

Abstract
A multiple microfermentor battery was designed for high-throughput recombinant protein production in Escherichia coli. This novel system comprises eight aerated glass reactors with a working volume of 80 ml and a moving external optical sensor for measuring optical densities at 600 nm (OD600) ranging from 0.05 to 100 online. Each reactor can be fitted with miniature probes to monitor temperature, dissolved oxygen (DO), and pH. Independent temperature regulation for each vessel is obtained with heating/cooling Peltier devices. Data from pH, DO, and turbidity sensors are collected on a FieldPoint (National Instruments) I/O interface and are processed and recorded by a LabVIEW program on a personal computer, which enables feedback control of the culture parameters. A high-density medium formulation was designed, which enabled us to grow E. coli to OD600 up to 100 in batch cultures with oxygen-enriched aeration. Accordingly, the biomass and the amount of recombinant protein produced in a 70-ml culture were at least equivalent to the biomass and the amount of recombinant protein obtained in a Fernbach flask with 1 liter of conventional medium. Thus, the microfermentor battery appears to be well suited for automated parallel cultures and process optimization, such as that needed for structural genomics projects.
Recent developments in structural biology, particularly the advent of structural genomics projects, have emphasized the need for high-throughput cultivation of clones producing recombinant proteins. The obvious way to achieve this goal is to reduce culture volumes and to combine reactors in a battery for multiple parallel cultures. In addition to a high throughput, such a battery should yield sufficient biomass for preparative purposes. Furthermore, the device should offer the opportunity to grow cultures in an automated manner while well-defined culture parameters are implemented. Thus, temperature, pH, dissolved oxygen (DO), and biomass should be monitored online, enabling feedback control and programming of automated sequences of events, such as temperature shifts and addition of an inducer, as a function of cell density.
Several designs for miniature, parallel cultures have been described previously (4, 8, 10, 11, 15-17, 21) and have even been commercialized (e.g., Sixfors [Infors, Switzerland] and fedbatch-pro [DASGIP AG, Germany]). However, none of these designs fully meets the requirements described above. Discounting systems whose volume is too small for preparative purposes (4, 8, 11, 15, 21) leaves the parallelized miniature bioreactors of Puskeiler et al. (17) and the commercial Sixfors and fedbatch-pro fermentor batteries. The volumes of the reactors of the commercial batteries (300 to 500 ml) are large enough for preparative production of recombinant proteins, but they are a bit cumbersome to handle. More importantly, although the fermentors can be equipped with devices for measuring cell density, the systems are not designed to integrate this parameter for closed-loop feedback control of the fermentation. In addition, commercially available systems for measuring optical density are designed to monitor cell density in single reactors, and fitting all reactors with independent devices would be impractical due to the high cost and space requirement. Thus, it is not possible to program automated sequences of events (temperature shifts, addition of inducer) that would be triggered when the cultures reach preset optical density values. The miniature bioreactors of Puskeiler et al. are an interesting combination of parallelized reactors with a liquid-handling robot, which collects samples from the bioreactors and monitors the pH and optical density using microtiter plate technology. The bioreactors are equipped with high-speed impellers that provide excellent oxygen transfer properties, and the robot also manages fed-batch addition of substrate, so that biomass densities of 39 g (dry weight)/liter can be obtained. With a working volume of 8 to 15 ml, this would be sufficient for preparative purification of well-produced recombinant proteins. However, the setup does not provide the opportunity to program independent temperature schedules for each reactor, which is important for optimizing culture parameters.
In this paper, we describe the design and basic operation of a multiple microfermentor (MMF) battery comprising eight miniature bioreactors whose culture parameters can be controlled independently. Each bioreactor consists of an 80-ml glass vessel equipped with pH, DO, and temperature probes to allow continuous measurement and control of these parameters. An external movable sensor provides online monitoring of the turbidity in each reactor without disturbing the process and eliminates the need to collect samples for optical density determinations. In order to obtain sufficient biomass for preparative purposes and to optimize the production of recombinant proteins in Escherichia coli, we developed an enriched high-density culture medium. The performance of the system was validated with a set of clones expressing genes from various organisms, including Mycobacterium tuberculosis, Mycobacterium leprae, Trypanosoma cruzi, and Staphylococcus aureus. For each clone, the yield of target recombinant protein obtained in microfermentors with high-density medium was compared with the yield obtained in Fernbach flasks containing 1 liter of modified Lennox medium. The results show that parallel cultures grown in microfermentors meet the needs for preparative, high-throughput production of recombinant proteins, such as the proteins required for structural genomics studies, and that process optimization can be automated with minimal intervention of the operator. Furthermore, parameters defined for 70-ml microfermentors could be successfully extrapolated to a 1.6-liter classical fermentor.
MATERIALS AND METHODS
Main components of the microfermentor battery.
