

Abstract
Spinobulbar muscular atrophy and Huntington's disease are caused by polyglutamine expansion in the androgen receptor and huntingtin, respectively, and their pathogenesis has been associated with abnormal nuclear localization and aggregation of truncated forms of these proteins. Here we show, in diverse cell types, that glucocorticoids can up- or down-modulate aggregation and nuclear localization of expanded polyglutamine polypeptides derived from the androgen receptor and huntingtin through specific regulation of gene expression. Wild-type glucocorticoid receptor (GR), as well as C-terminal deletion derivatives, suppressed the aggregation and nuclear localization of these polypeptides, whereas mutations within the DNA binding domain and N terminus of GR abolished this activity. Surprisingly, deletion of a transcriptional regulatory domain within the GR N terminus markedly increased aggregation and nuclear localization of the expanded polyglutamine proteins. Thus, aggregation and nuclear localization of expanded polyglutamine proteins are regulated cellular processes that can be modulated by a well-characterized transcriptional regulator, the GR. Our findings suggest approaches to study the molecular pathogenesis and selective neuronal degeneration of polyglutamine expansion diseases.
Polyglutamine expansion diseases include spinobulbar muscular atrophy (SBMA), Huntington's disease (HD), dentatorubro-pallidoluysian atrophy, and several spinocerebellar ataxias. Each is caused by CAG codon expansion within a unique gene producing a polyglutamine tract enlargement in the target protein. Each mutant protein causes selective neurodegeneration within the central nervous system (CNS). For example, polyglutamine expansion in the N terminus of the androgen receptor (AR) causes SBMA (1, 2), an X-linked progressive motor neuron disease, without affecting other neurons in the CNS in which it is also expressed. In contrast, polyglutamine expansion in the huntingtin protein (htt) causes selective degeneration of cortical and striatal neurons despite its expression throughout the nervous system (3, 4). Their common mutation (a CAG repeat expansion) suggests that all eight polyglutamine expansion diseases may be unified by a single pathologic mechanism. This mechanism may involve formation of nuclear and cytoplasmic aggregates that includes the expanded polyglutamine proteins, a characteristic feature of polyglutamine expansion diseases in patients (5, 6), transgenic animals (7–9), and cell culture models (10–12). The relationship of these protein aggregates to pathogenesis remains uncertain: several studies have reported dissociation of aggregate formation from cytotoxicity (9, 12–14), whereas others have drawn a close parallel (5, 8).
Formation of polyglutamine protein aggregates has been tied to polyglutamine protein ubiquitination (5, 6, 8, 12, 15), transglutaminase crosslinking of the glutamine tracts (16), overexpression of an htt-associated protein (17), as well as amino acid sequences outside of the polyglutamine domain (18). Others have demonstrated reduction of aggregate formation by overexpression of the chaperone HDJ-2 in tissue culture (10). Studies in cultured neurons and transgenic animals suggest that nuclear localization of polyglutamine proteins may underlie their pathogenicity (9, 12). Nuclear localization sequences have been identified in certain polyglutamine proteins (e.g., ataxin-1), but not in others (e.g., htt), and although small size may allow diffusion of certain small polyglutamine proteins into the nucleus, some larger polyglutamine proteins, such as ataxin-1 and ataxin-3, probably are actively transported (9, 19). However, the cellular control over the aggregation and nuclear localization of polyglutamine proteins that may underlie their regional specificity and neuronal toxicity in human disease is unknown. Here we demonstrate that a physiologic regulator of transcription can modulate polyglutamine protein aggregation and nuclear localization.
Materials and Methods
Plasmids.
