

Abstract
Biotin synthase is an iron-sulfur protein that utilizes AdoMet to catalyze the presumed radical-mediated insertion of a sulfur atom between the saturated C6 and C9 carbons of dethiobiotin. Biotin synthase (BioB) is aerobically purified as a dimer that contains [2Fe-2S]2+ clusters and is inactive in the absence of additional iron and reductants, and anaerobic reduction of BioB with sodium dithionite results in conversion to enzyme containing [4Fe-4S]2+ and/or [4Fe-4S]+ clusters. To establish the predominant cluster forms present in biotin synthase in anaerobic assays, and by inference in Escherichia coli, we have accurately determined the extinction coefficient and cluster content of the enzyme under oxidized and reduced conditions and have examined the equilibrium reduction potentials at which cluster reductions and conversions occur as monitored by UV/visible and EPR spectroscopy. In contrast to previous reports, we find that aerobically purified BioB contains ca. 1.2-1.5 [2Fe-2S]2+ clusters per monomer with ε452 = 8400 M-1 cm-1 per monomer. Upon reduction, the [2Fe-2S]2+ clusters are converted to [4Fe-4S] clusters with two widely separate reduction potentials of -140 and -430 mV. BioB reconstituted with excess iron and sulfide in 60% ethylene glycol was found to contain two [4Fe-4S]2+ clusters per monomer with ε400 = 30 000 M-1 cm-1 per monomer and is reduced with lower midpoint potentials of -440 and -505 mV, respectively. Finally, as predicted by the measured redox potentials, enzyme incubated under typical anaerobic assay conditions is repurified containing one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer. These results indicate that the dominant stable cluster state for biotin synthase is a dimer containing two [2Fe-2S]2+ and two [4Fe-4S]2+ clusters.
The final step in the biosynthesis of biotin is the insertion of a sulfur atom between the C6 and C9 carbons of dethiobiotin, a reaction catalyzed by biotin synthase. Although genetic analysis in Escherichia coli indicates that BioB is the sole protein component of biotin synthase (1, 2), in vitro activity with overexpressed enzyme has been low in all studies to date (3-5). However, biotin production is markedly increased in the presence of AdoMet1 and flavodoxin (5-8), and this has been interpreted as evidence that biotin synthase belongs to a growing family of AdoMet-dependent radical enzymes. Consistent with this view, BioB contains a conserved iron-sulfur cluster binding motif, CxxxCxxC, that is shared by other radical enzymes, including pyruvate formate-lyase activating enzyme (9), class III (anaerobic) ribonucleotide reductase (10), lysine 2,3-amino-mutase (11), lipoyl synthase (12), and benzylsuccinate synthase (13).
The mechanism of radical generation and role of the iron-sulfur cluster in AdoMet-dependent radical enzymes is poorly understood. The overall reaction catalyzed is the reduction of AdoMet to methionine and 5′-deoxyadenosine (14), with one electron for the reduction derived from flavodoxin in E. coli and the other electron derived by oxidation of a substrate or protein residue to an organic radical. The detailed mechanism is undoubtedly more complex. Our working hypothesis is that biotin synthase will share some mechanistic features with lysine 2,3-aminomutase, for which it has recently been shown that AdoMet coordinates to one Fe in a [4Fe-4S] cluster (15). In biotin synthase, flavodoxin would then deliver an electron into the iron-sulfur cluster/AdoMet complex, leading to spontaneous scission of the AdoMet C-S bond. The liberated 5′-deoxyadenosyl radical then abstracts a hydrogen atom from a proximal C-H bond, generating a carbon radical on the substrate. Using an allylic AdoMet analogue, Frey and co-workers have recently demonstrated the chemical competence of the 5′-deoxyadenosyl radical as an intermediate in LAM (16). Further, in both LAM and biotin synthase, direct abstraction of a hydrogen atom from the substrate by the 5′-deoxyadenosyl radical has been demonstrated by deuterium incorporation into the product 5′-deoxyadenosine (17, 18).
On the basis of this mechanistic analogy with lysine 2,3-aminomutase, one would expect that active biotin synthase should contain [4Fe-4S] clusters, but in contrast BioB is purified containing [2Fe-2S]2+ clusters under either aerobic or anaerobic conditions. However, the [2Fe-2S]2+ clusters can be converted to a [4Fe-4S]2+/+ cluster in the presence of chemical reductants alone (19-21), and the yield of [4Fe-4S]2+/+ clusters is increased with the addition of exogenous iron and sulfide (21, 22). Mossbauer studies confirm the conversion of [2Fe-2S]2+ to [4Fe-4S]2+ clusters and also indicate that this process is accompanied by the release of iron from the protein (20, 21). Maximal in vitro activity requires the addition of exogenous Fe3+ and DTT (4-6), conditions which may result in at least partial [4Fe-4S] cluster assembly (20-22), although it has not been demonstrated that BioB contains [4Fe-4S] clusters under either assay conditions or typical in vivo conditions.
