ADQ | 528–530 | + | |
| V | B–C | Y | 525 | − | E79 vw |
| V | B–C | Q | 530 | + | E78 bb |
| V/VI | D–A | K | 551 | + | |
| V/VI | D–A | R | 553 | + | |
| V/VI | D–A | S | 555 | + | E79 bb |
| VI | B–C (N) | YDG | 572–574 | − | |
| VI | B–C (C) | HTF | 575–577 | + | |
| VI | B–C | Y | 572 | − | L76 vw; G81 vw |
| VI | B–C | F | 577 | + | G81 vw |
| aThe six blades of the Kelch β-propeller are labeled I–VI. | |||||
| bThe four β-strands of each blade of the Kelch β-propeller are labeled A–D. | |||||
| cThe amino-acid number of the indicated residue in the human Keap1 protein. | |||||
| dThe ability of mutant Keap1 proteins containing an alanine substitution for the indicated amino acid to repress Nrf2-dependent transcription. A ‘+' indicates that the mutant Keap1 protein is equivalent to the wild-type Keap1 protein, whereas a ‘−' indicates that the mutant Keap1 protein is significantly impaired in terms of repression of Nrf2-dependent transcription. | |||||
| eAmino acids in Nrf2 contacted by the indicated residue in the human Keap1 protein. A ‘bb' indicates that the side chain of the Keap1 residue contacts the backbone of the Nrf2 peptide; an ‘sc' indicates that the side chain of the Keap1 residue contacts the side chain of the Nrf2 residue; a ‘vw' indicates that the side chain of the Keap1 residue participates in hydrophobic van der Waals interactions with the Nrf2 residue. | |||||
Solvent molecules mediate additional contacts between the peptide and the Kelch domain (Table II), enabling an additional contact between E79 and Arg415 and allowing E82 to interact with both Asn414 and Ser602. The carboxylate oxygen atom of D77 contacts two water molecules that make bridging hydrogen bonds with Arg415 and Arg380. The hydroxyl group of T80 utilizes a water molecule to interact with Arg380.
Table 2.
Amino acid contacts in the complex
| Peptide residue/atom | Kelch residue/atom | Distance (Å) | Bridging water | Distance (Å) | Kelch residue/atom | Distance (Å) |
|---|---|---|---|---|---|---|
| D77 OD2 | HOH 110 | 2.57 | Arg415 NH1 | 2.76 | ||
| HOH 125 | 2.80 | Arg380 NH1 | 2.90 | |||
| E78 O | Gln530 NE2 | 2.98 | ||||
| E79 O | Ser555 OG | 2.59 | ||||
| E79 OE1 | Ser508 OG | 2.63 | ||||
| Arg415 NH2 | 2.72 | |||||
| Arg415 NH1 | 3.04 | |||||
| E79 OE2 | Arg483 NH2 | 3.17 | ||||
| Arg483 NE | 2.70 | |||||
| HOH 176 | 2.98 | Arg415 NH1 | 3.14 | |||
| T80 O | Ser602 OG | 2.77 | ||||
| T80 OG1 | HOH 45 | 2.75 | Arg380 NH1 | 3.04 | ||
| E82 OE1 | Asn382 ND2 | 2.98 | ||||
| Arg380 NH1 | 2.82 | |||||
| Arg380 NE | 2.82 | |||||
| E82 OE2 | Ser363 OG | 2.63 | ||||
| HOH 33 | 2.97 | Asn414 OD1 | 2.80 | |||
| HOH 4 | 2.75 | Ser602 OG | 2.94 | |||
| F83 O | Asn382 ND2 | 2.99 | ||||
| F83 N | Try334 OH | 3.34 |
All six blades of the Kelch β-propeller contribute to complex formation (Table I). Kelch domain residues that contact the side chains of the peptide are concentrated on one side of the binding pocket in blades II, III, IV, and V, whereas residues that contact backbone atoms of the peptide or participate in van der Waals interactions are located on the other side of the binding pocket in blades V, VI, I, and II (Tables I and II).
Side chains from six residues in Keap1 (Ser363, Asn382, Arg380, Arg415, Arg483, and Ser508) participate in hydrogen bond interactions with the carboxylate oxygen atoms from E79 and E82 in the peptide (Figure 4A). Some of these six residues also participate in other interactions with the peptide. For instance, the backbone amide of Asn382 interacts with the carbonyl oxygen of F83, and the side chains of both Arg415 and Arg380 form a hydrogen bond to a solvent water molecule that bridges to the carboxylate oxygen of D77 (Table II). Side chains from five additional residues in Keap1 participate in hydrogen bonding with the peptide backbone: these are Tyr334, Asn382, Gln530, Ser555, and Ser602 (Figure 4B). Finally, the side chains from seven residues in Keap1 participate in van der Waals interactions with the peptide, including Tyr334, Asn387, Arg415, Ser508, Tyr525, Tyr572, and Phe577 (Table I).
Identification of amino acids within the binding pocket of Keap1 that are required for binding to Nrf2
An extensive alanine-scan mutagenesis of Keap1 was performed to determine the contribution of specific amino acids in Keap1 for binding of Nrf2. Twelve Keap1 mutants that contained alanine substitutions for three or four adjacent residues within each B–C loop were constructed (Table I). The ability of the mutant Keap1 proteins to bind Nrf2 was determined by co-immunoprecipitation and their ability to repress Nrf2-dependent transcription was determined by a reporter gene assay using an ARE-dependent luciferase gene. Eight of the 12 mutant Keap1 proteins were impaired in their ability to bind to Nrf2 and to repress Nrf2-dependent gene transcription (Figure 5A and B).