The equipment, which fits on a lab bench, comprises the eight-bioreactor battery proper (Fig. 1), a control cabinet containing equipment-specific control devices, and a personal computer unit for programming and monitoring the course of culture processes (2). The battery prototype was manufactured and assembled by Genomic SA, Collonges-sous-Salève, France.
FIG. 1.

General view of the microfermentor battery. The heat exchangers, infrared sensor, stepper motor, and positioning system are shown with the metal casing removed for clarity.
Bioreactor design.
Bioreactors were designed to scale down the working volume to 80 ml, to ensure nonlimiting oxygen transfer for bacterial growth, and to enable external online measurement of turbidity. As shown in Fig. 2, each microreactor consists of a 19-cm glass tube with a 2-cm square section for the external turbidity sensor, which requires plane surfaces for measurement. A Teflon headplate kept in place by a pierced bakelite screw cap has three screw adaptors for inserting the pH, DO, and temperature probes. Each reactor has ports for aeration, exhaust, filling, sampling, and injection of inoculum and additives, such as inducing compounds. The ports are connected to flexible tubing by means of fast-mount, sterilizable connectors. Prior to cultivation, the fully equipped microfermentors were autoclaved and filled with sterile medium.
FIG. 2.

Design of the bioreactor vessels. The gas inlet is fitted with 0.22-μm filters. The injection port is plugged by a septum through which an inducer can be injected by means of a syringe needle.
Each culture vessel is aerated through a glass sparger (Robu Technology; 16 to 40 μm) located at the bottom of the reactor in a corner to allow measurement of turbidity. The sparger ensures that there are high oxygen transfer rates, and microbubbling eliminates the need for mechanical stirring. When necessary, air is enriched with pure oxygen by means of a gas-mixing electrovalve system (Bürkert 078945 microelectrovalves). Automatic injection of an inducer through the septum of the injection port is triggered by software-controlled tubing microelectrovalves connected to a pressurized vessel containing the inducer.
Turbidity monitoring.
Online measurement of the turbidity in an MMF is performed by a noninvasive, external sensor, which comprises an infrared light-emitting diode (Honeywell LED SE5470-003) and a photodetector (Honeywell PhotoDarlington SD5410) facing one another on either side of the microfermentor. Turbidity monitoring is based on modulation of the voltage (Ve) fed into the emitting diode, which is recursively adjusted electronically to obtain a received light intensity corresponding to a predefined output current of the photodetector (5). The voltage is measured with an FP-AI-100 analog input module (National Instruments), and the sensitivity (output current) is fixed by an FP-AO-200 4- to 20-mA current output module. Signals are transmitted to or from the personal computer by an FP-1000 RS232 communication interface and recorded. The input voltage is recorded and serves as the primary signal. The signal can then be converted to the optical density at 600 nm (OD600) after feeding into the system an empirical equation derived from a calibration curve determined with appropriately diluted samples of the cultivated organism. By varying the set output current, it is possible to choose the sensitivity of the device, so that the turbidity can be measured for OD600 values ranging from 0.05 to 100. The sensor is calibrated against noninoculated medium before each culture cycle.
The diode and the photodetector are carried on a fork-like support, which is moved along a rail by a stepper motor (Sanyo Denki type 103H7126-0440 motor and PMM-MD-23221-10 driver) that positions it sequentially in front of each culture vessel for taking turbidity measurements. Ve signals are averaged from 20 readings taken at 100-ms intervals after a 3-s delay during which microbubbling of the aeration gas is stopped. The frequencies of the measurements and of the shifts of the measuring unit are computer controlled. Thus, the system is able to monitor and display online the simultaneous changes in turbidity in eight parallel cultures.
Regulation of temperature.
The temperature of each bioreactor is controlled individually by a PID CB100 thermal regulator (System C Industrie), which is coupled to a Peltier device (AMS Tech CP 1.0-127-05 LSP) for rapid heating and cooling of a square-section aluminum sheath into which the bioreactor is inserted. Real temperatures measured by the Pt100 microprobes (Thermo-electra P4706812008) in each unit are transmitted to the computer via an RS-232/RS-485 converter (B&B Electronics) and compared to the set temperatures in order to regulate the Peltier devices accordingly.
Monitoring of pH and DO.
The pH of the cultures is monitored using a miniature pH probe (Broadley James). DO is measured with a 6-mm polarographic oxygen probe (Meredos GmbH) with a time constant of 15 to 20 s. The probe is calibrated after sparging a 0.1 M KCl solution with nitrogen (0% saturation) or air (100% saturation). The oxygen transfer coefficient (kLa) is measured using the gassing out method (19). The MMF vessel is filled with 70 ml of a 0.1 M KCl solution or with medium. The solution is then purged of oxygen by bubbling nitrogen until the medium is oxygen-free. Finally, the gas is switched to air at a rate of 1 volume of gas per volume of medium per min, and the resulting increase in oxygen concentration is monitored. The value of kLa is derived after fitting the data to the equation.
