

Abstract
Cutaneous sporotrichosis is a chronic granulomatous fungal infection caused by Sporothrix schenckii with worldwide distribution. Its traditional diagnosis is time-consuming and difficult to differentiate from that of a clinical sporotrichoid lesion caused by various pathogens. In this study, a nested PCR assay for the detection of S. schenckii was evaluated by using a sequence of 18S rRNA gene as a target. For the examination of specificity and sensitivity, five clinical isolates with 1 ATCC 10213 strain of S. schenckii, 10 strains of clinical common fungi, 3 strains of Mycobacterium spp., Staphylococcus aureus, and normal human skin tissue were used. The expected fragment was amplified from six S. schenckii isolates in the first round and nested PCR but not from other microorganisms and human DNA. Their sequences were 100% identical to the S. schenckii 18S rRNA gene sequence deposited in GenBank. A detection limit of 40 fg of S. schenckii DNA extract was determined with ethidium bromide staining. Serial dilution studies demonstrated that the nested PCR could detect a DNA amount of 1 CFU of S. schenckii in tissue samples. We further investigated the nested PCR assay for the detection of S. schenckii from the tail tissues of 5 experimentally infected mice and from the clinical biopsy specimens of 12 patients with sporotrichosis confirmed by culture or histochemical staining. The nested PCR assay was positive in all 5 infected mice and in 11 of the 12 clinical specimens. The high sensitivity and specificity of this nested PCR indicate that the assay can provide rapid diagnosis with sufficient accuracy to be clinically useful for patients with sporotrichosis.
Cutaneous sporotrichosis is a subacute or chronic granulomatous fungal infection caused by the dimorphic fungus Sporothrix schenckii (6, 11). The organism occurs worldwide and is most commonly found in soil, sphagnum moss, and decaying vegetation. Although sporotrichosis is most often reported from Mexico and Central and South America, it is also common in Asia and more widespread in temperate and tropical zones (9, 16). Most often, the infection occurs following minor traumatic events leading to the implantation of the fungus onto the skin. Clinical sporotrichosis frequently presents in its cutaneous forms, lymphocutaneous and fixed cutaneous (7). Life-threatening hematogenous dissemination and systemic infection of the disease, however, occasionally occur in immunocompromised persons (1, 6).
Histologically, cutaneous sporotrichosis usually presents a nonspecific granulomatous reaction that tends to form concentric zones. In mammalian tissues, S. schenckii is yeast-like, appearing as spherical or cigar-shaped bodies measuring 4 to 6 μm in length that reproduce by budding (7). However, direct microscopic identification of the organism in biopsy sections is always difficult because of the paucity of the organism (2, 18). The conventional method for definitive diagnosis of sporotrichosis is based on time-consuming tissue cultures, and S. schenckii was grown easily on a Sabouraud medium in previous studies (2, 6). However, cultures of the biopsy specimens frequently yield negative results. Although the fluorescent antibody or immunohistochemical techniques also provide a rapid diagnosis of sporotrichosis, they are not available in most clinical laboratories (2, 6, 7). Therefore, the development of an easy, reliable, and specific assay for the detection of S. schenckii in biopsy specimens would be very useful.
Recently, PCR techniques have been introduced to detect systemic fungal infections (10, 21). A S. schenckii PCR assay may provide a more effective and rapid way to diagnosis sporotrichosis from clinical sporotrichoid infections that can be also caused by various other pathogens, including bacteria, fungi, leishmania, and atypical mycobacteria (12, 13, 14, 22). Hence, this may significantly minimize health risks. This may also minimize the cost of treatment as an alternative to uncertain clinical trials for negative-culture sporotrichoid infections. To our knowledge, this is the first description of the use of nested PCR to detect S. schenckii DNA from tissue samples of experimentally infected mice and from clinical specimens of patients with sporotrichosis.
MATERIALS AND METHODS
Microorganisms.