Figure 5.

(A) COS1 cells were cotransfected with expression vectors for HA-Nrf2 and mutant Keap1 proteins as indicated. Total cell lysates were analyzed by immunoblot with anti-HA and anti-Keap1 antibodies (bottom two panels). Anti-Keap1 immunoprecipitates (IP) were subjected to immunoblot analysis using anti-HA antibodies (top panel). (B) MDA-MB-231 cells were transfected with expression vectors for HA-Nrf2 (100 ng) and mutant Keap1 proteins (50 ng) as indicated, and with an ARE-dependent firefly luciferase reporter gene construct (100 ng). A plasmid encoding Renilla luciferase (10 ng) was included as a control for transfection efficiency. The data shown represent the means and standard deviation of results from three independent experiments. (C) HEK 293 T cells were transfected with expression vectors for Keap1-CBD and mutant HA-Nrf2 proteins as indicated. Total cell lysates were analyzed by immunoblotting with anti-HA and anti-CBD antibodies (bottom two panels). The lysates were incubated with chitin beads, washed, and proteins that remained associated with the chitin beads were analyzed by immunoblotting with anti-HA antibodies (top panel). An asterisk (*) indicates a nonspecific protein detected by the antibody. (D) Reporter assays were formed as described in (B). (E) Pulldown assays were performed as described for (C). (F) Reporter assays were performed as described in (B). (G) Co-immunoprecipitation assays were performed as described in (A), except that the Keap1 and Nrf2 expression vectors were separately transfected into cells and cell lysates were mixed before the immunoprecipitation after input amounts were normalized to Nrf2 levels. (H). Reporter assays were performed as described in (B).
The alanine-scan mutagenesis was refined in a second set of mutant Keap1 proteins, in which 13 amino acids in the B–C loops, each with a surface-exposed side chain, were individually mutated to alanine (Table I). Substitution of individual alanine residues for Tyr334, Asn382, His436, Tyr525, and Tyr572 significantly disrupted the ability of Keap1 to bind Nrf2 and repress Nrf2-dependent transcription (Figure 5C and D). With the exception of His436, all of these residues were observed in the crystal structure to participate in contacts with the Nrf2-derived peptide (Figure 4 and Tables I and II). It is not clear why alanine substitution at His436 perturbed the ability of Keap1 to bind Nrf2, as the circular dichroic (CD) spectrum of the His436A Kelch domain is nearly identical to the CD spectrum of the wild-type Kelch domain (Supplementary Figures 1 and 2). An additional residue, Phe478, was not required for binding to Nrf2 (Figure 5C), but was required for repression of Nrf2-dependent gene expression (Figure 5D). Further analysis revealed that this mutant was defective for directing ubiquitination onto Nrf2 (data not shown).
A third set of mutant Keap1 proteins were constructed in which six arginine residues and one lysine residue, all with surface-exposed side chains, were substituted with alanine residues (Table I). This analysis identified three arginine residues (Arg380, Arg415, Arg483) that were required for binding to Nrf2 and for repression of Nrf2-dependent gene expression (Figure 5E and F). As seen in the crystal structure, the side chains of these three arginine residues are involved in multiple contacts with the Nrf2-derived peptide (Figure 4 and Tables I and II).
A final set of mutant Keap1 proteins were constructed that contained alanine residues individually substituted for four serine residues located in the D–A loops, Ser363, Ser508, Ser555, and Ser602 (Table I). Although the side chains of these serine residues are involved in hydrogen bond interactions with the Nrf2-derived peptide (Figure 4), individual alanine substitutions of these serine residues did not significantly reduce the ability of Keap1 to bind to Nrf2 or to repress Nrf2-dependent gene expression (Figure 5G and H).
Keap1 forms a homodimer capable of binding two Nrf2 molecules
The structure of the human Kelch domain bound to a 16-mer Nrf2-derived peptide and the recently published structure of the murine Kelch domain bound to a shorter Nrf2-derived peptide (Padmanabhan et al, 2006) indicate that the Kelch domain of Keap1 contains a single binding site for the DxETGE motif in Nrf2. As Keap1 dimerizes via its N-terminal BTB domain (Zipper and Mulcahy, 2002), the structural data are consistent with the notion that Keap1 and Nrf2 form a complex with a 2:2 stoichiometry. However, several recent reports have suggested that the Keap1 dimer binds one Nrf2 polypeptide, resulting in a 2:1 stoichiometry (Eggler et al, 2005; Tong et al, 2006). As these in vitro biochemical experiments used high concentrations of purified proteins obtained from prokaryote expression systems, the stoichiometry of the Keap1:Nrf2 complex in the more complex environment of a eukaryote cell remains an open question.
To confirm that Keap1 exists as a dimer in eukaryote cells, a CBD-tagged Keap1 protein was coexpressed in HEK 293 T cells along with an untagged Keap1 protein. The untagged Keap1 protein was pulled down with chitin beads only in the presence of CBD-tagged Keap1 (Figure 6A, second panel from top, compare lane 13 with lane 14). This result is consistent with a prior report that protein–protein interactions involving the N-terminal BTB domain enable Keap1 to form a homodimer (Zipper and Mulcahy, 2002), although the possibility that Keap1 forms higher order oligomeric structures in cells cannot be ruled out. As expected, Nrf2 was pulled down with chitin beads in the presence of the CBD-tagged wild-type Keap1 protein (Figure 6A, top panel, lanes 11 and 12).