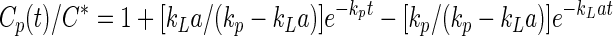 |
where C* is the concentration of DO at equilibrium, Cp(t) is the concentration of DO determined as a function of time t, and kp is the mass transfer coefficient of the probe (inverse of the probe time constant [6]).
Electronic equipment.
The electronic parts directly involved in monitoring and controlling culture parameters consist of E/S FieldPoint interfaces and a PCI-7344 card stepper motion controller for trajectory and point-to-point control of the turbidity sensor. The remaining parts of the Fieldpoint interface are an FP-1000 (RS232) communication module and four FP-RLY-420 modules, each comprising eight relays. Two of these modules control the functioning of the miniature three-way electrovalves responsible for mixing and injecting the gases used for aeration. The remaining two modules may be used to control two-way electrovalves for automatic injection of inducer into the reactors or for programmed withdrawal of culture samples.
Automated programmable run control.
The user interface for monitoring and programming cultivation parameters is provided by the Multigen program, an in-house-developed LabVIEW software routine running in a Microsoft Windows XP environment. For each bioreactor, the program manages the implementation of programmed settings and continuous recording of cultivation parameters (turbidity, pH, DO, temperature). Values are stored automatically as Microsoft Excel tables and are displayed graphically on the computer screen. For each culture, the user can independently program, as a function of time or turbidity, sequences of different temperature settings or the opening of electrovalves controlling gas (aeration) or fluid fluxes (sampling or addition of inducer).
Test genes and constructs.
The recombinant proteins whose production was tested originated from a set of genes of various organisms, including genes of M. tuberculosis and M. leprae chosen as targets for a structural genomics project. Except for the gene encoding tobacco etch virus protease (7), which was cloned into pMal-C2, most genes were inserted into vectors, such as pET28 (EMD Biosciences), pIVEX3 (Roche), or pDEST17 (Invitrogen), designed for conditional expression of target genes under control of a T7 RNA polymerase-dependent promoter. The plasmids encode an N- or C-terminal His6 tag fused to the target polypeptide. The expression plasmids were introduced into BL21(DE3) (20), in which synthesis of T7 RNA polymerase is controlled by the lac repressor. This strain harbored pDIA17 (13), a plasmid containing the lacI gene cloned at the BamHI site of pACYC184 for tighter control of target gene expression.
Culture media.
Modified Lennox medium contains twice as much tryptone and yeast extract as Lennox medium and the same amount of NaCl; thus, it contains (per liter) 20 g Bacto tryptone, 10 g Bacto yeast extract, and 5 g NaCl. 20-40-20 medium contains (per liter) 20 g glycerol, 40 g yeast extract, 20 g tryptone, and 100 ml of a buffer solution containing (per liter) 70 g KH2PO4, 70 g Na2HPO4, and 50 g NaCl. The high-density medium (HDM medium) used for cultures in microfermentors contains (per liter) 40 g glycerol, 40 g yeast extract, 20 g tryptone, 3 g NH4Cl, 0.021 g thiamine hydrochloride, 1.4 ml Struktol J673 antifoam (Schill & Seilacher, Hamburg, Germany), 56 ml buffer solution containing (per liter) 26.6 g KH2PO4, 80 g K2HPO4, 26.7 g NH4Cl, 13.3 g Na2SO4, and 2 ml of a trace element solution containing (per liter) 7.44 mg H3BO3, 16 mg MnCl2 · 4H2O, 24 mg CaCl2 · 2H2O, 64 mg FeCl3 · 6H2O, 280 mg MgCl2 · 6 H2O, 0.200 mg AlCl3 · 6H2O, 0.213 mg CrCl3 · 6H2O, 0.952 mg CoCl2 · 6H2O, 0.136 mg CuCl2 · 2H2O, 0.190 mg NiCl2 · 6H2O, 0.194 mg Na2MoO4 · 2H2O, 0.139 mg Na2SeO3 · 5H2O, 0.125 mg VCl3, and 0.544 mg ZnCl2 (1). Antibiotics were added to the growth medium to the following concentrations, depending on the plasmid used: 30 μg/ml chloramphenicol, 100 μg/ml ampicillin, and 50 μg/ml kanamycin.
Fernbach flask cultivation.