Five clinical isolates of S. schenckii obtained from the Department of Clinical Microbiology of Chang Gung Memorial Hospital and one ATCC 10213 strain of S. schenckii were used and grown on a Sabouraud medium at 25°C for 1 week. The identification of S. schenckii was confirmed by standard morphological studies (15). Mycelial colonies were scraped off the agar, suspended in sterile water, frozen, and stored at −20°C. The closely related saprophytic fungus, Ophiostoma stenoceras (ATCC 22433), obtained from American Type Culture Collection, was also used and grown on a Sabouraud medium at 25°C for 1 week. Isolates of Candida albicans, Candida glabrata, Candida parapsilosis, Candida tropicalis, Trichosporon asahii, Trichophyton rubrum, Fonsecaea spp., Cladosporum spp., Penicillium spp., and Aspergillus flavus, obtained from the Department of Clinical Microbiology, were grown on Sabouraud agar and identified by standard methods. All fungal suspensions were prepared as described above. The DNA extracts of the bacterial microorganisms, including Mycobacterium tuberculosis, Mycobacterium marinum, Mycobacterium chelonae, and Staphylococcus aureus, were directly provided by the Department of Clinical Pathology for the PCR assay.
Animal model and experimental infection.
ICR mice (female, 5 to 10 weeks old) were obtained from the National Laboratory Animal Breeding and Research Center (Taipei, Taiwan, Republic of China). The animal studies were performed under the guidelines set forth in the 1996 Guide for the Care and Use of Laboratory Animals of the National Research Council (8a). We adapted an animal model of sporotrichosis as previously described by Tachibana et al. (19) with some modifications. Briefly, ATCC 10213 strain of S. schenckii was cultivated in brain heart infusion broth (Difco Laboratories, Detroit, Mich.) with shaking at 37°C for 7 days to obtain yeast forms. Conversion to yeast phase was recorded under a light microscope, and over 90% yeast form was observed. The pellet of cultures was resuspended and 10-fold serially diluted in sterile phosphate-buffered saline. A volume of 100 μl of each dilution containing organisms was cultured on Sabouraud agar at 25°C for 5 days, and the CFU counts of cultures were enumerated. For experimental infection, five mice were injected subcutaneously at four points on their tails with 0.05 ml of the yeast form suspension (106 CFU/0.05 ml). Two mice injected with sterile phosphate-buffered saline served as the negative controls. The inflammatory tail skin tissues of the five experimental mice, 35 days after inoculation, and those of the control group were subjected to histochemical examination and DNA extraction.
DNA extraction from cultured strains.
To 100 μl of each fungal suspension in sterile water, equal volumes of DNA extraction buffer were added, which contained the following: 10 mM Tris-HCl (pH 8.0), 10 mM EDTA, 0.15 M NaCl, 2% sodium dodecyl sulfate, and proteinase K (Sigma Chemical Co., St. Louis, Mo.) to a final concentration of 0.5 mg/ml. The mixture was incubated at 55°C overnight, and then proteinase K was inactivated by heating the mixture to 95°C for 10 min. In the next step, the samples were exposed to three cycles of freezing in liquid nitrogen for 1 min and boiling for 5 min afterwards to disrupt the fungal cells. After cooling to room temperature, an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1 ratio) was added and the mixture was centrifuged at 12,000 × g for 5 min. The supernatant was transferred to a fresh tube, and the same procedure was repeated with chloroform-isoamyl alcohol (24:1). The DNA was precipitated with 2.5 volumes of ethanol at −20°C and centrifuged at 12,000 × g for 20 min at 4°C. The pellet was then allowed to dry. After rinsing with 70% ethanol at 4°C, the extracted DNA was dissolved in 50 μl of distilled water and 5 μl of suspension was used for PCR.
DNA extraction from clinical samples and infected mice.
From January 2000 to September 2001, 12 cutaneous biopsy specimens from patients with cutaneous sporotrichosis at the Department of Dermatology of Chang Gung Memorial Hospital were selected. In all of the cases, the diagnoses were confirmed by a positive histochemical stain (periodic acid-Schiff [PAS]) or by culturing for S. schenckii. All of the selected fresh biopsy specimens were stored frozen (at −70°C) for up to 1 year before DNA extraction was done. A biopsy specimen from normal human skin tissue was used as a control. All clinical samples and infected mouse tissues (about 0.1 g) were cut into pieces and suspended in 400 μl of DNA extraction buffer containing 0.5 mg of proteinase K (Sigma Chemical Co.)/ml. DNA extraction of the clinical samples and the infected mouse tissues was performed as described above.
Primer design.