Figure 6.
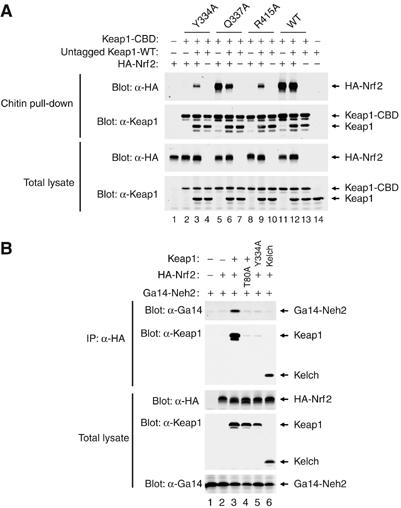
(A) HEK 293 T cells were transfected with expression vectors for the indicated Keap1 and Nrf2 proteins. Total cell lysates were analyzed by immunoblotting with anti-Keap1 and anti-HA antibodies (bottom two panels). Equivalent amounts of cell lysates were incubated with chitin beads, washed, and proteins that remained associated with the chitin beads were analyzed by immunoblotting with anti-HA and anti-Keap1 antibodies (top two panels). (B) HEK 293 T cells were transfected with expression vectors for the indicated Keap1 and Nrf2 proteins. Total cell lysates were analyzed by immunoblotting with anti-HA, anti-Keap1, and anti-Gal4 antibodies (bottom three panels). Anti-HA immunoprecipitates were analyzed by immunoblotting with anti-Gal4 and anti-Keap1 antibodies (top two panels).
Next, we generated heterodimeric Keap1 complexes containing one wild-type Kelch domain and one mutant Kelch domain to determine if the presence of a single functional Kelch domain in a Keap1 dimer would be sufficient to bind Nrf2. A CBD tag was appended to the mutant Keap1 protein such that two types of Keap1 dimers would be isolated on chitin beads: a Keap1 homodimer containing two mutant Kelch domains and a Keap1 heterodimer containing one wild-type and one mutant Kelch domain. Three mutant Keap1 proteins were used for this analysis, including two that were unable to bind Nrf2 on their own (Y334A and R415A) and one (Q337A) that was able to bind Nrf2 as well as the wild-type Keap1 protein. The ability of the homodimeric and heterodimeric complexes to bind Nrf2 was assessed by chitin pulldown followed by immunoblot analysis. As expected, no Nrf2 was pulled down with chitin beads in the presence of either the Keap1-Y334A and Keap1-R415A proteins (Figure 6A, upper panel, lanes 2 and 8). However, coexpression of the wild-type untagged Keap1 protein with either of these two mutant Keap1 proteins partially restored pulldown of Nrf2 (Figure 6A, upper panel, compare lanes 3, 9, and 12). Partial restoration of Nrf2 pulldown is expected, as only a portion of the dimeric Keap1 complexes isolated on chitin beads would have one wild-type Kelch domain with the remainder of the Keap1 complexes containing two non-functional Kelch domains. These results demonstrate that a single functional Kelch domain within the context of a Keap1 dimer is sufficient to bind Nrf2.
Finally, we asked if the Keap1 dimer is capable of binding two different Nrf2 proteins. The Neh2 domain of Nrf2 was fused onto the Gal4 DNA binding domain and coexpressed with HA-tagged full-length Nrf2 protein in the absence or presence of coexpressed Keap1. Cell lysates were immunoprecipitated with anti-HA antibodies and the resultant immunoprecipitates were probed for the presence of the Gal4-Neh2 protein using anti-Gal4 antibodies. The Gal4-Neh2 protein was only present in the anti-HA immunoprecipitates in the presence of coexpressed Keap1 (Figure 6B, upper panel, compare lanes 2 and 3). Thus, the Keap1 protein functions as a bridge between the HA-Nrf2 protein and the Gal4-Neh2 protein. A single alanine substitution for T80 within the DxETGE motif of HA-Nrf2 abolished co-immunoprecipitation of the Gal4-Neh2 protein (Figure 6B, upper panel, lane 4). The Keap1-Y334A protein, which is not able to bind either HA-Nrf2 or Gal4-Neh2, is not able to bridge the two Nrf2 proteins (Figure 6B, upper panel, lane 5). Likewise, the isolated Kelch domain, which is not able to form a homodimer, is not able to bridge the two Nrf2 proteins although it readily binds to the HA-Nrf2 protein (Figure 6B, upper panel, lane 6). These results indicate that a Keap1 dimer is able to bridge two different DxETGE-containing proteins, consistent with the notion that Keap1 and Nrf2 can form a 2:2 stoichiometric complex.
Phosphorylation of the conserved threonine within the DxETGE motif in Nrf2 disrupts binding to Keap1
The crystal structure of the Nrf2-derived peptide bound to the Kelch domain revealed the presence of two overlapping type I β-turns, residues 77–80 and 78–81, in which the side chains of D77 and T80, respectively, form hydrogen bonds with the peptide backbone. The threonine residue T80 was of particular interest, given the possibility that phosphorylation of this residue might interfere with formation of the β-turn and prevent binding of Nrf2 to Keap1.
Peptide competition experiments were carried out using 14-mer peptides that contained either an alanine or phosphothreonine in place of T80. A 14-mer peptide corresponding to amino acids 74–87 of Nrf2 that contained an alanine in place of T80 was unable to displace HA-Nrf2 from Keap1-CBD even at input peptide levels 100-fold higher than that needed for the wild-type peptide to displace Nrf2 from Keap1 (Figure 7A, compare lanes 3–6 with lanes 7–10). Likewise, a 14-mer peptide that contained a phosphothreonine residue in place of T80 was also unable to displace Nrf2 from Keap1 (Figure 7A, lanes 11–14).