Hemagglutinin (HA)-tagged ARN127 was created initially by subcloning human AR cDNA (kind gift of Diane Merry, Thomas Jefferson University, Philadelphia) downstream of the HA sequence in a yeast expression vector pRD54 (kind gift of Joachim Li, University of California, San Francisco). The ARN127 sequence then was PCR-amplified by using the following primers: 5′, TAAGGATCCGTCGACAAAATGGCCTACCCATATGATGTTCCA and 3′, AGCCGGGACCTCACGGTGGGGCTCTCTCCATCTAGAACTCAGCGCCTAGGAA. The resultant protein contains seven linker amino acids between the HA and AR sequences. This fragment was subcloned into a mammalian expression vector, pIRES.NEO (CLONTECH), which uses a cytomegalovirus promoter, in both its wild-type (wt) (25 glutamines) and mutant (65 glutamines) forms. An HA-tagged htt exon 1 fragment was created by using genomic DNA from a HD patient with 62 CAG repeats. PCR primers were used to engineer an HA epitope on the amino terminus of the protein: 5′, CCGGAATTCATGGCCTACCCATATGATGTTCCAGATTACGCTTCTTTGACCATGGCGACCCTGGAAAAGCTG, and 3′, GGAGGATCCTCACGGTCGGTGCAGCGGCTC. This fragment was cut with BamHI and EcoRI and cloned into pIRES.NEO. For cell count studies, an htt-green fluorescent protein (GFP) construct was used to facilitate sample processing by PCR-amplifying HD genomic DNA with the following primers: 5′, TGGGATCCGTCGACATGGCGACCCTGGAAAAGCTGATGAAGG, and 3′, TGGGATCCCTAGAATTCCGGTCGGTGCAGCGGCTCCTCAGCCACAGC. This fragment was cloned in frame upstream of a mammalian expression plasmid (p6R) that has been described (20) containing GFP sequence driven by the Rous sarcoma virus promoter. Steroid receptor expression plasmids [rat GR, rat mineralocorticoid receptor (MR), and human AR] also were constructed in the p6R vector backbone. All constructs were sequenced.
Cell Culture and Immunofluorescence.
Human embryonic kidney (HEK) 293 cells were grown in DME-H21 with 10% FBS on polyornithine-coated acid-washed glass coverslips in a 24-well plate and were transfected with 200 ng of ARN127 expression plasmid (pHA.ARN127.ires.neo) and 50 ng of rat GR expression plasmid (p6RGR) using Lipofectamine Plus (GIBCO/BRL). Transfection efficiency averaged approximately 70%. Cells were treated with 100 nM dexamethasone (dex) or with EtOH vehicle control at the time of transfection and were harvested 48 h after transfection. Cells were fixed in 4% paraformaldehyde/PBS and stained with mouse monoclonal anti-HA antibody (Babco, Richmond, CA) and goat anti-mouse rhodamine-linked secondary antibody (Molecular Probes), along with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). Subcellular localization was determined by visual inspection and colocalization of signal from DAPI and rhodamine. N2a cells were grown on glass coverslips coated with matrigel (1:50 dilution) in DMEM-H21 with 10% FBS. Transfection was performed with Effectene reagent (Qiagen, Chatsworth, CA), and cells were harvested and immunostained 48 h later.
Determination of Percentage of Cells with Aggregates.
Relative effects on aggregation were determined as follows. After immunostaining as above, cells were visualized at ×40. The number of cells displaying nuclear or cytoplasmic aggregates was divided by the total number of transfected cells (as determined by the presence of expressed protein). This percentage was compared with the percentage of identically transfected cells showing aggregates after hormone stimulation (dex for GR, cortisol for MR, and GMM, the GR N terminus replacing that of MR) at 100 nM to give a “relative” aggregation value. At least 300 transfected cells were counted for each data point. Typical percent aggregate formation varied somewhat from experiment to experiment, but averaged 15% of transfected cells in the absence of activated GR. Relative effects in the presence and absence of hormone were averaged over at least three separate transfections. For constitutively active derivatives of GR (i.e., GRN525), effects were determined by comparing cells expressing p6RGRN525 to those carrying the empty expression vector p6R.
Results
A Truncated Form of Expanded AR Forms Aggregates in HEK293 Cells.
Studies in human tissue and cell culture suggest that the toxic form of AR is a short fragment of the receptor containing the expanded polyglutamine tract (6, 11, 21–23). Therefore we expressed HA-tagged C-terminal truncation mutants of wt (25 glutamines) and expanded (65 glutamines) AR and monitored their intracellular distribution and ability to form aggregates when expressed in HEK293 cells (Fig. 1).
Figure 1.