BL21(DE3)(pDIA17) competent cells were freshly transformed with the expression plasmids. Clones growing from the selection plates were resuspended and inoculated into precultures of Lennox broth up to the late log phase, chilled, and stored overnight at 4°C. Cells were decanted or centrifuged and inoculated into 2-liter Fernbach flasks containing 1 liter of modified Lennox medium at an OD600 of 0.1 to 1, depending on the temperature of cultivation. The flasks were agitated at 200 rpm. At an OD600 of 1.7 to 2.4, protein production was induced by adding isopropyl-β-d-thiogalactoside (IPTG) to a final concentration of 1 mM. Cultivation was continued at a temperature and for a period of time calculated to optimize for each clone the yield of soluble target polypeptide, depending on the solubility of the polypeptide produced. Typically, induction was carried out at 37°C for 1.5 h or at 30°C for 3 h for soluble proteins; for poorly soluble proteins, the culture was cooled to 14°C or 23°C before addition of IPTG and further incubation for 15 h or 5 h, respectively, at the same temperature.
High-cell-density cultures in microfermentors.
High-cell-density cultivation in microfermentors was performed in 70 ml HDM medium supplemented with appropriate antibiotics. Bioreactors were inoculated directly using a starting OD600 of 0.4 to 0.8 with freshly transformed cells resuspended from the selection plates in 1 ml Luria-Bertani (LB) medium (12). The gas flow rate was maintained at 1 volume of gas per volume of medium per min, and pure oxygen was progressively mixed with the inlet air stream in order to maintain the dissolved oxygen concentration above 80% of air saturation. For soluble proteins, cultivation was carried out at 37°C or 30°C and IPTG was added to a final concentration of 1 mM once the OD600 of the cultures reached 40 to 45. Cultures were further incubated either for 1.5 h at 37°C or for 3 h at 30°C. For poorly soluble or toxic proteins, cultivation was performed at 30°C until the OD600 of the cultures reached 20 to 25, and the temperature was rapidly lowered to 14°C or 16°C before addition of the inducer (1 mM [final concentration] IPTG). The culture was further incubated overnight (15 to 18 h) at the same temperature. At the end of cultivation, the temperature was lowered to 4°C before the cells were harvested.
High-cell-density cultures in classical fermentors.
Cultures were grown in 2-liter fermentors (Biolafitte or Bioflo3000 fermentors from New Brunswick). Freshly transformed clones growing from the selection plates were used to inoculate precultures of 20-40-20 medium, which were grown to the early stationary phase, chilled, and decanted overnight at 4°C. Cells were inoculated into 1 or 1.5 liter HDM medium at a starting OD600 of 0.7 to 1.3. The cultivation protocols were derived from conditions defined in microfermentors. Aeration was regulated in order to maintain 80% of air saturation by varying the speed of stirring and by progressively mixing pure oxygen with the air inlet stream.
Quantification of soluble recombinant protein production.
Cells from 1-ml samples (microfermentors and classical fermentors) or 10-ml samples (Fernbach flasks) were harvested by centrifugation, stored at −20°C, and resuspended in 1 ml buffer A (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole; pH 7.0). Bacteria were disrupted by sonication, and the particulate fraction containing cell debris and insoluble recombinant protein was removed by centrifugation for 20 min at 16,100 × g. One milliliter of the supernatant, diluted with buffer A in order to saturate 10 to 90% of the binding capacity of the resin, was added to 200 μl (bed volume) BD TALON Co2+ resin (BD Biosciences) equilibrated with buffer A. The suspension was agitated for 60 min at room temperature on a rotary shaker before the nonadsorbed fraction was separated by centrifugation. The resin was washed three times for 10 min with 1 ml buffer A, and the recombinant protein was eluted by washing the resin repeatedly with buffer B (70 mM sodium phosphate, 420 mM NaCl, 209 mM imidazole; pH 7.0) until protein could no longer be detected in the eluted fractions. The total protein in the eluted fractions was quantified by the method of Bradford (3) using a Bio-Rad protein assay kit with bovine serum albumin as a standard. The purity of the eluted His6-tagged protein and its absence from the nonadsorbed and wash fractions were assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (9).
Assay of AMP kinase activity.
AMP kinase activity was assayed as described by Munier-Lehmann et al. (14).
RESULTS
Online turbidity monitoring.
Compared to other parameters (dry cell weight, cell count), measurement of turbidity is the easiest method to determine the density of biomass in microorganism cultures. For this purpose, we developed a fully automated external sensor which precludes contamination and the need to withdraw samples, with the concomitant loss of material. Hence, there is practically no limit to the number of measurements that can be taken during the course of cultivation.