Figure 7.
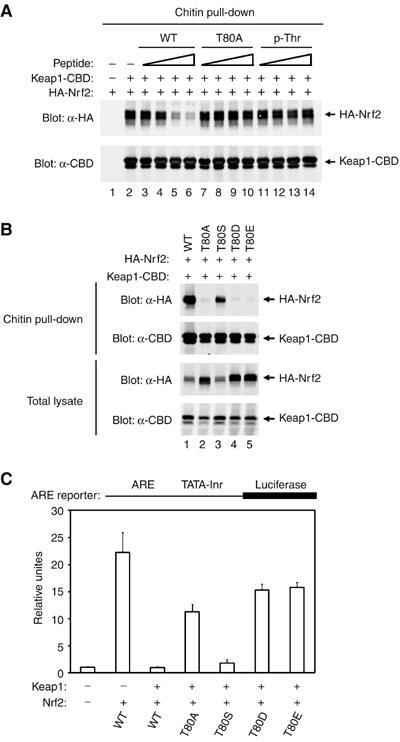
(A) COS1 cells were transfected with expression vectors for Keap1-CBD and HA-Nrf2 and the Keap1:Nrf2 complex isolated on chitin beads. The beads were incubated with the indicated peptides, washed, and proteins that remained bound to the beads were analyzed by immunoblot with anti-HA and anti-CBD antibodies. The amounts of peptides added to each sample were 10 ng (lanes 3, 7, 11), 100 ng (lanes 4, 8, 12), 1 μg (lanes 5, 9, 13), and 10 μg (lanes 6, 10, 14). (B) HEK 293 T cells were transfected with expression vectors for the indicated Keap1 and Nrf2 proteins. Total cell lysates were analyzed by immunoblotting with anti-HA and anti-CBD antibodies (bottom two panels). Equivalent amounts of cell lysates were incubated with chitin beads, washed, and proteins that remained associated with the chitin beads were analyzed by immunoblotting with anti-HA and anti-CBD antibodies (top two panels). (C) Reporter assays were performed as described in Figure 5B.
To examine the importance of T80 for Keap1-mediated repression of Nrf2-dependent gene expression in cells, we characterized several mutant Nrf2 proteins in which T80 was altered to alanine, serine, or to the phosphomimetic glutamate or aspartate. Consistent with the peptide competition experiments, the T80A Nrf2 mutant protein did not bind Keap1 and was resistant to Keap1-mediated repression of Nrf2-dependent gene expression (Figure 7B, top panel, lane 2, and Figure 7C). Nrf2 proteins containing either of the phosphomimetic substitutions (T80D and T80E) were also impaired in binding to Keap1 and were not subject to Keap1-mediated repression (Figure 7B, top panel, lanes 4 and 5, and Figure 7C). In contrast, the T80S mutant Nrf2 protein behaved essentially like the wild-type Nrf2 protein, with only a slight reduction of binding of Nrf2 to Keap1 and Keap1-mediated repression (Figure 7B, top panel, lane 3, and Figure 7C). These results indicate that the hydroxyl group of the serine residue is an effective substitute for the hydroxyl group of the threonine residue in stabilizing the β-turn of the DxETGE motif. In contrast, either the absence of the threonine hydroxyl group or the presence of a negative charge disrupts the β-turn region and likely prevents the flanking glutamate residues from interacting with their cognate residues in Keap1.
Discussion
The ability of cullin-dependent ubiquitin ligase complexes to carry out protein ubiquitination in a highly specific and regulated manner derives from the ability of substrate adaptor proteins to recognize their substrates with high affinity and specificity. The substrate adaptor proteins for the Cul1 and Cul2 proteins have been intensively studied and structural analyses have provided a detailed understanding of their ability to recognize specific substrates (Hon et al, 2002; Min et al, 2002; Orlicky et al, 2003; Wu et al, 2003). Recent work has suggested that the BTB-Kelch proteins, defined by an N-terminal BTB domain and a C-terminal Kelch domain, function as substrate adaptor proteins for Cul3 (Cullinan et al, 2004; Kobayashi et al, 2004, 2006; Zhang et al, 2004, 2005; Furukawa and Xiong, 2005; Wang et al, 2005). In this report, we have determined the structure of the Kelch domain of human Keap1 bound to a 16-mer peptide derived from its substrate, Nrf2. Together with a structure of the Kelch domain of murine Keap1 bound to a 9-mer Nrf2-derived peptide published while this manuscript was under review (Padmanabhan et al, 2006), these structural studies provide a detailed picture of how Keap1 binds Nrf2.
Comparison of amino-acid contacts between the Kelch domain and the Nrf2-derived peptide with functional analysis of mutant Keap1 proteins containing individual alanine substitutions indicates that charged residues and hydrophobic residues located in the binding pocket of Keap1 are the major contributors to the stability of the complex. In particular, the side chains of Tyr334, Arg380, Asn382, Arg415, Arg483, Tyr525, and Tyr572 in Keap1 each contact the Nrf2-derived peptide and individual alanine substitutions for these residues significantly impair binding to Nrf2 and repression of Nrf2-dependent gene expression. In contrast, individual alanine substitutions of four serine residues (Ser363, Ser508, Ser555, and Ser602) that directly contact the Nrf2-derived peptide did not significantly impair binding of Nrf2 or repression of Nrf2-dependent transcription. Thus, hydrogen bond contacts between these serine residues and Nrf2 do not contribute significantly to the overall stability of the Keap1:Nrf2 complex. Nevertheless, the overall agreement between our structural and functional studies indicates that the crystal structure of the complex between the Kelch domain of Keap1 and the Nrf2-derived peptide is an accurate representation of amino-acid contacts between the full-length Keap1 and Nrf2 proteins.