Only a truncated form of expanded polyglutamine AR [ARN127(65)] spontaneously forms intracellular aggregates. Diagram of AR C-terminal deletion mutations and their effect on expanded polyglutamine protein aggregation. DBD denotes the DNA binding domain; LIGAND BINDING denotes the C-terminal ligand binding domain.
After 48 h, the polyglutamine expansion did not induce aggregation of AR derivatives lacking the C terminus and DNA binding domains. However, a shorter AR fragment containing the expanded tract of glutamines [ARN127(65)] formed primarily cytoplasmic and some nuclear aggregates in approximately 15% of transfected cells, whereas the wt fragment [ARN127(25)] localized primarily in the cytoplasm without detectable aggregates (Figs. 1 and 2A). This finding is consistent with reports that ARN127(65) forms cytoplasmic and nuclear aggregates and produces cytotoxicity in cultured COS cells (11), and in a form with 112 glutamines, produces a neurologic phenotype in a transgenic mouse model of spinobulbar muscular atrophy (D. Merry, personal communication).
Figure 2.

Characteristics of ARN127(65) aggregation in HEK293 cells and suppression by GR derivatives. (A) Immunofluorescence of HEK293 cells cotransfected with 200 ng of ARN127 expression plasmid and 50 ng of wtGR or GRΔ108–317 expression plasmid after 48 h. Cells cotransfected with ARN127(25) and wtGR in the absence (a) or presence (b) of dex, and with GRΔ108–317 plus dex (c) all show diffuse distribution of ARN127(25). ARN127(65) cotransfected with wtGR forms cytoplasmic aggregates in the absence of dex (e), distributes diffusely in the presence of dex (f), and forms nuclear aggregates when cotransfected with GRΔ108–317 plus dex (g). A dying cell is seen with cotransfected ARN127(65) and GRΔ108–317 plus dex when viewed under immunofluorescence (d) or phase (h) microscopy. Note nuclear aggregates visible under phase microscopy. HA-tagged ARN127 is displayed in red and DAPI nuclear stain in green. (Magnifications: ×100.) (B) GR DNA binding domain and N terminus are required for suppression of polyglutamine protein aggregation. Relative suppression of ARN127(65) aggregation is shown for various GR mutants, MR, and GR-MR fusions. This valve represents the ratios of the percentage of transfected cells with aggregates minus and plus hormone administration, scored by counting >300 cells for each data point for at least three transfections. For constitutively active receptor, this value represents the ratio of the percentage of cells with aggregates in the presence (GRN525) and absence (p6R) of the receptor. Average percentage of cells with aggregates was ≈15% for wtGR − dex and ≈3.5% for wtGR + dex. Error bars represent the SEM. (C) Diagram of various GR mutants and fusions to MR. GRΔ108–317 has amino acids 108–317 deleted from its N terminus. GR(AF1–14) contains 16 amino acid substitutions within amino acids 108–317. MGG, chimera with MR N terminus replacing that of GR; GMM, chimera with GR N terminus replacing that of MR.
GR Mediates Suppression of ARN127(65) Aggregation.
Remarkably, in the course of experiments testing the recruitment of steroid receptors into polyglutamine protein aggregates, we discovered that cotransfected GR reduced aggregate formation in ARN127(65)-transfected HEK293 cells upon addition of dex, a synthetic glucocorticoid. In contrast, GR had no effect on ARN127(25) distribution or solubility (Fig. 2A). How might GR produce such effects? GR is a hormone-dependent transcriptional regulator (24): upon hormone binding, GR dissociates from an hsp90-containing molecular chaperone complex and translocates from the cytoplasm into the nucleus, where it binds to genomic sites termed glucocorticoid response elements and modulates expression of nearby genes. The glucocorticoid effects that we observed depended on exogenously added GR, as transfection of empty expression vector (p6R) produced no hormonal regulation (Fig. 2B).
Upon hormone stimulation, GR decreased aggregate formation by approximately 75%. GR R466K, a point mutant that is defective for DNA binding (25), was ineffective, suggesting that DNA binding is required to reduce aggregation (Fig. 2B; see Fig. 2C for diagram of various GR mutants). Other closely related steroid receptors such as MR and full-length AR, which bind identical DNA sites, failed to prevent ARN127(65) aggregation (see Fig. 2B for MR; AR data not shown). Thus, a glucocorticoid-specific pathway appears to modulate aggregation of the mutant ARN127(65) protein.