An important specification of the system was that it should be able to provide a reliable estimate of the biomass over a wide range of turbidities. Two factors contribute to achievement of this goal. First, the sensitivity of the sensor can be regulated by adjusting the value of the reference current of the photodetector; low intensity is chosen for densities in the high range and vice versa. Second, for high densities, the optical density does not follow Beer's law, since secondary back scattering redirects light that would be scattered off the path to the detector at low density. Hence, even at very high densities, a significant fraction of the emitted light reaches the detector. Figure 3 shows the relationship between the actual OD600 measured manually for appropriately diluted samples and the voltage required to maintain the output current of the photodetector at the set value. The Ve-versus-OD600 relationship was independent of the recombinant strain being cultivated, since the same calibration curve could be used to fit points derived from cultures of BL21(DE3)(pDIA17) clones carrying six different genes. For OD600 values greater than 10, the points are adequately fitted by a straight line (R2 = 0.974; standard deviation of the residuals, 3.86), which can serve as a calibration curve for converting the Ve signal into OD600. For OD600 values less than 10, the linear relationship is no longer valid, leading to underestimation of the true OD600. Thus, for low densities, a logarithmic equation should be used as the relationship between turbidity and biomass approaches Beer's law for diluted suspensions. However, the linear calibration was satisfactory for the purposes of the experiments described below, which involved high cell densities.
FIG. 3.

Correlation between the voltage of the emitting diode (Ve) and the actual OD600. Cultures of BL21(DE3)(pDIA17) clones which harbored pDEST17 plasmids carrying genes of M. tuberculosis were cultivated in microfermentors while the voltage of the light-emitting diode was recorded continuously. At defined times, samples were withdrawn and appropriately diluted (OD600, <1), and the OD600 was determined with a Digilab Hitachi U-1800 spectrophotometer. The values were derived from six different clones whose behavior was very similar.
Temperature regulation.
The architecture of the temperature control system proved to be well adapted for programming independent sequences of temperature settings, an essential feature for optimization of bacterial cultures, particularly for producing recombinant proteins in a soluble form. Monitoring the temperature inside the bioreactor ensures that the set temperature is controlled accurately without interference due to problems of heat transfer or to generation of metabolic heat. Operating performance was characterized by a mean heating slope of 5°C/min, a mean cooling slope of 3°C/min in the 15 to 50°C range (minimum, −5°C; maximum, 65°C), and ±0.5°C stability.
Aeration.
Adequate aeration is a key parameter in order to obtain high cell densities in bioreactors. The sintered glass sparger produced a draft of uniform bubbles that resulted in both mixing of the reactor content and efficient oxygen transfer. A kLa value of 750 h−1 was obtained in 0.1 M KCl, using a gassing rate of 1 volume of gas per volume of medium per min. Using oxygen-supplemented aeration, it was possible to maintain the DO concentration at at least 80% of air saturation even at a high cell density.
High-density medium formulation.
In order to use microfermentors for preparative production of recombinant proteins, it was necessary to optimize growth conditions so that E. coli could be grown to high cell densities. To keep the procedure as simple and robust as possible, we chose a batch process using HDM medium, a balanced complex medium. Experiments with increasing concentrations of glycerol showed that 40 g/liter was optimum in terms of the specific growth rate and maximum biomass. At 40 g/liter glycerol, the acetate concentration remained below 2 g/liter, indicating that aeration was not limiting, whereas the acetate concentration reached 5 g/liter for glycerol concentrations of 60 and 80 g/liter, which resulted in lower growth yields (data not shown). Monitoring the pH indicated that moderate acidification down to pH 5.8 occurred at high cell densities toward the end of the culture. Nevertheless, using HDM medium as described above, cultures routinely achieved OD600 of 50 to 100 (10 to 20 g [dry weight]/liter), depending on the cultivation protocol and the toxicity of the target protein, which shows that tight pH monitoring and regulation were not essential. Thus, the biomass obtained in a 70-ml reactor was 1.5- to 2-fold higher than that obtained in a Fernbach flask with 1 liter of modified Lennox medium, whose final OD600 was about 3 to 4. The doubling time was 35 to 45 min for most clones grown at 37°C in HDM medium in the absence of inducer.
Validation of microfermentor technology for recombinant protein production.
In order to verify that high-cell-density cultivation was accompanied by a concomitant increase in recombinant protein production, we compared the amounts of recombinant protein produced in a Fernbach flask containing 1 liter of modified Lennox medium and in a 70-ml microfermentor reactor containing HDM medium for 26 different clones. The set of proteins tested included mostly, but not exclusively, M. tuberculosis and M. leprae polypeptides. For each clone, cultures were grown at the same temperature (30°C or 37°C), and the induction protocol was adapted for either system; IPTG was added to a final concentration of 1 mM during the late exponential growth phase as described in Materials and Methods. The His6-tagged soluble recombinant proteins produced were quantified after analytical purification on Co2+-loaded agarose beads. The reproducibility of the data is shown by the fact that, for three independent MMF cultures of the clone expressing Rv2543, an average of 2.3 mg/ml culture (standard deviation, 0.17 mg/ml) was recovered as purifiable protein. The experimental results are shown in Fig. 4. The yields obtained in 70-ml MMF reactors and in Fernbach flasks containing 1 liter of medium were equivalent within a factor of 2 for most proteins, in spite of the fact that the amount of soluble recombinant protein produced varied from 4 to 168 mg/culture, depending on the construct expressed. On average, the levels produced were somewhat higher (147%) in the microfermentor cultures than in the Fernbach flask cultures.