An alternate role for iron-sulfur clusters in biotin synthase has also been proposed. Marquet and co-workers denatured and refolded BioB in the presence of [34S]sulfide and found that, upon incubation with substrates, the resulting enzyme produced ∼0.2 equiv of biotin with 70% label incorporation (23). On the basis of these results, the authors suggest that the iron-sulfur cluster in biotin synthase also serves as the sulfur donor for formation of the biotin thioether ring. Studies by Gibson and co-workers of biotin synthesis by uniformly 35S-labeled enzyme are also consistent with this proposal (24). This dual role for iron-sulfur clusters is clearly in contrast with other AdoMet-dependent radical enzymes and suggests that biotin synthase catalyzes two reactions requiring successive access of AdoMet and dethiobiotin to an iron-sulfur cluster.
In an effort to identify the active cluster state in biotin synthase and to further understand the role of the iron-sulfur clusters in AdoMet cleavage and sulfur insertion, we have determined the Fe and S content, and by inference the cluster content, of BioB under various oxidized and reduced conditions. We have also examined the electrochemical potentials required for iron-sulfur cluster reduction and for cluster interconversion in BioB. We show that the aerobically purified protein contains ∼1.2-1.5 [2Fe-2S]2+ clusters per monomer, but this number is increased to ∼1.9 clusters upon brief incubation with iron. Electrochemical titration of as-purified BioB containing [2Fe-2S]2+ clusters with dithionite shows two reduction waves at -140 and -430 mV, presumably for reduction of opposing clusters in the monomer. BioB is artificially reconstituted with excess FeCl3, Na2S, and dithionite in anaerobic 60% ethylene glycol to a protein containing two [4Fe-4S]2+ clusters per monomer; reduction of these clusters proceeds with two midpoint potentials of -440 and -505 mV. On the basis of these electrochemical analyses, we propose that the predominant equilibrium form of BioB under both in vitro assay conditions and in vivo aerobic growth conditions contains one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer. UV/visible spectra and Fe and S analyses of enzyme incubated under in vitro assay conditions support this proposal.
MATERIALS AND METHODS
Materials. All reagents were obtained from commercial sources and used without further purification. BioB and His6-BioB were purified as previously described (21). His6-BioB was used for all data reported in this paper, although in selected titrations we have shown that similar results are obtained with WT BioB. In general, the initial estimated Fe and S content of aerobically purified WT BioB is lower than His6-BioB; we believe this is due to the prolonged purification procedures and the decreased purity of WT preparations (21). Removal of the His tag by selective proteolysis does not noticeably alter the spectral properties or activity of His6 BioB (unpublished results), and we therefore conclude that the His tag is not significantly involved in Fe ligation. Unless otherwise stated, all protein purification steps were performed under aerobic conditions, and all protein reduction, reconstitution, and analyses were performed under oxygen-free argon or nitrogen atmosphere. The buffers used for potentiometric measurements were 50 mM Tris-HCl and 100 mM NaCl at pH = 7.5-8.5 or 50 mM Bis-Tris propane HCl and 100 mM NaCl at pH = 6.5-7.5.
Determination of the Extinction Coefficient and Fe and S Content of Oxidized BioB. Four separate preparations of His6-BioB were performed as previously described (21) and prepared as ∼2 mg/mL stock solutions. UV/visible spectra were recorded, and then protein content was determined using the Bradford assay (Bio-Rad) with BSA (2 mg/mL, Pierce Chemical Co.) as a standard. Samples were also analyzed by quantitative amino acid analysis (AAA Service Laboratory, Boring, OR). Iron was determined using bathophenanthroline under reductive conditions after digestion of the protein in 0.8% KMnO4/0.2 N HCl as described by Fish (25). Standards were prepared by dilution of a commercial Fe atomic absorption standard (Aldrich). ICP analysis was performed (Department of Earth and Environmental Science, University of Pennsylvania) on several oxidized BioB samples to assess the accuracy of these results. Sulfide was determined using the Beinert method (26) as modified by Broderick (27). Standards were prepared gravimetrically from Na2S.9H2O as described by Beinert (26).
Reconstitution of [4Fe-4S]2+ Clusters in BioB. To obtain the midpoint potentials of the [4Fe-4S]2+ clusters in biotin synthase, the initial [2Fe-2S]2+ protein was converted to protein containing either one or two [4Fe-4S]2+ clusters. To obtain BioB containing ca. one [4Fe-4S]2+ cluster per monomer, BioB (100 μM final concentration in 1 mL) was diluted into 60% ethylene glycol and 100 mM Tris-HCl, pH 7.5, in an anaerobic cuvette and made anaerobic by passing a steady argon stream over the solution for 60 min with continuous stirring. DTT (5 mM), FeCl3 (500 μM), and dithionite (5 mM) were added, and the UV/visible spectrum was monitored over 4 h. Since no additional sulfide was added, the yield of [4Fe-4S]2+ protein is limited by the sulfide available from the initial protein sample. BioB containing two [4Fe-4S]2+ clusters was obtained in a manner similar to that described above, except that anaerobic solutions of DTT (5 mM), FeCl3 (500 μM), and Na2S (500 μM) were added to the protein in ethylene glycol buffer prior to addition of dithionite (5 mM). In each case, the buffer components were removed by passing the sample through a Sephadex G25 desalting column (1 × 20 cm, Amersham-Pharmacia) equilibrated with anaerobic 50 mM Tris-HCl and 100 mM NaCl, pH 7.5, in a nitrogen glovebox. The samples were transferred to an anaerobic cuvette outfitted with a sealed array of microelectrodes.