Our structural and functional studies provide strong support for the notion that the DxETGE motif is the principal Keap1 binding site in Nrf2. The lysine residues in Nrf2 that are targeted for ubiquitination are located at a distance of 10–30 amino acids on the N-terminal side of the DxETGE motif (Zhang et al, 2004). Binding of Nrf2 to Keap1 via the DxETGE motif would position these residues for ubiquitin transfer. A second Keap1 binding site in Nrf2, containing the consensus sequence LxxQDxDLG and located at a distance of approximately 50 amino acids on the N-terminal side of the DxETGE motif, has recently been identified (Tong et al, 2006). The LxxQDxDLG motif binds in the substrate binding pocket of the Kelch domain although the affinity of Keap1 for the LxxQDxDLG motif is approximately two orders of magnitude less than for the DxETGE motif. Yamamoto and co-workers (Tong et al, 2006) have proposed a model in which one Nrf2 polypeptide, via both the DxETGE and the LxxQDxDLG motifs, bridges the two Kelch domains in the Keap1 homodimer. This model is supported by in vitro biochemical experiments using purified proteins that suggest a 2:1 stoichiometry of the Keap:Nrf2 complex (Eggler et al, 2005; Tong et al, 2006). In contrast, our experiments, performed by ectopic expression of Keap1 and Nrf2 proteins in the complex intracellular milieu of a eukaryote cell, indicate that the two Kelch domains of a Keap1 homodimer are capable of binding independently to two different Nrf2 polypeptides, consistent with a 2:2 stoichiometry for the Keap:Nrf2 complex. A structure-based model of the Keap1:Cul3:Rbx1 complex suggests that the two Kelch domains in the Keap1 dimer are spatially separated (Stogios et al, 2005), consistent with the notion that each Kelch domain can independently present a substrate protein to the E2 conjugating enzyme recruited into the E3 ubiquitin ligase complex by Rbx1. As this structural model is largely based on computational modeling, detailed structural and functional analysis of the entire ubiquitin ligase complex is needed to provide further insight into how Keap1 presents Nrf2 as a substrate for ubiquitination.
Keap1 constitutively targets Nrf2 for ubiquitin-dependent degradation under basal conditions of cell growth. Following exposure of cells to electrophilic chemicals or overt oxidative stress, Nrf2 is able to escape Keap1-mediated degradation, accumulate in the nucleus, and activate gene expression. The prevailing model for activation of Nrf2 is that reactive chemicals and oxidative stress modify one or more cysteine residues in Keap1 that are located in the BTB and linker domains (Dinkova-Kostova et al, 2002; Zhang and Hannink, 2003; Wakabayashi et al, 2004; Eggler et al, 2005; Hong et al, 2005a, 2005b). As a result, the ability of Keap1 to assemble into a functional E3 ubiquitin ligase complex with Cul3 is perturbed and Nrf2 is no longer efficiently targeted for ubiquitin-dependent degradation although Nrf2 remains associated with Keap1 (Zhang and Hannink, 2003; Zhang et al, 2004; Kobayashi et al, 2006). However, our present results indicate that phosphorylation of threonine residue T80 within the DxETGE motif markedly decreases the ability of an Nrf2-derived peptide to compete with Nrf2 for binding to Keap1 in vitro. Furthermore, phosphomimetic mutants of Nrf2 escape Keap1-mediated repression in cells. As the side chain of T80 participates in hydrogen bonds that stabilize the β-turn, which, in turn, enables the DxETGE motif to fit into the binding pocket of Keap1, phosphorylation of T80 is likely to perturb the conformation of the peptide so that it is unable to fit into the binding site on Keap1. The bulky phosphoryl group may also provide steric hindrance. Although phosphorylation of T80 has not been demonstrated in vivo, an intriguing hypothesis is that phosphorylation of T80 may enable activation of Nrf2 in the absence of overt exposure to electrophilic compounds or oxidative stress. A detailed investigation of the phosphorylation status of T80 in Nrf2 in vivo will provide novel insights into the multiplicity of pathways that can lead to activation of Nrf2-dependent gene expression.
Keap1 is one of 49 BTB-Kelch proteins encoded by the human genome. Point mutations in the Kelch domain of several BTB-Kelch proteins, including Keap1, gigaxonin, and ENC1, have been associated with human diseases (Figure 1A). In the case of Keap1, one gene variant (G364C) and one somatic mutation (G430C) have been associated with lung cancer (Padmanabhan et al, 2006). The G364C mutation is located in the A–D loop of blade I and likely disrupts the substrate binding pocket, whereas the G430C mutation is adjacent to the B strand in blade III and likely disrupts the structure of the Kelch domain. As expected, both of these mutant Keap1 proteins are impaired in their ability to bind Nrf2 (Padmanabhan et al, 2006). A number of disease-associated point mutations within the Kelch domains of both gigaxonin and ENC1 have been described (Bomont et al, 2000, 2003; Kuhlenbaumer et al, 2002; Liang et al, 2004) and it is likely that these mutations will perturb the association of gigaxonin and ENC1 with their respective substrate proteins. Furthermore, as the overall dimension of the substrate binding pocket, formed by the D–A and the B–C loops, is likely to be very similar in other BTB-Kelch proteins, substrate(s) for other BTB-Kelch proteins may also utilize β-turn motifs to bind their cognate substrate adaptor. The Keap1 protein has emerged as a representative prototype for the BTB-Kelch proteins and a detailed understanding of the structure and function of Keap1 will continue to illuminate our understanding of this large and important family of human proteins.