The design of oligonucleotides used in this study was based on comparison of the sequence of 18S rRNA gene of S. schenckii (accession no. M85053) and those of other fungi in the GenBank database (National Center for Biotechnology Information, National Library of Medicine, Bethesda, Md.). In order to ensure the specificity of the PCR assay, a BLAST search was performed on the designed primers. After alignment and visual assessment, two sets of primers binding to the sequence of 18S rRNA gene of S. schenckii, but not to the corresponding human DNA and other common human pathogens, were selected for the nested PCR assay. The outer primer set was SS1, 5′-CTC GTT CGG CAC CTT ACA CG-3′, and SS2, 5′-CGC TGC CAA AGC AAC GCG GG-3′, which were complementary to positions 1007 to 1026 and 1311 to 1292 of the GenBank sequence, respectively, defining a 305-nucleotide amplicon. The inner primer set consisted of SS3, 5′-ACT CAC CAG GTC CAG ACA CGA TG-3′, and SS4, 5′-CGC GGG CTA TTT AGC AGG TTA AG-3′, which were complementary to the nucleotide positions 1146 to 1168 and 1297 to 1275 of the deposited sequence, respectively. They delimit a 152-nucleotide sequence. The primer pairs were synthesized on an oligonucleotide synthesizer (Perkin-Elmer Cetus, Norwalk, Conn.).
PCR assay.
The reaction mixture of the first-round PCR consisted of 5 μl of DNA extract in a total volume of 50 μl, with final concentrations of 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 0.1% Triton X-100, 1.5 mM MgCl2, 0.4 μM concentrations of primers SS1 and SS2, 1.5 U of Taq polymerase (Promega, Madison, Wis.), and a 200 μM concentration of each deoxynucleotide triphosphate (Promega). The reaction mixture of the nested PCR was identical, except that 3 μl of the first reaction product and the inner primer pair SS3 and SS4 were used. The PCR was performed in 0.5-ml thin-wall polypropylene tubes in a Gene Amp PCR System 9600 (Perkin-Elmer Cetus) programmed as follows: 95°C for 5 min; 40 cycles of 95°C for 1 min, 68°C for 1 min, 72°C for 1 min; and final extension at 72°C for 10 min. PCR products (5 μl) were analyzed by electrophoresis on a 2% (wt/vol) agarose gel, stained with ethidium bromide, and visualized on a UV transilluminator.
To determine the lower limit of detection by the nested PCR, assays were performed by using the serially diluted genomic DNA of S. schenckii ATCC 10213 in a given concentration ranging from 0.4 fg to 4 ng. In addition, the nested PCR assay also tested the fungal DNA solutions extracted from 0.1 g of normal human skin premixed with serial dilutions of 1, 101, 102, 103, or 104 cells of S. schenckii.
Controls.
To ensure overall quality and reproducibility of data, certain quality control procedures were applied. A volume of 5 μl containing 100 fg of S. schenckii DNA extracted from cultural ATCC 10213 strain was used in every PCR assay as a positive control. All steps of preparation of PCR mixes were carried out in a laminar flow hood with aseptic techniques. In order to monitor crossover contaminations, 450 μl of sterile water was included in the DNA extraction procedure and was used as a negative control after every fifth sample in the nested PCR assay. Reaction mixtures without DNA were run in the first and nested PCRs to detect contamination. To screen for PCR inhibitors, 100 fg of S. schenckii DNA was added to 5 μl of DNA extract from a PCR-negative sample, and the corresponding PCR assay was repeated.
Automated sequencing and analysis of amplified DNA.
The amplified DNA was purified to remove excess primer using microconcentrators (Amicon Inc., Beverly, Mass.). The DNA fragments were directly sequenced by using the PCR primers with the Dye-Deoxy Terminator Cycle Sequencing kit in an automated DNA Sequencer (model 373A; Applied Biosystems, Foster City, Calif.). Data analysis was carried out with the ABI Seqed and/or Sequence Navigator programs. Sequences generated from both strands were edited, aligned, and used for a BLAST search in GenBank (National Center for Biotechnology Information).
RESULTS
Specificity of nested PCR for S. schenckii.
All six S. schenckii isolates, including the ATCC strain, resulted in positive results in the first-round and nested PCR. No amplification was observed by the PCR assays with DNA extracts of the other 10 strains of common fungi, 3 Mycobacterium spp., one strain of S. aureus, and normal human skin tissue. As shown in Fig. 1, a specific approximately 150-bp fragment was amplified from S. schenckii but not from other microorganisms and human DNA in the nested PCR step with the inner primer pair SS3 and SS4. The closely related fungus, O. stenoceras, which shared high homology of 18S rRNA gene sequences with S. schenckii, was also tested in the specificity studies. Amplicons with similar sizes were found in the PCR assay (data not shown).