GR N Terminus Is Required For Suppression of Aggregation.
To identify functional domains of GR responsible for this activity, we assayed several receptor derivatives. Deletion of the N terminus of GR, which harbors a transcriptional regulatory activity (GR407C) (26), abolished GR suppression of ARN127(65) aggregation (Fig. 2B), whereas deletion of the C-terminal ligand binding domain of GR (GRN525), produced constitutive, hormone-independent suppression of ARN127(65) aggregation (Fig. 2B). Thus, the GR N terminus appears to be necessary for this activity, whereas the C-terminal ligand binding domain (which mediates association with a chaperone complex) is not.
In an alternate approach, we studied a GR-MR fusion in which the MR N terminus is replaced by that of GR (GMM) (20). This chimeric receptor (GMM) produced mineralocorticoid regulation of ARN127(65) aggregation; in contrast, the reciprocal construct with the GR N terminus replaced by that of MR (MGG) had no effect after dex stimulation (Fig. 2B). Together, these results are consistent with the view that specific regulation of gene expression mediated by the N terminus of GR underlies its activity.
Deletion Within the GR N Terminus Increases Aggregation.
We analyzed further the role of the GR N terminus by using GRΔ108–317 (Fig. 2C), a deletion of amino acids 108–317, which encompasses an N-terminal transcriptional regulatory domain (AF1). Remarkably, cotransfection with GRΔ108–317 produced a 2-fold increase in ARN127(65) aggregation upon hormone stimulation, and virtually all of these cells had exclusively nuclear distribution of ARN127(65) (Figs. 2Ag and 3A). By contrast, there was no change in the aggregation or subcellular localization of ARN127(25) (Fig. 2Ac), demonstrating that these effects are specific to the expanded polyglutamine protein. Cells with predominantly nuclear localization of ARN127(65) occasionally showed condensed or fragmented DNA and rounded cell bodies, consistent with cellular toxicity (see Fig. 2A d and h), but we noted no overt effects on cell growth and survival. Western blots confirmed that neither wtGR nor GRΔ108–317 significantly changed ARN127(65) protein levels (data not shown). Lastly, to test the possibility that GRΔ108–317 was exerting its effects by a mechanism unrelated to wtGR function, we performed a competition using increasing amounts of wtGR. A 5-fold excess of transfected wtGR (250 ng) counteracted the effects of GRΔ108–317 in HEK293 cells (Fig. 3A), suggesting that wtGR and GRΔ108–317 inversely modulate expression of a gene that regulates aggregation and nuclear localization of ARN127(65).
Figure 3.

GRΔ108–317 increases ARN127(65) aggregation in HEK293 and N2a cells; GRN525(AF1–14) fails to suppress aggregation. (A) In HEK293 cells, GRΔ108–317 increases ARN127(65) aggregation by 2-fold upon hormone stimulation (first bar). Increasing amounts of cotransfected wtGR plasmid block this activity (last three bars). (B) In HEK293 cells, constitutively active GRN525(AF1–14) (50 ng) fails to suppress ARN127(65) aggregation (first bar), yet it blocks GRN525 suppression of ARN127(65) aggregation at 50 ng and 250 ng (last three bars). (C) In N2a cells, cotransfected GRΔ108–317 (50 ng) increases ARN127(65) (200 ng) aggregation after hormone addition (first bar) and 4-fold excess of wtGR (200 ng) blocks this effect (second bar). (D) In N2a cells, immunofluorescence shows diffuse cytoplasmic distribution and nuclear aggregates of ARN127(65) at 48 h. Cells were cotransfected with GRΔ108–317 (50 ng) and ARN127(65) (200 ng) plasmid and grown in the absence (a) or presence (b) of dex. HA-tagged ARN127(65) is displayed in red and DAPI nuclear stain in green. (Magnification: ×100.)
GR N-Terminal Transcriptional Activity Parallels its Suppression of ARN127(65) Aggregation.