Redox Potentiometry. BioB containing either [2Fe-2S]2+ or [4Fe-4S]2+ clusters (∼50-100 μM monomer) was reduced by titration with dithionite (10 mM stock, 1-2 μL per addition) in an anaerobic cuvette. Methyl viologen, benzyl viologen, anthroquinone, and naphthoquinone (5 μM each) were added as redox mediators to facilitate equilibration of protein and electrodes. The redox potential of the cuvette was monitored by an adaptation of the method described by Dutton (28), using a platinum wire working electrode and Ag/AgCl reference electrode. To ensure complete equilibration of the sample with the electrodes, the protein was vigorously stirred for 2-5 min with a small Teflon stir bar that had been soaked in 0.5 M dithionite overnight to remove bound oxygen. Potentials are reported vs the standard hydrogen electrode (SHE). It should be noted that the reduction potentials monitored by the above method are not readily reversible; addition of an oxidant such as O2 or K3Fe(CN)6 to the reduced protein does not reproduce the same cluster state as the original protein, presumably due to the spontaneous cluster conversions that occur upon reduction and oxidation (19, 20). Thus the potentials reported should be considered an upper limit to the true reversible midpoint potentials.
Potentiometric data were fit to either one or two summed Nernst curves (eq 1), where n is the number of electrons
| 1 |
transferred in each step and F/RT = 60 mV. Since iron-sulfur cluster reduction is a one-electron process, we forced n = 1.0 and allowed the fitting routine to alter the midpoint potentials and absorbance changes associated with each reduction wave. For low-potential reductions, where the endpoint absorbance is not observed due to the prohibitively low potentials (<-530 mV), we have forced the curve fit to a reasonable range of final absorbance values (∼30-50% of the initial absorbance) and report the range of midpoint potentials consistent with our data. The uncertainty reported in the text represents primarily variability between successive experiments and enzyme preparations.
EPR Spectroscopy. Electron paramagnetic resonance (EPR) spectroscopy was performed using a Bruker ESP300E spectrometer operating at X-band frequencies. Temperature control was maintained by an Oxford ESR 900 continuous flow liquid helium cryostat interfaced with an Oxford ITC4 temperature controller. Microwave frequency was measured by a Hewlett-Packard 5350B frequency counter. Typical EPR parameters were as follows: sample temperature, 5-50 K; microwave frequency, 9.423 GHz; microwave power, 20 mW; modulation frequency, 100 kHz; modulation amplitude, 6.4 G; time constant, 164 ms.
RESULTS
Aerobically Purified BioB Contains Maximally Two [2Fe-2S]2+ Clusters per Monomer. BioB is purified as a dimer containing [2Fe-2S]2+ clusters, as indicated by an absorption maximum at 452 nm in the visible spectrum (19, 29) and as demonstrated by Mossbauer spectroscopy (20). Initial estimates suggested that there was one [2Fe-2S]2+ cluster per monomer (19). The predicted polypeptide sequence has cysteines at residues 54, 57, and 60 that share a similar spacing with other AdoMet-dependent radical enzymes; mutagenesis of these residues results in protein that is purified without bound iron (30), and it has been concluded that in the aerobically purified protein these cysteines bind a [2Fe-2S]2+ cluster. However, BioB has several other potential metal ligands, including conserved cysteines at residues 97, 128, and 188, and we had observed heterogeneous behavior in the aerobically purified protein that suggested that more than one type of [2Fe-2S]2+ cluster might be present. To further investigate this possibility, we analyzed four different preparations of aerobically purified BioB for Fe and S content and calculated extinction coefficients for the protein as purified and following reconstitution. Using quantitative amino acid analysis of several samples from four different enzyme preparations, we calculate an extinction coefficient at 452 nm of 8400 ± 800 M-1 cm-1 per monomer for the initial [2Fe-2S]2+ protein. The expected extinction coefficient for one [2Fe-2S]2+ cluster with complete cysteinyl ligation is ca. 8000-10 000 M-1 cm-1 (31), although recent studies suggest that incomplete cysteinyl ligation, as is found in BioB (19), may result in a lowered extinction coefficient at ∼460 nm of 5300-6300 M-1 cm-1 (32, 33). On the basis of these latter values, our experimental extinction coefficient of 8400 M-1 cm-1 translates to ca. 1.3-1.6 [2Fe-2S]2+ clusters per monomer. Protein determination using the Bradford assay with BSA as a standard essentially confirmed these results, and comparison with amino acid analysis indicates that the Bradford assay overestimates the BioB concentration by ∼10%. Iron and sulfide analyses indicate 2.5-3 Fe and 3-4 S atoms per monomer, with the range indicating variability between different enzyme preparations. These chemical analyses are consistent with ca. 1.2-1.5 [2Fe-2S]2+ clusters per monomer, and one might expect that the addition of Fe3+ might result in reconstitution to protein containing two [2Fe-2S]2+ clusters per monomer. Indeed, when we incubate BioB with FeCl3 for 15 min under argon, followed by anaerobic gel filtration chromatography to remove excess iron, we see an increase in the iron content to 3.7 ± 0.4 Fe atoms per monomer. We also see an increase in the extinction coefficient to ε452 = 11 300 M-1 cm-1 per monomer, suggesting increased saturation of the [2Fe-2S]2+ cluster binding sites. Taken together, these data suggest that the BioB monomer contains two iron-sulfur cluster binding sites and is purified with only partial saturation of at least one of these sites.