Materials and methods
Recombinant DNA molecules and peptides
Eukaryote expression plasmids for the wild-type Keap1, CBD-tagged Keap1, HA-tagged Nrf2 and Gal4-Neh2 proteins, and the ARE-dependent reporter plasmid have been described previously (Zhang et al, 2004). The pET15b-based expression vector for the His-tagged Kelch domain of Keap1 has been described previously. Mutant Keap1 and Nrf2 cDNAs were generated by site-specific mutagenesis with standard overlap-extension techniques. All the genes used in this study were sequenced in the context of the expression vectors used for the experiments. Peptides were purchased from Sigma-Genosys and from Cell-tek and were supplied at greater than 95% purity following HPLC purification.
Cell culture, transfections, and reporter gene assays
COS1, MDA-MB-231, and HEK-293 T cells were purchased from ATCC. Cells were maintained in either Dulbecco's modified Eagle's medium or Eagle's minimal essential medium in the presence of 10% fetal bovine serum. Transfections were performed with Lipofectamine Plus (Gibco BRL). Reporter gene assays were performed using the Promega Dual-Light assay system.
Antibodies, immunoprecipitation, peptide competition, and immunoblot analyses
The anti-Keap1 antibody has been described (Zhang and Hannink, 2003). Antibodies against Nrf2 (Santa Cruz), chitin binding domain (New England Biolabs), and the HA epitope (Covance) were purchased from commercial sources. For immunoprecipitation, cell extracts were prepared in RIPA buffer (10 mM sodium phosphate (pH 7.9), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% Triton SDS) containing 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (Sigma). Soluble cell lysates were incubated with 2 μg of affinity-purified antibodies for 2 h at 4°C followed by incubation at 4°C with protein A-agarose beads (Sigma) for 2 h. Unbound proteins were removed by washing four times with lysis buffer. The immunoprecipitated proteins were eluted in sample buffer by boiling for 5 min, electrophoresed through SDS–polyacrylamide gels, transferred to nitrocellulose membranes, and subjected to immunoblot analysis.
For peptide competition assay, COS1 cells were transfected with expression vectors for HA-Nrf2 and Keap1-CBD. The transfected cells were exposed to proteasome inhibitor MG132 for 4.5 h before cell lysis. Cell lysates were collected in ELB buffer (50 mM Hepes (pH 7.9), 250 mM NaCl, 0.1% Triton X-100, 5 mM EDTA) containing 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail, and phosphatase inhibitors (1 mM EGTA, 1 mM Na3VO4, 100 mM NaF, 20 mM sodium pyrophosphate). The lysates (200 μg) were incubated with chitin bead at 4°C, pelleted by centrifugation (3000 g), and washed in ELB buffer. Proteins that remained associated with the chitin beads were further incubated with the peptides (10 ng, 100 ng, 1 μg, or 10 μg) at 4°C and, after several washes in ELB buffer, were analyzed by immunoblotting with anti-HA and anti-CBD antibodies.
Isothermal titration calorimetry
Isothermal titration calorimetry experiments were conducted at 25°C in a MicroCal VP-ITC. The Kelch protein was dialyzed to equilibrium against the reaction buffer (50 mM Tris, pH 8.0, 100 mM NaCl, 5 mM DTT). An aliquot of the dialysis reservoir was retained for preparing dilutions of the Kelch domain and peptide. Immediately before the experiment, the protein and peptide solutions were briefly degassed under vacuum. Additions (10 μl) of the peptide were made, at 4 min intervals, to samples of the Kelch domain contained in the 1.41 ml sample cell. In three experiments, a 5 μM solution of the Kelch domain was titrated with 50 μM peptide. In a fourth experiment, a 6.25 μM solution of the Kelch domain was titrated with 88 μM peptide. The integrated data from the four experiments were analyzed globally, employing a singe-site binding model, to extract estimates for the binding constant and binding enthalpy.
Crystallization and data collection
Purification of the Kelch domain was carried out as previously described (Li et al, 2004). Crystals were grown by hanging drop vapor diffusion at 4°C from 2 μl of a 1:3 molar ratio mixture of protein to peptide and 2 μl of well buffer. Diffraction quality crystals grew within 2 days from a well buffer containing 0.2 M MgCl2, 0.1 M Tris–HCl, and 30% w/v PEG 4000, at a protein concentration of 10 mg/ml. A single crystal was used to collect a 1.55 Å data set at −180°C on beamline MBC 4.2.2 at the Advanced Light Source of Lawrence Berkeley National Laboratory. All data were indexed and integrated with d*Trek (Pflugrath, 1999). Data collection and refinement statistics are given in Table III.
Table 3.