If the N terminus of GR mediates its effects on polyglutamine protein aggregation through its transcriptional regulatory activity, mutations within this domain should yield parallel effects on transcription and suppression of aggregation. Thus, we examined eight AF1 mutants that reduce GRN525 transcription activation function (26). The predicted correlation was observed, with the four most severe mutants completely unable to suppress ARN127(65) aggregation. One such mutant, GRN525(AF1–14), contains 16 amino acid substitutions scattered through the N terminus between amino acids 108 and 317. This mutant actually increased aggregation approximately 40%, yielding patterns of nuclear aggregates similar to those conferred by GRΔ108–317, and a 5-fold excess of GRN525(AF1–14) blocked wtGRN525 activity (Fig. 3B). GR AF1 transcriptional activation function thus correlates with GR suppression of ARN127(65) aggregation.
Glucocorticoid Effects Manifest in Neural Cells.
To test whether our findings could be extended to cells other than HEK293, we carried out parallel experiments in N2a cells, a mouse neuroblastoma line. In the absence of cotransfected GR, ARN127(65) was diffusely distributed, with only a small percentage (1–2%) of transfected cells showing nuclear or cytoplasmic aggregates at 48 h (Fig. 3Da). As with HEK293 cells, cotransfection of 200 ng ARN127(65) with 50 ng GRΔ108–317 dramatically increased nuclear and cytoplasmic aggregation of ARN127(65) by about 6-fold after hormone stimulation (Fig. 3 C and Db), whereas a 4-fold excess of wtGR plasmid blocked aggregate formation induced by GRΔ108–317 (Fig. 3C). These values differed quantitatively, but not qualitatively, from those observed in HEK293 cells. Thus GR's effects on polyglutamine protein aggregation and nuclear localization are consistent in cells derived from different species and tissue types.
Glucocorticoid Effects Apply to Expanded htt.
To test whether the observed effects could be extended to other polyglutamine proteins, we studied htt, which has been well characterized in both transgenic animal and in vitro systems (8, 27). GR dramatically reduced aggregation of a htt-green fluorescent protein fusion by 75% in transfected HEK293 cells (Fig. 4A) and GRΔ108–317 increased aggregation by 2-fold, again with a dramatic appearance of nuclear aggregates (shown with HA-tagged htt in Fig. 4B). We conclude that GR may regulate gene expression important to the pathogenesis of spinobulbar muscular atrophy, HD, and possibly other polyglutamine expansion diseases.
Figure 4.
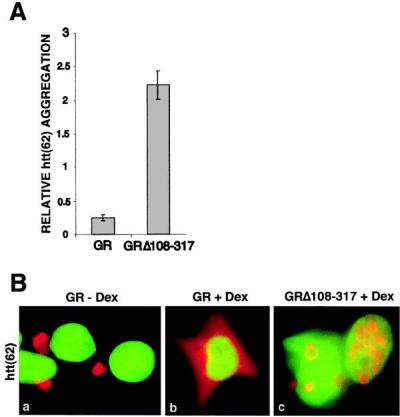
(A) GR modulates htt aggregation in HEK293 cells. wtGR (50 ng) suppresses aggregation of htt(62)-GFP (200 ng) (first bar), whereas GRΔ108–317 promotes its aggregation (second bar) after dex stimulation. (B) Immunofluorescence of HEK293 cells cotransfected with HA-tagged htt(62) (200 ng) and wtGR (50 ng) shows cytoplasmic aggregates minus dex (a), whereas dex stimulation causes diffuse distribution of htt (b). Cotransfection with GRΔ108–317 increases the formation of nuclear aggregates after dex stimulation (c). HA-tagged htt(62) is displayed in red and DAPI nuclear stain in green. (Magnification: ×100.)
Discussion
This study demonstrates that the aggregation and nuclear localization of expanded polyglutamine proteins derived from AR and htt are regulated within the cell and suggests that GR-controlled gene expression can modulate this process. GR activity in this regard correlates entirely with the transcriptional activation function of its N terminus, because deletion of or mutation within this region abolishes this function. Moreover, GRΔ108–317 appears to modulate inversely the same cellular pathway, because it promotes aggregation and nuclear localization and is antagonized by wtGR. Thus, GRΔ108–317 likely functions as a dominant negative, blocking the activity of endogenous GR, or of other transcription factors that may bind nearby promoter sites. Indeed, we and others (28) have found that GRΔ108–317 inhibited wtGR-mediated activation of a glucocorticoid-responsive reporter gene (data not shown).