Our previous report of cluster conversions in biotin synthase (21) cited an extinction coefficient significantly lower than that reported in this paper. This value was estimated for extensively purified WT BioB using the Bradford assay to determine the protein concentration. We attribute the lower extinction coefficient to several factors, including the loss of Fe during the extensive aerobic purification, the lower purity of WT enzyme, and the overestimation of the protein concentration by the Bradford assay. Since all protein concentrations in our previous paper (21) were based upon the initial spectrum of the oxidized protein, the concentration of protein was consistently over-estimated. The present analysis is based upon rapidly purified His6-BioB; no other significant changes have been made to the protocols or results previously described.
Reduction of [2Fe-2S]2+ BioB Proceeds with Two Widely Disparate Potentials. Although BioB is purified containing 1.2-1.5 [2Fe-2S]2+ clusters per monomer, in vitro activity requires additional Fe3+ and DTT, as well as an enzymatic reducing system, and therefore we presume that reduction and potential reassembly of the [2Fe-2S]2+ clusters is required for conversion to the active enzyme. Reduction of BioB containing [2Fe-2S]2+ clusters with dithionite in 60% ethylene glycol is a slow process that results in apparent conversion to [4Fe-4S]2+ clusters (19, 21), while reduction in aqueous buffer is more rapid and results in formation of [4Fe-4S]+ clusters (21). We have proposed a mechanism for this process in which initial reduction to [2Fe-2S]+ clusters leads to dissociation of iron and sulfide from the protein over 10-15 min, while slower reassociation of preformed clusters results in direct conversion to [4Fe-4S]2+/+ protein over 1-2 h (21). In this scheme, the disappearance of the absorption band at 452 nm reports the initial reduction of [2Fe-2S]2+ clusters, while in aqueous solution the appearance of the product [4Fe-4S]+ cluster is largely invisible by UV/visible spectroscopy. We followed the UV/visible spectrum as the [2Fe-2S]2+ enzyme was titrated with small aliquots of dithionite (Figure 1A) and simultaneously monitored the cell potential with platinum and Ag/AgCl electrodes. The solution was allowed to equilibrate for 5 min with vigorous stirring following each addition of dithionite to ensure complete equilibration of protein with electrodes. We found that the reduction of [2Fe-2S]2+ clusters occurred in two waves: approximately 20-40% of the absorbance at 452 nm associated with [2Fe-2S]2+ clusters disappeared with a relatively high reduction potential of -140 ± 20 mV (Figure 1B). At this point, spectral changes plateau until the remainder of [2Fe-2S]2+ clusters are reduced with a reduction potential of -430 ± 20 mV. The spectral changes associated with the first and second waves of reduction are similar (Figure 1A, inset), with the major feature a loss of the absorption band at ca. 460 nm. However, it appears that a weaker absorption band observed in the initial protein sample at ca. 550 nm is lost predominantly upon reduction of the second low-potential cluster, suggesting that the two [2Fe-2S]2+ clusters are not spectroscopically identical. The intermediate product after reduction to Eh = -300 mV is EPR silent, even after 1 h of incubation at this potential, and an increase in free iron was detected in the EPR spectra (g ≈ 4.7), suggesting dissociation of the first [2Fe-2S]2+ cluster from the protein upon reduction. The product of reduction of both [2Fe-2S]2+ clusters at Eh < -500 mV (>2 h incubation) is ∼0.7 [4Fe-4S]+ clusters, as observed by EPR spectroscopy, consistent with previous reports of reductive cluster conversion (19, 21). The observation of two waves of reduction is consistent with the presence of two electrochemically distinct [2Fe-2S]2+ clusters; the total change in absorbance associated with reduction of the low-potential cluster (ΔA452 = -0.5) corresponds to ∼1 cluster per monomer, while that associated with the high-potential cluster (ΔA452 = -0.35) corresponds to ∼0.7 cluster per monomer. The magnitude of the absorbance changes associated with the high-potential cluster varies between enzyme preparations, while the magnitude associated with the low-potential cluster is invariant. We suggest that aerobically purified BioB contains one saturated [2Fe-2S]2+ cluster binding site, as well as one unsaturated, perhaps partially degraded, cluster binding site.