FIG. 4.

(Left panel) Yields of recombinant proteins obtained after cultivation of the production clones in 70-ml microfermentors with HDM medium. (Right panel) Ratios of the yields of recombinant proteins in 70-ml microfermentors containing HDM medium to the yields of recombinant proteins in Fernbach flasks containing 1 liter of modified Lennox medium. Analytical purification and quantification of His6-tagged soluble recombinant proteins were performed as described in Materials and Methods. Cultivation protocols, including temperature and timing of IPTG addition, were adapted to the systems, and the best production in each system was used to calculate the ratios. The genes encoding the proteins tested are indicated between the panels. Designations beginning with Rv and ML are designations for the reading frames of the M. tuberculosis and M. leprae genomes, respectively, whose descriptions are available at the websites http://genolist.pasteur.fr/TubercuList/ and http://genolist.pasteur.fr/Leproma/. The following other proteins were included: Trypanosoma cruzi proline racemase (TcPRAC), tobacco etch virus protease fused to maltose-binding protein (MBP-TEV), and Vga(A) of Staphylococcus aureus (VGA).
The available evidence indicates that the microfermentor cultivation protocol does not jeopardize proper protein folding in vivo. In the case of M. tuberculosis AMP kinase, encoded by Rv0733, the specific activities of the enzyme purified from cultures grown either in MMF containing HDM medium or in Fernbach flasks containing LB medium were the same (150 ± 10 U/mg). Furthermore, 12 proteins originating from MMF cultures have been crystallized so far, and the structures of 10 of them have been determined by X-ray diffraction; the results confirmed that the microfermentor cultivation protocol does not jeopardize proper protein folding in vivo (A. Haouz and P. Alzari, personal communication).
Fully automated MMF process: application to production of poorly soluble protein.
In many cases, high-level production of recombinant proteins results in the formation of inclusion bodies that are not suitable for enzymatic or structural studies. Significant improvement can often be obtained by inducing expression after the culture is shifted to a low temperature (18). As shown in Fig. 5, the MMF technology is well suited to perform the required steps in a fully automated manner. The recombinant E. coli strain BL21(DE3)(pDIA17) expressing the Rv0289 gene of M. tuberculosis cloned in pDEST17 was grown at 30°C until the OD600 reached a preselected value, 30. At this point, the Peltier device automatically shifted the culture to 14°C. Following temperature stabilization at 14°C, IPTG (final concentration, 1 mM) was automatically injected into the culture, and incubation was continued for 18 h at the same temperature. The whole process was monitored and displayed graphically, which showed the temperature downshift and the concomitant decrease in the growth rate (Fig. 5A). The results of a sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of the total soluble and insoluble fractions and of the Co2+ affinity-purified soluble material are shown in Fig. 5b. Although the majority of the recombinant protein was still in the insoluble fraction, the yield of soluble product was improved a great deal compared to the yield in a culture in which induction was performed without a decrease in the temperature.
FIG. 5.

Programming and monitoring optimized cultivation protocols. (A) Monitoring of growth and temperature in a culture of BL21(DE3)(pDIA17) expressing the Rv0289 gene of M. tuberculosis. The culture was grown in automated mode with preprogrammed temperature shifts and addition of inducer. ▪, temperatures; ▴, OD600. The arrow indicates the time of IPTG addition. (B) Analysis of soluble recombinant protein produced without and with a temperature downshift to 14°C. Cells harvested at the end of the culture were subjected to analytical purification as described in Materials and Methods. Lanes 1 to 5, fractions from a culture maintained at 30°C for 3 h after induction; lanes 6 to 10, fractions from the culture shown in panel A, which was shifted to 14°C, induced, and then incubated overnight at the same temperature; lanes 1 and 6, total soluble extract; lanes 2 and 7, insoluble fraction; lanes 3 to 5 and 8 to 10, fractions purified by metal affinity chromatography.
Scale-up from MMF to conventional fermentors.
For some purposes (e.g., large-scale drug screening), the amount of recombinant protein recovered from MMF bioreactors may not be sufficient. Thus, we assessed whether recombinant protein production could be reliably scaled up by transposing culture protocols established in MMF to larger, classical fermentors. For this, we compared the yields of five proteins obtained in an MMF bioreactor and in a classical fermentor, using three different induction protocols that had been previously optimized in MMF for each protein. As shown in Table 1, the amounts of recombinant protein recovered per ml of culture were comparable. However, in some cases (for example, Rv2238c and Rv1827), the yield of target protein relative to total cell mass (μg/OD600) was lower for the larger-scale cultures grown in classical fermentors. A likely explanation for this is that synthesis of these proteins is detrimental to growth, even in the absence of inducer, which results in selection pressure favoring the appearance of variants with a higher growth rate and lower productivity. Indeed, Rv1827 produces heterogeneous colonies, and the production phenotype is unstable (data not shown). Thus, cultures grown in larger volumes, which require more cell cycles, should be affected more by drifting of the production phenotype, leading to higher biomass but lower specific productivity.