It seems unlikely that GR's activity results from direct interaction with ARN127(65). First, although rat GR (the subject of this study) contains a tract of 17 glutamines in its N terminus, human GR, with only three glutamines in the same location, affects polyglutamine proteins similarly. Second, upon hormone stimulation virtually all of the GR appears to move to the cell nucleus, whereas most ARN127(65) remains in the cytoplasm, and we have not observed association of GR with aggregated ARN127(65) in colocalization studies. Additionally, GR has identical effects on htt, despite lack of homology between htt and AR outside of the polyglutamine region. Last, if GRN525 directly bound ARN127(65) to reduce its aggregation, it should not be blocked by GRN525(AF1–14), which by this model should have a reduced affinity for ARN127(65). We tried to test whether cycloheximide, a protein synthesis inhibitor, blocked the GR effects, but the drug was toxic over the 48-h time course of the experiment and actually increased polyglutamine protein aggregation. Thus, although our evidence remains somewhat circumstantial and correlative, we conclude that the simplest interpretation is that the GR effects are transcriptionally mediated.
We have demonstrated dramatic effects on polyglutamine protein behavior in two distinct cell types (human kidney and mouse neuronal). However, although many aspects of the cell biology of expanded polyglutamine proteins have shown remarkable similarity among cultured cells, transgenic animals, and patients (5–11, 18, 29), cell culture systems clearly do not reflect accurately all aspects of polyglutamine expansion diseases. For example, it is particularly difficult to model accurately regional central nervous system neurotoxicity or the extended time course of neuronal dysfunction, which takes place over months to years. Thus, testing glucocorticoid effects in mice transgenic for polyglutamine expansion proteins will be crucial in the future.
In addition to its implications for nuclear trafficking, other disorders of misfolded proteins, and GR regulation of specific gene expression, this study opens an avenue of investigation into the molecular pathogenesis and selective neuronal vulnerability of polyglutamine expansion diseases. First, we have demonstrated that the nuclear localization and aggregation of expanded polyglutamine proteins is a regulated cellular process, and second, that we can manipulate this process by using a very well-characterized regulator of gene expression, the GR. It will be interesting to identify the genes that underlie these cellular pathways, and thus in turn to gain insight into the determinants of specific neuronal dysfunction and degeneration in the polyglutamine expansion diseases.
Acknowledgments
We thank Ivan Diamond, Jorge Iñiguez-Lluhí, Brian Freeman, Jonathan Weissman, Steven Finkbeiner, William Welch, Diane Merry, Kenneth Fischbeck, and Ethan Signer for helpful comments on the manuscript. Support for this work comes from the National Institutes of Health and National Science Foundation (K.R.Y.) and the National Institutes of Health and the American Philosophical Society (M.I.D.).