Figure 1:
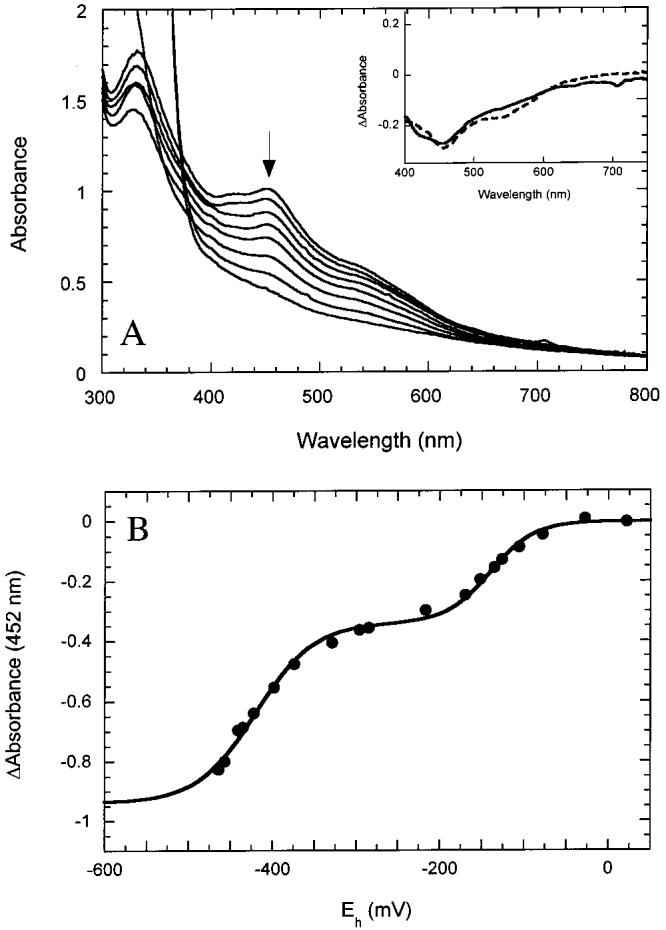
Reduction of BioB containing [2Fe-2S]2+ clusters. (A) UV/visible spectra of BioB (120 μM monomer) in anaerobic 50 mM Tris-HCl and 100 mM NaCl, pH 8.0, were recorded as the cell potential was lowered by titration with dithionite. The decrease in absorbance at 452 nm indicates reduction of the [2Fe-2S]2+ clusters. At the intermediate stage of reduction (Eh ≈ -300 mV) no product was detectable by EPR or UV/visible spectroscopy, and a decrease in the Fe and S content of the protein indicates dissociation of the reduced [2Fe-2S]+ cluster. The product of complete reduction was BioB containing ∼0.7 [4Fe-4S]+ clusters per monomer (21). (Inset) Difference spectra associated with the first wave of reduction (solid curve) and the second wave of reduction (dashed curve) have maxima at 460 nm, suggesting the predominant reaction observed is reduction of a [2Fe-2S]2+ cluster. (B) The absorbance change at 452 nm was followed as a function of the measured cell potential. The titration of two [2Fe-2S]2+ clusters is fit by two summed Nernst curves (eq 1) with midpoint potentials of -140 ± 40 and -430 ± 30 mV. The uncertainty represents variability between experiments.
BioB Can Be Reconstituted to Protein Containing Two [4Fe-4S]2+ Clusters per Monomer. Reduction of BioB with dithionite in 60% ethylene glycol results in the conversion of [2Fe-2S]2+ clusters to [4Fe-4S]2+ clusters (19). Addition of FeCl3 and Na2S results in an increase in the yield of [4Fe-4S]2+ clusters. We incubated BioB with FeCl3, Na2S, and DTT in anaerobic 60% ethylene glycol and 100 mM Tris-HCl, pH 8, and added dithionite. Following a 4 h incubation, we removed excess reagents by gel filtration chromatography in a nitrogen glovebox. The resulting [4Fe-4S]2+ protein has a broad absorption band from 380 to 420 nm with an extinction coefficient of ε400 = 30 000 ± 2000 M-1 cm-1 per monomer. Fe and S analyses indicate the presence of 7.5 ± 1.0 Fe atoms and 8.2 ± 0.5 S atoms per monomer. We propose that artificial reconstitution under the conditions described results in both of the initial [2Fe-2S]2+ cluster sites becoming filled with [4Fe-4S]2+ clusters.