TABLE 1.
Production of recombinant proteins in MMF reactors and in classical fermentors
| Protein | Induction protocol | Bioreactor | Vol | Final OD600 | Concn of purified protein
|
|
|---|---|---|---|---|---|---|
| mg/ml culture | μg/OD600 | |||||
| Rv2543 | 37°C, 2 h | Fermentor | 1.6 liters | 80 | 2.1 | 26 |
| 37°C, 2 h | MMF | 80 ml | 70 | 2.3 | 32.7 | |
| T. cruzi racemase | 20°C, 3 h | Fermentor | 1.6 liters | 48 | 0.26 | 5.4 |
| 20°C, 4.5 h | MMF | 60 ml | 58 | 0.33 | 5.7 | |
| Rv2238c | 14°C, overnight | Fermentor | 1 liter | 82 | 0.49 | 6.0 |
| 14°C, overnight | MMF | 66 ml | 57 | 0.63 | 11 | |
| Rv1827 | 14°C, overnight | Fermentor | 1 liter | 51 | 0.36 | 7.0 |
| 14°C, overnight | MMF | 62 ml | 21 | 0.29 | 13.8 | |
| Rv2256c | 14°C, overnight | Fermentor | 1 liter | 97 | 1.3 | 13.4 |
| 14°C, overnight | MMF | 70 ml | 72 | 0.91 | 12.6 | |
DISCUSSION
The distinguishing feature of the microfermentor battery described in this report is that it allows rigorous, independent monitoring of culture parameters in each reactor, including online monitoring of cell density. The latter feature means that sophisticated cultivation protocols involving shifts of temperature and addition of inducer can be programmed as a function of cell density and implemented without intervention of the operator. This is particularly useful for difficult proteins, whose production may require long protocols that extend beyond working hours.
Unlike commercial devices for online monitoring of cell density (e.g., Mettler Toledo Trb 8300 + probe InPro 8000; Cerex Max), the turbidity sensor of the MMF battery is entirely external. Therefore, it is well suited for monitoring a set of miniature reactors, since the sensor occupies no internal reactor space and does not need to be sterilized. In addition, a single movable sensor can monitor the growth of all cultures quasisimultaneously, avoiding the need to calibrate multiple sensors against each other. The adjustable sensitivity offers a wide dynamic range up to the high cell densities needed to obtain sufficient biomass for preparative purposes.
The design of the bioreactors provides highly efficient stirring and aeration without a requirement for mechanical agitation devices. The high rate of oxygen transfer, combined with a suitably enriched buffered medium, was a key feature in enabling batch cultivation of recombinant E. coli clones to very high cell densities. Indeed, cultures in HDM medium in Fernbach flasks failed to grow to OD600 greater than 20 (data not shown). A high biomass concentration was not detrimental to recombinant protein production, so that the 80-ml volume of the bioreactors was sufficient for preparative purposes in most cases, which facilitated handling of the cultures and harvesting of the cells. Furthermore, accurate monitoring and control of culture parameters make it possible to scale up cultures to larger, classical fermentors if necessary.
While in this study we dealt with the production of unlabeled recombinant protein in E. coli, the equipment is also a powerful tool for optimizing the composition of specialized culture media (e.g., media for labeling recombinant proteins with 13C, 15N, or selenomethionine). Likewise, it may be used to study the growth and physiology of other microorganisms, and we are currently using it to optimize culture conditions for Mycobacterium smegmatis and Legionella pneumophila. In the future, we will integrate an automated fluid handling station to obtain a complete device with plug-and-pump fluidic connections to address filling with sterile medium, the inoculation step of the microfermentor battery, and automated sampling during bacterial growth for analytical purification analysis.
Acknowledgments
We thank P. M. Alzari and S. T. Cole for their support and for their interest in the development of the microfermentor battery, Y. Archambeau for assembling the electronic control elements of the battery, N. Berger, F. Proux, O. Chesneau, and P. Minoprio for providing the recombinant clones used in this study, M. Nowakowski and A. Meier for technical assistance, and P. Béguin for editorial assistance with the manuscript.
This work was funded by grants from the National Genopole Network, France (contract RNG-2002-008), the Ministry of Research, France (contract 01B0095), and the European Union SPINE (EC grant QLG2-CT-2002-00988) and X-TB (EC grant QLK2-CT-2001-02018).