Data collection and refinement statistics
| Data collection statistics | |
| Wavelength (Å) | 1.239 |
| Space group | P21212 |
| Cell (Å) | a=76.76, b=92.07, c=46.31 |
| No. of molecules per asuc | 1 |
| Resolution (Å) | 36.42–1.50 |
| Mosaicity (deg) | 0.34 |
| No. of observations | 321 494 |
| No. of unique reflections | 50 551 |
| Redundancy | 6.3 (3.3) |
| Rmerge (%) | 7.7 (45.6) |
| Mean I/σI | 11.1 (2.1) |
| Completeness (%) | 94.7 (64.6) |
| Refinement statistics | |
| Resolution range (Å) | 36.0–1.50 |
| Rcryst a | 17.9 |
| Rfree b | 19.9 |
| R.m.s.d. bond distance (Å) | 0.009 |
| R.m.s.d. bond angle (deg) | 1.211 |
| Total no. of non-H atoms in asuc | 2728 |
| No. of solvent molecules | 333 |
| Avg. peptide B-value (Å2) | 18.1 |
| Avg. protein B-value (Å2) | 16.9 |
| Avg. solvent B-value (Å2) | 30.2 |
| Numbers in parentheses refer to statistics for outer resolution shell (1.55–1.50 Å). | |
| aRcryst=∑∣Fo−Fc∣/∑∣Fc∣, where Fo and Fc are observed and calculated structure factors, respectively. | |
| bRfree is the R factor calculated from 5% of the reflections not included in refinement. No σ-cutoff of the data was used. | |
| casu, asymmetric unit. | |
Structure solution and refinement
The structure of Kelch-peptide complex was determined by molecular replacement using MOLREP (Murshudov et al, 1997) and the structure of the Kelch domain (1U6D) as the search model. Refinement was performed with REFMAC 5.0 (Murshudov et al, 1999); progress was monitored by use of Rfree and 5% of the data were set aside for crossvalidation before refinement. Water molecules were placed in peaks greater than 3.0σ in Fo−Fc maps and within hydrogen bonding distance to nitrogen or oxygen atoms of the protein, or other solvent atoms. TLS refinement (Winn et al, 2001) was performed with the protein and peptide as separate rigid bodies. Model building was performed interactively using Coot (Emsley and Cowtan, 2004).
The final model of the protein consisted of 285 residues in the Kelch domain, 16 residues in the peptide, and 333 water molecules (Table III). Residues are numbered according to the sequence of the intact Keap1 protein and Nrf2 proteins. Clear electron density was present for the backbone of all residues of the peptide; residues at the termini of the peptide are involved in crystal contacts with other molecules in the unit cell. Density for the side chains of five residues in the Kelch domain (Glu446, Arg447, Glu493, Gln528, and Lys551) and two in the peptide (T72 and E78) was not well defined and is represented as alanine residues. A total of 17 residues have been modeled in two conformations, including one in the peptide (Q75). The model was evaluated by SFCHECK (Vaguine et al, 1999) and WHAT_CHECK (Hooft et al, 1996). Figures were prepared with PYMOL (De Lano, 2002). The coordinates have been deposited in the PDB with code 2FLU. The model has good geometry, with 91.6% of its residues in the most favored regions of the Ramachandran plot and 0.0% in the disallowed regions (Laskowski et al, 1993).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We are indebted to Dr Jay Nix and the staff of beamline 4.2.2 at the Advanced Light Source of Lawrence Berkeley Laboratory for collection of the synchrotron data. We thank Dr Jack Tanner and Dr Shrikesh Sachdev for helpful discussions. This work was supported by a research grant from NIH (AT003899) and a development project in P50 CA103130.
References
- Ames BN, Shigenaga MK (1993) DNA and free radicals. In Oxidants are a Major Contributor to Cancer and Aging, Halliwell B, Aruoma OI (eds) pp 1–15. New York, NY [Google Scholar]
- Bloom D, Dhakshinamoorthy S, Jaiswal AK (2002) Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 21: 2191–2200 [DOI] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M (2000) The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 26: 370–374 [DOI] [PubMed] [Google Scholar]
- Bomont P, Ioos C, Yalcinkaya C, Korinthenberg R, Vallat JM, Assami S, Munnich A, Chabrol B, Kurlemann G, Tazir M, Koenig M (2003) Identification of seven novel mutations in the GAN gene. Hum Mutat 21: 446. [DOI] [PubMed] [Google Scholar]
- Bonnefont-Rousselot D (2002) Glucose and reactive oxygen species. Curr Opin Clin Nutr Metab Care 5: 561–568 [DOI] [PubMed] [Google Scholar]
- Bruno C, Bertini E, Federico A, Tonoli E, Lispi ML, Cassandrini D, Pedemonte M, Santorelli FM, Filocamo M, Dotti MT, Schenone A, Malandrini A, Minetti C (2004) Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology 62: 13–16 [DOI] [PubMed] [Google Scholar]
- Cash AD, Perry G, Smith MA (2002) Therapeutic potential in Alzheimer disease. Curr Med Chem 9: 1605–1610 [DOI] [PubMed] [Google Scholar]
- Ceconi C, Boraso A, Cargnoni A, Ferrari R (2003) Oxidative stress in cardiovascular disease: myth or fact? Arch Biochem Biophys 420: 217–221 [DOI] [PubMed] [Google Scholar]
- Chan JY, Han XL, Kan YW (1993) Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc Natl Acad Sci USA 90: 11371–11375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA (2004) The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol 24: 8477–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KJ (2000) Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 50: 279–289 [DOI] [PubMed] [Google Scholar]
- De Lano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific [Google Scholar]
- Dhakshinamoorthy S, Jaiswal AK (2001) Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 20: 3906–3917 [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA 99: 11908–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD (2005) Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci USA 102: 10070–10075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247 [DOI] [PubMed] [Google Scholar]
- Furukawa M, Xiong Y (2005) BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the cullin 3-Roc1 ligase. Mol Cell Biol 25: 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanbari HA, Ghanbari K, Harris PL, Jones PK, Kubat Z, Castellani RJ, Wolozin BL, Smith MA, Perry G (2004) Oxidative damage in cultured human olfactory neurons from Alzheimer's disease patients. Aging Cell 3: 41–44 [DOI] [PubMed] [Google Scholar]