When we then titrated BioB containing two [4Fe-4S]2+ clusters per monomer with dithionite (Figure 2A), we found that the clusters were reduced with two barely distinguishable midpoint potentials of -440 and -505 mV (Figure 2B). The presence of two separate midpoint potentials is apparent when the spectral changes from 420 to 480 nm are compared to those from 550 to 625 nm. For comparison, the spectral changes at 420 and 550 nm are normalized in Figure 2B; the decrease in absorbance at 550 nm (squares) is fit to a single low-midpoint potential of -505 mV, while the decrease in absorbance at 420 nm (triangles) is best fit to two potentials of -440 and -505 mV. The difference between data at these two wavelengths (circles) emphasizes the presence of two electrochemically distinct clusters. The spectral changes associated with the first and second wave of reduction (Figure 2A, inset) again show slight spectroscopic differences between the two [4Fe-4S]2+ clusters in the region from 525 to 650 nm. It should be noted that methyl viologen, used as an electrochemical mediator, contributes slightly to the spectrum at low potential with a sharp band at 395 nm and a broad band centered around 600 nm. However, the spectral changes due to cluster reduction are much larger than those predicted from the low concentrations of methyl viologen present in these titrations.
Figure 2:
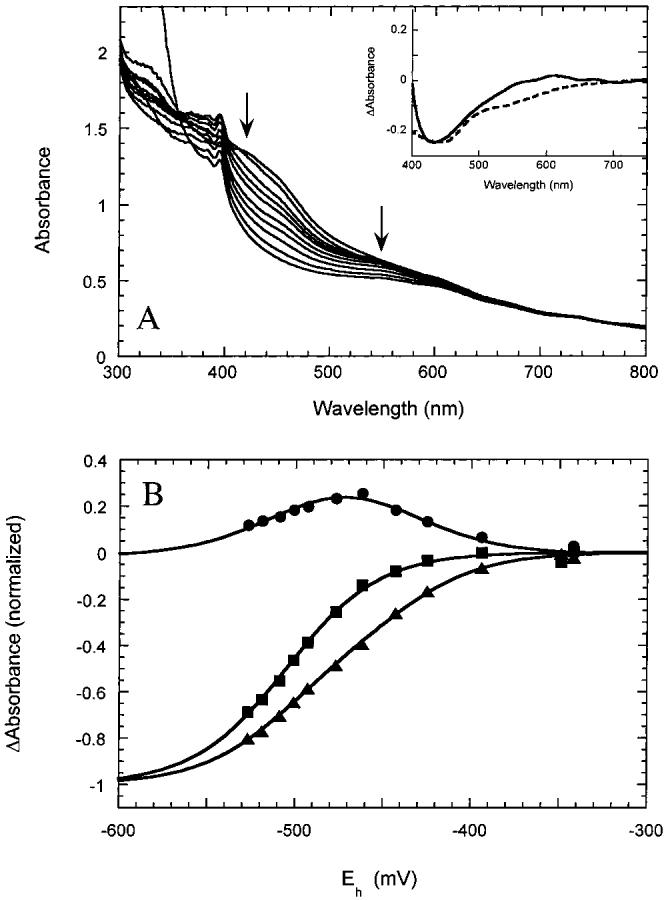
Reduction of BioB containing [4Fe-4S]2+ clusters. (A) UV/visible spectra of BioB (∼70 μM monomer) containing two [4Fe-4S]2+ clusters per monomer in anaerobic 50 mM Tris-HCl and 100 mM NaCl, pH 8.0, were recorded as the potential is lowered by titration with dithionite. The [4Fe-4S]2+ clusters were initially generated by reductive reassembly in the presence of iron and sulfide as described in Materials and Methods. The decrease in absorbance from 400 to 550 nm is associated with reduction of [4Fe-4S]2+ clusters to [4Fe-4S]+ clusters. Small peaks observed at 395 and 590 nm are due to the presence of reduced methyl viologen. (Inset) Difference spectra associated with the first wave of reduction (solid curve) and the second wave of reduction (dashed curve). (B) The spectral changes at 420 nm (triangles) and 550 nm (squares) are plotted after normalizing by dividing by the total extrapolated absorbance change after complete reduction. The data at 550 nm (squares) are fit to a single Nernst curve with a midpoint potential of -505 mV, while the data at 420 nm (triangles) are fit to the sum of two Nernst curves with potentials of -440 and -505 mV. The difference between these spectral changes (circles) emphasizes the presence of two waves of reduction, again fit to potentials of -440 and -505 mV.
BioB can also be prepared that contains approximately one [4Fe-4S]2+ cluster per monomer by incubation of the oxidized protein with dithionite in anaerobic 60% ethylene glycol (19, 21). When we titrated BioB containing one [4Fe-4S]2+ cluster, we observed a single midpoint potential of -440 ± 20 mV (Figure 3). This potential is identical to the higher potential observed when two clusters are present, suggesting that only one of the two potential cluster binding sites preferentially binds the [4Fe-4S]2+ cluster.