Abbreviations
- GR
glucocorticoid receptor
- HD
Huntington's disease
- AR
androgen receptor
- htt
huntingtin protein
- HA
hemagglutinin
- MR
mineralocorticoid receptor
- DAPI
4′,6-diamidino-2-phenylindole
- dex
dexamethasone
- wt
wild type
- HEK
human embryonic kidney
References
- 1.Kennedy W R, Alter M, Sung J H. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 2.La Spada A R, Wilson E M, Lubahn D B, Harding A E, Fischbeck K H. Nature (London) 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 3.Strong T V, Tagle D A, Valdes J M, Elmer L W, Boehm K, Swaroop M, Kaatz K W, Collins F S, Albin R L. Nat Genet. 1993;5:259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- 4.The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 5.DiFiglia M, Sapp E, Chase K O, Davies S W, Bates G P, Vonsattel J P, Aronin N. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Miwa S, Kobayashi Y, Merry D E, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck K H, Sobue G. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 7.Warrick J M, Paulson H L, Gray-Board G L, Bui Q T, Fischbeck K H, Pittman R N, Bonini N M. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 8.Davies S W, Turmaine M, Cozens B A, DiFiglia M, Sharp A H, Ross C A, Scherzinger E, Wanker E E, Mangiarini L, Bates G P. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 9.Klement I A, Skinner P J, Kaytor M D, Yi H, Hersch S M, Clark H B, Zoghbi H Y, Orr H T. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 10.Cummings C J, Mancini M A, Antalffy B, DeFranco D B, Orr H T, Zoghbi H Y. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 11.Merry D E, Kobayashi Y, Bailey C K, Taye A A, Fischbeck K H. Hum Mol Genet. 1998;7:693–701. doi: 10.1093/hmg/7.4.693. [DOI] [PubMed] [Google Scholar]
- 12.Saudou F, Finkbeiner S, Devys D, Greenberg M E. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 13.Kim M, Lee H S, LaForet G, McIntyre C, Martin E J, Chang P, Kim T W, Williams M, Reddy P H, Tagle D, et al. J Neurosci. 1999;19:964–973. doi: 10.1523/JNEUROSCI.19-03-00964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodgson J, Agopyan N, Gutekunst C-A, Hayden M. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 15.Paulson H L, Perez M K, Trottier Y, Trojanowski J Q, Subramony S H, Das S S, Vig P, Mandel J L, Fischbeck K H, Pittman R N. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 16.Kahlem P, Green H, Djian P. Mol Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- 17.Sittler A, Walter S, Wedemeyer N, Hasenbank R, Scherzinger E, Eickhoff H, Bates G P, Lehrach H, Wanker E E. Mol Cell. 1998;2:427–436. doi: 10.1016/s1097-2765(00)80142-2. [DOI] [PubMed] [Google Scholar]
- 18.Burright E N, Davidson J D, Duvick L A, Koshy B, Zoghbi H Y, Orr H T. Hum Mol Genet. 1997;6:513–518. doi: 10.1093/hmg/6.4.513. [DOI] [PubMed] [Google Scholar]
- 19.Tait D, Riccio M, Sittler A, Scherzinger E, Santi S, Ognibene A, Maraldi N M, Lehrach H, Wanker E E. Hum Mol Genet. 1998;7:991–997. doi: 10.1093/hmg/7.6.991. [DOI] [PubMed] [Google Scholar]
- 20.Pearce D, Yamamoto K R. Science. 1993;259:1161–1165. doi: 10.1126/science.8382376. [DOI] [PubMed] [Google Scholar]
- 21.Ellerby L M, Hackam A S, Propp S S, Ellerby H M, Rabizadeh S, Cashman N R, Trifiro M A, Pinsky L, Wellington C L, Salvesen G S, et al. J Neurochem. 1999;72:185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- 22.Wellington C L, Ellerby L M, Hackam A S, Margolis R L, Trifiro M A, Singaraja R, McCutcheon K, Salvesen G S, Propp S S, Bromm M, et al. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 23.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim S U, et al. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 24.Beato M, Chavez S, Truss M. Steroids. 1996;61:240–251. doi: 10.1016/0039-128x(96)00030-x. [DOI] [PubMed] [Google Scholar]
- 25.Schena M, Freedman L P, Yamamoto K R. Genes Dev. 1989;3:1590–1601. doi: 10.1101/gad.3.10.1590. [DOI] [PubMed] [Google Scholar]
- 26.Iniguez-Lluhi J A, Lou D Y, Yamamoto K R. J Biol Chem. 1997;272:4149–4156. doi: 10.1074/jbc.272.7.4149. [DOI] [PubMed] [Google Scholar]
- 27.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates G P, Davies S W, Lehrach H, Wanker E E. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 28.Delaunay F, Liden J, Gustafsson J A, Okret S. Eur J Biochem. 1996;242:839–845. doi: 10.1111/j.1432-1033.1996.0839r.x. [DOI] [PubMed] [Google Scholar]
- 29.Chai Y, Koppenhafer S L, Shoesmith S J, Perez M K, Paulson H L. Hum Mol Genet. 1999;8:673–682. doi: 10.1093/hmg/8.4.673. [DOI] [PubMed] [Google Scholar]