FIG. 1.

Specificity of the nested PCR assay. (A) DNA extracted from common fungi. (B) DNA extracted from bacteria, mycobacteria, and normal human tissue. Lanes: M, molecular size marker (100-bp ladder [Promega]); 1, distilled water; 2, S. schenckii; 3, C. albicans; 4, C. glabrata; 5, C. parapsilosis; 6, C. tropicalis; 7, T. asahii; 8, T. rubrum; 9, Fonsecaea spp.; 10, Cladosporum spp.; 11, Penicillium spp.; 12, A. flavus; 13, S. schenckii; 14, M. tuberculosis; 15, M. marinum; 16, M. chelonae; 17, S. aureus; 18, normal human skin.
Sensitivity of nested PCR for S. schenckii.
The sensitivity of the PCR test was evaluated through the following means. First, by using 10-fold serial dilutions of DNA extracted from S. schenckii, first-round PCR with the specific primers SS1 and SS2 gave amplified products with about 300 bp in length, as expected. The detection limit of S. schenckii target DNA with ethidium bromide staining by the first-round PCR was 4 pg, whereas as little as 40 fg could be detected by the nested PCR (Fig. 2). The sensitivity of the nested PCR was 100 times higher than that of the first-round PCR. Furthermore, according to the serial dilution studies, the DNA amount of 1 CFU of S. schenckii per sample gave a positive result by the nested PCR assay.
FIG. 2.
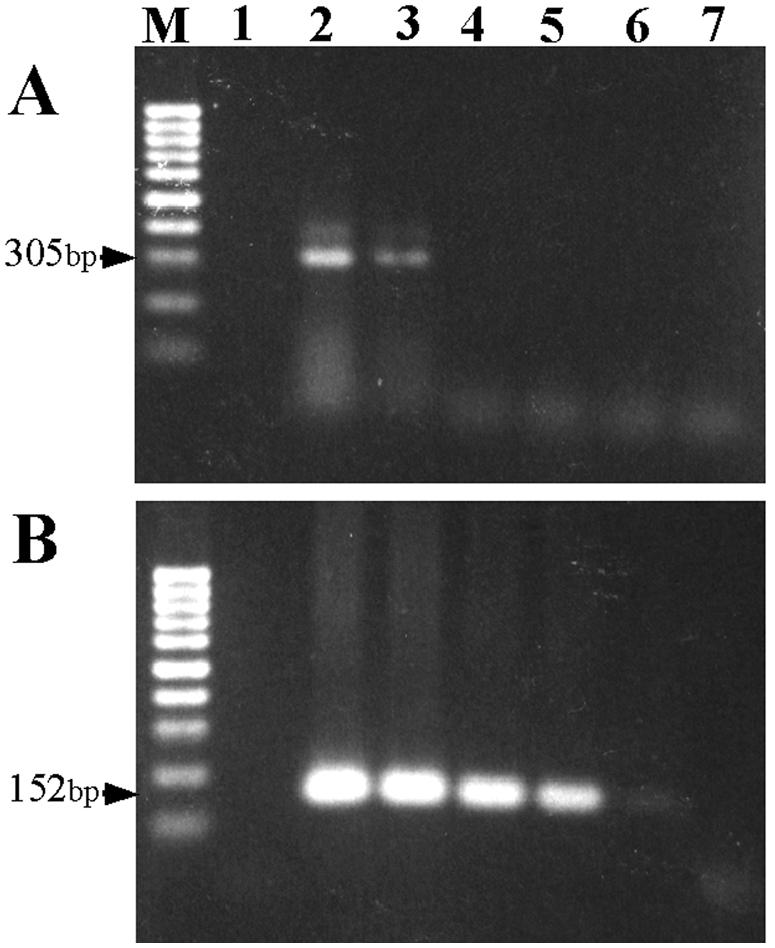
Sensitivity of the PCR assay with DNA from S. schenckii. (A) First-round PCR assay. (B) Nested PCR assay. Lanes: M, molecular size marker (100-bp ladder [Promega]); 1, distilled water; 2, 4 × 10−11 g of S. schenckii DNA; 3, 4 × 10−12 g of S. schenckii DNA; 4, 4 × 10−13 g of S. schenckii DNA; 5, 4 × 10−14 g of S. schenckii DNA; 6, 4 × 10−15 g of S. schenckii DNA; 7, 4 × 10−16 g of S. schenckii DNA.