Figure 3:
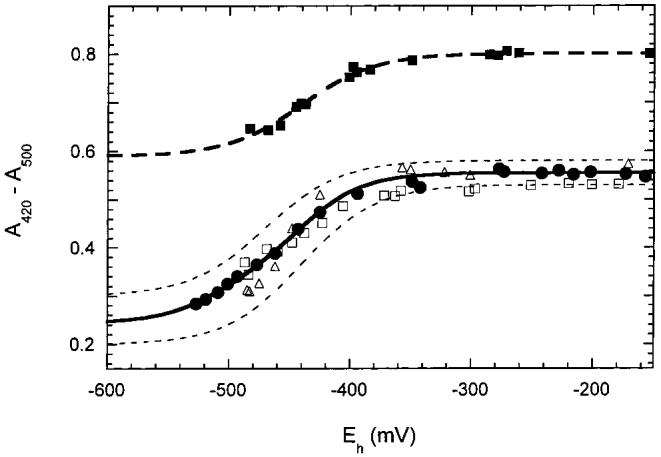
The absorbance changes due to reduction of [4Fe-4S]2+ clusters were followed at varying pH and for partially reconstituted protein. In this case, the difference in absorbance between 420 and 500 nm is plotted to reduce error due to instrument instability over the prolonged titration times required. The lower curve shows data for reduction of two [4Fe-4S]2+ clusters per monomer at pH 7.5 (open triangles), 8.0 (open squares), and 8.5 (solid circles). The upper curve shows data for one [4Fe-4S]2+ cluster per monomer at pH 7.5 (solid squares) shifted arbitrarily along the y-axis to facilitate replotting on the same graph. The titration of two [4Fe-4S]2+ clusters is fit by the solid curve with two midpoint potentials of -440 and -505 mV; dashed curves show an approximate error of ± 20 mV as well as a range of final absorbance values. The titration of one [4Fe-4S]2+ cluster per dimer is fit by the dashed curve with a single midpoint potential of -440 mV.
Reduction of redox cofactors is often accompanied by protonation of the cofactor or a nearby protein residue to compensate for a change in the electrostatic charge of the cofactor. This can lead to a pH dependence of the reduction potential; examples of pH-dependent midpoint potentials for iron-sulfur clusters include the Rieske protein from the bc1 complex and one cluster in complex I (34, 35). Since we envision that the [4Fe-4S]2+ to [4Fe-4S]+ reduction is critical to the mechanism of radical generation in biotin synthase, we examined whether the reduced state was stabilized by protonation over the physiologic pH range. We generated BioB containing two [4Fe-4S]2+ clusters at pH 7.5 and then exchanged into buffers of varying pH by passing the protein sample through desalting columns equilibrated with Bis-Tris propane or Tris-HCl buffers over the pH range 6.5-9.0. We find no evidence of a pH-dependent midpoint potential. Representative data for pH 7.5, 8.0, and 8.5 are shown in Figure 3, and outside the typical scatter of the data, there are no consistent changes. Interestingly, we did observe differences in the stability of [4Fe-4S]2+ clusters over this pH range. When the sample was exchanged into low-pH buffer (pH <7), it was generally much more prone to oxidative cluster destruction, even in the low oxygen environment of the nitrogen glovebox. Conversely, exchange into high-pH buffer (pH >8) resulted in improved stability.
Effect of 60% Ethylene Glycol on Observed Reduction Potentials. Ethylene glycol and glycerol are commonly used as cryoprotectants and glassing agents in the preparation of frozen protein samples for spectroscopy. It was the preparation of frozen glasses for resonance Raman and magnetic circular dichroism spectroscopy that initially led to the observation of reductive cluster conversion in BioB (19). We routinely observe that incubation with excess dithionite in 60% ethylene glycol results in formation of predominantly [4Fe-4S]2+ clusters, while in aqueous buffer incubation with dithionite results in complete reduction to [4Fe-4S]+ clusters. We reasoned that this could be due to an effect of ethylene glycol upon the midpoint potential of the [4Fe-4S]2+ protein. To examine this, we produced BioB containing two [4Fe-4S]2+ clusters per monomer and, after desalting to remove excess iron and sulfide, reintroduced the protein into 60% ethylene glycol, 50 mM Tris-HCl, and 100 mM NaCl, pH 7.5. This protein sample was then titrated with dithionite, allowing ∼5 min equilibration between additions due to the high viscosity of the buffer and the resulting slow electrochemical equilibration. We observed only slight reduction of the [4Fe-4S]2+ clusters present in the protein sample down to a cell potential of -500 mV (Figure 4). In control experiments, methyl and benzyl viologen in 60% ethylene glycol, 50 mM Tris-HCl, and 100 mM NaCl, pH 7, were titrated with dithionite, and the midpoint potentials obtained were within ± 20 mV of the literature values in the absence of ethylene glycol, indicating that ethylene glycol does not significantly impair the performance of the electrodes. The failure to observe reduction of the [4Fe-4S]2+ clusters is consistent with a shift in the midpoint potentials to at least -550 mV in 60% ethylene glycol. We suggest that this shift of >100 mV in the midpoint potential is due to one of two potential effects. First, BioB may exist in at least two alternate conformations, and it is possible that ethylene glycol has altered the equilibrium population of these conformations. A similar effect has been observed for methionine synthase, where glycerol favors a conformation that is inactive for methionine production but is associated with flavodoxin binding and electron transfer (36). Alternately, the iron-sulfur cluster in BioB is bound by only three cysteine ligands, with the fourth ligation site potentially occupied by water (19), and it is possible that ethylene glycol binds to this coordination site as a glycolate ligand and directly alters the physical properties of the [4Fe-4S]2+ cluster.