- Hamilton CA, Miller WH, Al-Benna S, Brosnan MJ, Drummond RD, McBride MW, Dominiczak AF (2004) Strategies to reduce oxidative stress in cardiovascular disease. Clin Sci (London) 106: 219–234 [DOI] [PubMed] [Google Scholar]
- Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY (2002) Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 417: 975–978 [DOI] [PubMed] [Google Scholar]
- Hong F, Freeman ML, Liebler DC (2005a) Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 18: 1917–1926 [DOI] [PubMed] [Google Scholar]
- Hong F, Sekhar KR, Freeman ML, Liebler DC (2005b) Specific patterns of electrophile adduction trigger keap1 ubiquitination and NRF2 activation. J Biol Chem 280: 31768–31775 [DOI] [PubMed] [Google Scholar]
- Hooft RW, Sander C, Vriend G (1996) Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures. Proteins 26: 363–376 [DOI] [PubMed] [Google Scholar]
- Imlay JA (2003) Pathways of oxidative damage. Annu Rev Microbiol 57: 395–418 [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13: 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Loeb LA (2001) The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res 477: 7–21 [DOI] [PubMed] [Google Scholar]
- Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, McMahon M, Hayes JD, Itoh K, Yamamoto M (2005) Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys 433: 342–350 [DOI] [PubMed] [Google Scholar]
- Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M (2001) Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 6: 857–868 [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24: 7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M (2006) Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol 26: 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, Takagi Y, Yamamoto M (2002) Identification of the interactive interface and phylogenic conservation of the Nrf2–Keap1 system. Genes Cells 7: 807–820 [DOI] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Young P, Oberwittler C, Hunermund G, Schirmacher A, Domschke K, Ringelstein B, Stogbauer F (2002) Giant axonal neuropathy (GAN): case report and two novel mutations in the gigaxonin gene. Neurology 58: 1273–1276 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067 [DOI] [PubMed] [Google Scholar]
- Leung L, Kwong M, Hou S, Lee C, Chan JY (2003) Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J Biol Chem 278: 48021–48029 [DOI] [PubMed] [Google Scholar]
- Li W, Jain MR, Chen C, Yue X, Hebbar V, Zhou R, Kong AN (2005) Nrf2 possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem 280: 28430–28438 [DOI] [PubMed] [Google Scholar]
- Li X, Zhang D, Hannink M, Beamer LJ (2004) Crystal structure of the Kelch domain of human Keap1. J Biol Chem 279: 54750–54758 [DOI] [PubMed] [Google Scholar]
- Liang X-Q, Avraham HK, Jiang S, Avraham S (2004) Genetic alterations of the NRP/B gene are associated with human brain tumors. Oncogene 23: 5890–5990 [DOI] [PubMed] [Google Scholar]
- Lo SC, Hannink M (2006) CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol Cell Biol 26: 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers J, Fraser JA, McMahon M, Saunders RD, Hayes JD, McLellan LI (2004) Antioxidant and cytoprotective responses to redox stress. Biochem Soc Symp 71: 157–176 [DOI] [PubMed] [Google Scholar]
- McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD (2004) Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem 279: 31556–31567 [DOI] [PubMed] [Google Scholar]
- Mhatre M, Floyd RA, Hensley K (2004) Oxidative stress and neuroinflammation in Alzheimer's disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. J Alzheimers Dis 6: 147–157 [DOI] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr, Pavletich NP (2002) Structure of an HIF-1alpha-pVHL complex: hydroxyproline recognition in signaling. Science 296: 1886–1889 [DOI] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA 91: 9926–9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, O'Connor T, Katsuoka F, Engel JD, Yamamoto M (2002) Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 294: 1–12 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D 55 (Part 1): 247–255 [DOI] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F (2003) Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112: 243–256 [DOI] [PubMed] [Google Scholar]
- Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M (2006) Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 21: 689–700 [DOI] [PubMed] [Google Scholar]
- Pflugrath JW (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr D 55 (Part 10): 1718–1725 [DOI] [PubMed] [Google Scholar]
- Stogios PJ, Downs GS, Jauhal JJS, Nandra SK, Prive GG (2005) Sequence and structural analysis of BTB domain proteins. Genome Biol 6: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M (2006) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 26: 2887–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaguine AA, Richelle J, Wodak SJ (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D 55 (Part 1): 191–205 [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA 101: 2040–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35: 202–204 [DOI] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR (1995) The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell 83: 1047–1058 [DOI] [PubMed] [Google Scholar]
- Wang W, Ding J, Allen E, Zhu P, Zhang L, Vogel H, Yang Y (2005) Gigaxonin interacts with tubulin folding cofactor B and controls its degradation through the ubiquitin–proteasome pathway. Curr Biol 15: 2050–2055 [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D 57: 122–133 [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP (2003) Structure of a beta-TrCP1–Skp1–beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell 11: 1445–1456 [DOI] [PubMed] [Google Scholar]
- Yu X, Kensler T (2005) Nrf2 as a target for cancer chemoprevention. Mutat Res 591: 93–102 [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23: 8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24: 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M (2005) Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem 280: 30091–30099 [DOI] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT (2002) The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem 277: 36544–36552 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2