Detection of DNA specific for S. schenckii in infected mouse tissues and in clinical samples.
A summary of the results for detection of S. schenckii from tail tissues of mice is shown in Table 1. Both the histological stain and first-round PCR showed evidence of sporotrichosis in 2 of the 5 experimentally infected mice. By comparison, the nested PCR revealed positive results for all 5 experimental mice. The first-round and nested PCRs of the two control mice were negative. Although 12 clinical specimens were negative by the first-round PCR (data not shown), all clinical samples, except one, were positive by the nested PCR assay (Fig. 3). Cultures of clinical specimens were positive for 8 of 12 patients (66.6%), and histochemical PAS staining was positive for 7 of 12 patients (58.3%) (Table 2). The clinical sample with negative nested PCR results remained negative after 100 fg of the positive control DNA was added, demonstrating the presence of specific inhibitors in the sample. A second DNA extraction could not be done due to a limited sample source.
TABLE 1.
Detection of S. schenckii in tail tissues of experimentally infected micea
| Mouse no. | Result by:
|
||
|---|---|---|---|
| Histology (PAS stain) | First-round PCR | Nested PCR | |
| 1 | + | + | + |
| 2 | − | − | + |
| 3 | − | − | + |
| 4 | − | − | + |
| 5 | + | + | + |
| C1 | ND | − | − |
| C2 | ND | − | − |
C, control; +, positive; −, negative; ND, not done.
FIG. 3.

Detection of S. schenckii in DNA extracts of 12 clinical samples by nested PCR assay. Lanes: M, molecular size marker (100-bp ladder [Promega]); 1, positive control of S. schenckii; 2, distilled water; 3 to 14, patient no. 1 to 12, respectively. Lane 8 (patient no. 6) showed a negative result.
TABLE 2.
Detection of S. schenckii in clinical samples by nested PCR assay
| Patient no. | Resulta by:
|
Clinical presentation (location) | ||
|---|---|---|---|---|
| Culture | PAS stain | Nested PCR | ||
| 1 | + | + | + | Sporotrichoid nodules (right arm) |
| 2 | + | + | + | Ulcerated nodules (neck) |
| 3 | + | − | + | Ulcerated nodules (right hand) |
| 4 | + | − | + | Sporotrichoid nodules (left arm) |
| 5 | − | + | + | Verrucous plaques (nose) |
| 6 | + | − | − | Ulcerated nodules (left hand) |
| 7 | + | − | + | Indurated papules (right hand) |
| 8 | + | − | + | Sporotrichoid nodules (right arm) |
| 9 | − | + | + | Verrucous plaques (face) |
| 10 | − | + | + | Verrucous plaques (left foot) |
| 11 | + | + | + | Indurated papulo plaques (right hand) |
| 12 | − | + | + | Sporotrichoid nodules (left arm) |
+, positive; −, negative.
Sequence identification of PCR products.
All of the nested PCR products, including amplicons of six S. schenckii isolates, five experimentally infected mouse tails, and 11 clinical samples, were double-strand sequenced by using SS3 and SS4 primers with the Dye-Deoxy Terminator Cycle Sequencing kit. The obtained sequences were subjected to BLAST alignment. All of the sequences of amplicons showed 100% identity to the gene fragment (i.e., nucleotide position 1146 to 1297) of S. schenckii 18S rRNA gene (GenBank accession no. M85053).
DISCUSSION
This is the first report demonstrating the use of a nested PCR assay to detect S. schenckii DNA in tissue samples of mice serving as a model of sporotrichosis and in clinical samples from patients with the same mycotic disease. The capability of this technique to identify S. schenckii from clinical specimens makes it useful for rapid diagnosis of sporotrichosis with high sensitivity and specificity, instead of using conventional methods that often yield negative results (2, 18). In our experiences, the entire experimental procedures, excluding the overnight proteinase K incubation, could be done within 10 h. Using a method of physical destruction of fungal cells (4), the released DNA is sufficient for detection by the nested PCR assay with high sensitivity. The phenol-chloroform-isoamyl alcohol extraction method used in this study is technically simple, inexpensive, and broadly applicable in clinical microbiology laboratories.