REFERENCES
- 1.Bellalou, J., E. Frachon, and R. Longin. June 2005. Milieux de cultures bactériennes à moyenne ou haute densité se révélant être compatibles avec un mélange d'auto-induction et leurs applications. French patent application 0506707.
- 2.Bellalou, J., E. Frachon, R. Longin, and A. Meier. June 2003. Robotized platform for cell cultures in miniature reactor batteries, equipped with a system for real-time measurement of cellular turbidity or other optical properties. Canadian patent CA 2491166.
- 3.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 4.Doig, S. D., A. Diep, and F. Baganz. 2005. Characterization of a novel miniaturized bubble column bioreactor for high throughput cell cultivation. Biochem. Eng. J. 23:97-105. [Google Scholar]
- 5.Gaillon, L., R. Longin, and T. D. Luu. April 2004. Apparatus and method for measuring optical properties by feedback control. United States patent 6,723,554.
- 6.Gupta, A., and G. Rao. 2003. A study of oxygen transfer in shake flasks using a non-invasive oxygen sensor. Biotechnol. Bioeng. 84:351-358. [DOI] [PubMed] [Google Scholar]
- 7.Kapust, R. B., J. Tozser, J. D. Fox, D. E. Anderson, S. Cherry, T. D. Copeland, and D. S. Waugh. 2001. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14:993-1000. [DOI] [PubMed] [Google Scholar]
- 8.Kensy, F., H. F. Zimmermann, I. Knabben, T. Anderlei, H. Trauthwein, U. Dingerdissen, and J. Buchs. 2005. Oxygen transfer phenomena in 48-well microtiter plates: determination by optical monitoring of sulfite oxidation and verification by real-time measurement during microbial growth. Biotechnol. Bioeng. 89:698-708. [DOI] [PubMed] [Google Scholar]
- 9.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 10.Lamping, S. R., H. Zhang, B. Allen, and P. Ayazi Shamlou. 2003. Design of a prototype miniature bioreactor for high throughput automated bioprocessing. Chem. Eng. Sci. 58:747-758. [Google Scholar]
- 11.Maharbiz, M. M., W. J. Holtz, R. T. Howe, and J. D. Keasling. 2004. Microbioreactor arrays with parametric control for high-throughput experimentation. Biotechnol. Bioeng. 85:376-381. [DOI] [PubMed] [Google Scholar]
- 12.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 13.Munier, H., A. M. Gilles, P. Glaser, E. Krin, A. Danchin, R. Sarfati, and O. Barzu. 1991. Isolation and characterization of catalytic and calmodulin-binding domains of Bordetella pertussis adenylate cyclase. Eur. J. Biochem. 196:469-474. [DOI] [PubMed] [Google Scholar]
- 14.Munier-Lehmann, H., S. Burlacu-Miron, C. T. Craescu, H. H. Mantsch, and C. P. Schultz. 1999. A new subfamily of short bacterial adenylate kinases with the Mycobacterium tuberculosis enzyme as a model: a predictive and experimental study. Proteins 36:238-248. [PubMed] [Google Scholar]
- 15.Page, R., K. Moy, E. C. Sims, J. Velasquez, B. McManus, C. Grittini, T. L. Clayton, and R. C. Stevens. 2004. Scalable high-throughput micro-expression device for recombinant proteins. BioTechniques 37:364-368. [DOI] [PubMed] [Google Scholar]
- 16.Puskeiler, R., K. Kaufmann, and D. Weuster-Botz. 2005. Development, parallelization, and automation of a gas-inducing milliliter-scale bioreactor for high-throughput bioprocess design (HTBD). Biotechnol. Bioeng. 89:512-523. [DOI] [PubMed] [Google Scholar]
- 17.Puskeiler, R., A. Kusterer, G. T. John, and D. Weuster-Botz. 2005. Miniature bioreactors for automated high-throughput bioprocess design (HTBD): reproducibility of parallel fed-batch cultivations with Escherichia coli. Biotechnol. Appl. Biochem. 42:227-235. [DOI] [PubMed] [Google Scholar]
- 18.Schein, C. H. 1989. Production of soluble recombinant proteins in bacteria. Bio/Technology 7:1141-1148. [Google Scholar]
- 19.Stanbury, P. F., and A. Whitaker. 1984. Principles of fermentation technology, 1st ed. Pergamon Press, Oxford, United Kingdom.
- 20.Studier, F. W. 1991. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol. 219:37-44. [DOI] [PubMed] [Google Scholar]
- 21.Szita, N., P. Boccazzi, Z. Zhang, P. Boyle, A. J. Sinskey, and K. F. Jensen. 2005. Development of a multiplexed microbioreactor system for high-throughput bioprocessing. Lab. Chip 5:819-826. [DOI] [PubMed] [Google Scholar]