A common target of diagnostic fungal PCR assay is the 18S rRNA gene because its frequency in the genome yields a high sensitivity of the PCR (17). Although the 18S rRNA gene of S. schenckii is composed of many conserved regions, screening of DNA sequences deposited in GenBank for other clinically relevant fungi and human genomic DNA reveals low homology with the designed primer pairs. The specificity of this highly sensitive nested PCR assay was demonstrated by the negative results for DNA extracts of 10 common fungi, 3 Mycobacterium spp., and S. aureus as well as normal human skin tissue (Fig. 1). Furthermore, we have also tested the assay with many S. schenckii-negative skin specimens, including those which are culture positive for mycobacteria, with all of them yielding negative results (data not shown), indicating a real clinical specificity. The high annealing temperature of the nested PCR further increased the stringency, reducing the possibility of cross-amplification. One out of 12 PAS stains or culture-positive clinical samples was negative according to the nested PCR assay (Fig. 3). In this negative sample, specific inhibitors were demonstrated.
S. schenckii has been previously suggested to be the anamorphic species for O. stenoceras. O. stenoceras is a cause of sapstain on softwood and did not appear to be a human pathogen (8). O. stenoceras and S. schenckii have been reported to be very closely related, and their 18S rRNA genes are differ only at 3 sites among the more than 1,700 nucleotides (3). By employing the nested PCR assay, genomic DNA from ATCC 22433 O. stenoceras gave a positive amplicon sharing 100% sequence identity with 18S rRNA gene of S. schenckii at position 1007 to 1311. Since O. stenoceras is usually not found to be present in clinical samples, the specificity of this assay for the diagnosis of sporotrichosis is considered to be effective, especially when appropriate environmental controls are employed.
Nested PCRs are notorious for both DNA and environmental contaminations. However, there was no detectable contamination in this study since the control samples employed in every assay, comprising 5 samples, were always negative. Environmental contamination is another problem to be concerned about, although S. schenckii or O. stenoceras is not usually present in the environment as are Aspergillus and Penicillium species. Nevertheless, none of the control reagents and normal human skin tested in this study were positive by the PCR assay, indicating that if environmental contamination does occur, it is less than the threshold of this assay.
A sporotrichoid infection is a common cutaneous infectious disease that can be caused by various pathogens, including bacteria, fungi, leishmania, and atypical mycobacteria (12, 13, 14, 22). Among the causative pathogens, atypical mycobacteria, especially M. marinum, and S. schenckii are the leading causes of a sporotrichoid lesion. The paucity of those pathogens in clinical sporotrichoid lesions frequently results in a negative tissue culture and histochemical staining that is often a diagnostic challenge (5). The highly sensitive nested PCR assay can also detect S. schenckii DNA from specimens of patients with clinical sporotrichosis in the absence of histochemical and culture-positive evidence of S. schenckii (data not shown). Combining this nested PCR for detection of S. schenckii with a well-established mycobacterium PCR assay (20), a rapid and effective differential diagnosis with an accurate decision for therapy, instead of empirical therapy, can be made when physicians face sporotrichoid infections without positive histochemical or culture results.
In conclusion, using the nested PCR assay, we were successful in detecting DNA specific for S. schenckii in clinical samples from patients with sporotrichosis. This assay will not only allow a rapid diagnosis of sporotrichosis but will also provide a practical solution to the difficulties encountered in the identification of pathogens from histochemical and culture-negative sporotrichoid infection.
Acknowledgments
We thank Kuei-Lan Liu and Ching-Mei Hsu for technical assistance and expertise.
This study was supported by grants CMRP1241 and CMRPG2004 from the Chang Gung Memorial Hospital and NSC-88-2314-B-182-078 from the National Science Council, Taipei, Taiwan, Republic of China.
REFERENCES
- 1.Aronson, N. E. 1992. Disseminated sporotrichosis. JAMA 268:2021. [PubMed] [Google Scholar]
- 2.Belknap, B. S. 1989. Sporotrichosis. Dermatol. Clin. 7:193-202. [PubMed] [Google Scholar]
- 3.Berbee, M. L., and J. W. Taylor. 1992. 18S ribosomal RNA gene sequence characters place the human pathogen Sporothrix schenckii in the genus Ophiostoma. Exp. Mycol. 16:87-91. [Google Scholar]
- 4.Bialek, R., A. Ibricevic, C. Aepinus, L. K. Najvar, A. W. Fothergill, J. Knobloch, and J. R. Graybill. 2000. Detection of Paracoccidioides brasiliensis in tissue samples by a nested PCR assay. J. Clin. Microbiol. 38:2940-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd, J., D. R. Mehregan, and D. A. Mehregan. 2001. Utility of anti-bacillus Calmette-Guerin antibodies as a screen for organisms in sporotrichoid infections. J. Am. Acad. Dermatol. 44:261-264. [DOI] [PubMed] [Google Scholar]
- 6.Davis, B. A. 1996. Sporotrichosis. Dermatol. Clin. 14:69-76. [DOI] [PubMed] [Google Scholar]
- 7.de Araujo, T., A. C. Marques, and F. Kerdel. 2001. Sporotrichosis. Int. J. Dermatol. 40:737-742. [DOI] [PubMed] [Google Scholar]
- 8.Dixon, D. M., R. A. Duncan, and N. J. Hurd. 1992. Use of a mouse model to evaluate clinical and environmental isolates of Sporothrix spp. from the largest U.S. epidemic of sporotrichosis. J. Clin. Microbiol. 30:951-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Institute of Laboratory Animal Resources. 1996. Guide for the care and use of laboratory animals, 7th ed. National Research Council, National Academy Press, Washington, D.C.
- 9.Itoh, M., S. Okamoto, and H. Kariya. 1986. Survey of 200 cases of sporotrichosis. Dermatologica 172:209-213. [DOI] [PubMed] [Google Scholar]
- 10.Kan, V. L. 1993. Polymerase chain reaction for the diagnosis of candidemia. J. Infect. Dis. 168:779-783. [DOI] [PubMed] [Google Scholar]
- 11.Kauffman, C. A. 1999. Sporotrichosis. Clin. Infect. Dis. 29:231-237. [DOI] [PubMed] [Google Scholar]
- 12.Knox, J. M., S. G. Gever, R. G. Freeman, and F. Whitcomb. 1961. Atypical acid-fast organism infection of the skin. Arch. Dermatol. 84:386-391. [DOI] [PubMed] [Google Scholar]
- 13.Owens, D. W., and M. E. McBride. 1969. Sporotrichoid cutaneous infection with Mycobacterium kansasii. Arch. Dermatol. 100:54-58. [PubMed] [Google Scholar]
- 14.Philpott, J. A., Jr., A. R. Woodburne, W. B. Schaefer, and C. S. Mollohan. 1963. Swimming pool granuloma: a study of 290 cases. Arch. Dermatol. 88:158-162. [DOI] [PubMed] [Google Scholar]
- 15.Rippon, J. W. 1988. Sporotrichosis, p. 325-352. In J. W. Rippon (ed.), Medical mycology: the pathogenic fungi and the pathogenic actinomycetes, 3rd ed. W. B. Saunders, Co. Philadelphia, Pa.
- 16.Rivitti, E. A., and V. Aoki. 1999. Deep fungal infections in tropical countries. Clin. Dermatol. 17:171-190. [DOI] [PubMed] [Google Scholar]
- 17.Sandhu, G. S., B. C. Kline, L. Stockman, and G. D. Roberts. 1995. Molecular probes for diagnosis of fungal infections. J. Clin. Microbiol. 33:2913-2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segal, R. J., and P. H. Jacobs. 1979. Sporotrichosis. Int. J. Dermatol. 18:639-644. [DOI] [PubMed] [Google Scholar]
- 19.Tachibana, T., T. Matsuyama, and M. Mitsuyama. 1998. Characteristic infectivity of Sporothrix schenckii to mice depending on routes of infection and inherent fungal pathogenicity. Med. Mycol. 36:21-27. [PubMed] [Google Scholar]
- 20.Telenti, A., F. Marchesi, M. Balz, F. Bally, E. C. Bottger, and T. Bodmer. 1993. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 31:175-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Burik, J. A., D. Myerson, R. W. Schreckhise, and R. A. Bowden. 1998. Panfungal PCR assay for detection of fungal infection in human blood specimens. J. Clin. Microbiol. 36:1169-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh, D. S., M. V. Balagon, R. M. Abalos, E. S. Tiongco, R. V. Cellona, T. T. Fajardo, and G. P. Walsh. 1997. Multiple lesions of sporotrichoid leishmaniasis in a Filipino expatriate. J. Am. Acad. Dermatol. 36:847-849. [DOI] [PubMed] [Google Scholar]