Figure 4:
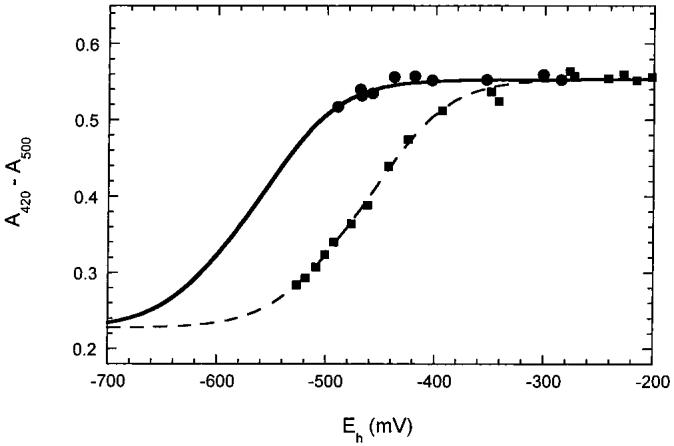
Titration of BioB (∼70 μM) containing two [4Fe-4S]2+ clusters with dithionite in 60% ethylene glycol (solid circles). Spectral changes due to cluster reduction were followed at 420 nm and corrected as in Figure 3 by subtraction of the absorbance at 500 nm. The [4Fe-4S]2+ clusters were not significantly reduced even at -500 mV; the slight decrease in absorbance allows us to estimate an upper limit for the midpoint potential of -550 mV. For comparison, the data from Figure 3 at pH 8.5 are shown fit to midpoint potentials of -440 and -505 mV. We were not able to obtain lower cell potentials in 60% ethylene glycol, despite using a large excess of dithionite.
Biotin Synthase Contains both [2Fe-2S]2+ and [4Fe-4S]2+ Clusters under in Vitro Assay Conditions. Typical in vitro biotin synthase assays contain both DTT and NADPH as potential reductants (5). When we perform our typical in vitro assay (37) in an electrochemical cell with the appropriate mediators, we observe an electrochemical potential of -330 mV, varying by less than ± 20 mV over the full time course of the assay. The electrochemical titrations described in the sections above suggest that one of the [2Fe-2S]2+ clusters in BioB is stable at potentials as low as -400 mV, and thus should be observable during an assay by characteristic features in the UV/visible spectrum, while the other cluster will be reductively converted to a [4Fe-4S]2+ cluster. We incubated BioB dimer initially containing [2Fe-2S]2+ clusters with catalytic amounts of flavodoxin, flavodoxin reductase, and excess AdoMet, NADPH, DTT, FeCl3, and Na2S for 30 min at 25 °C. Dethiobiotin was not added in order to prevent turnover of the enzyme. The UV/visible spectrum of the protein was altered over several minutes with a general increase in absorbance at all wavelengths and the appearance of an absorption band at 410 nm that is consistent with conversion of a [2Fe-2S]2+ cluster to a [4Fe-4S]2+ cluster. The protein was repurified under anaerobic conditions by ion-exchange chromatography and retains this mixed cluster spectrum (Figure 5, solid curve), with absorption bands at ∼410 and 460 nm that suggest the presence of both [4Fe-4S]2+ and [2Fe-2S]2+ clusters, respectively. The extinction coefficients, ε410 = 14 900 M-1 cm-1 and ε460 = 13 200 M-1 cm-1, are consistent with the presence of one of each cluster per monomer, and this spectrum is reasonably well fit with a spectrum simulated to a 1:1 mixture of [2Fe-2S]2+ and [4Fe-4S]2+ clusters (Figure 5, dotted curve). Fe and S analyses indicate the presence of 5.8 ± 0.3 Fe and 6.2 ± 0.2 S atoms per monomer. As expected, EPR spectroscopy did not show any EPR-detectable cluster forms.
Figure 5:
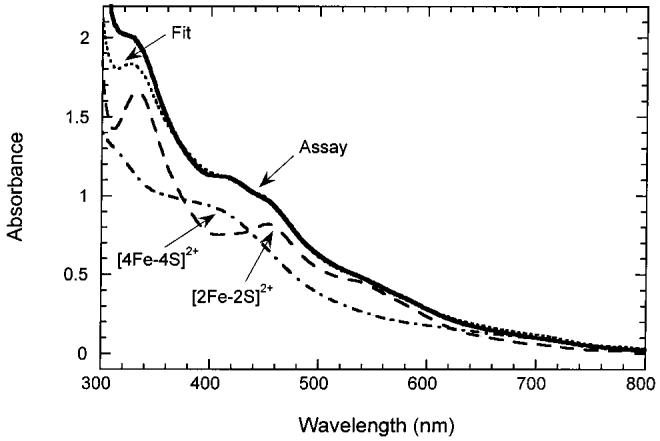
UV/visible spectrum of BioB before (long dash) and after (solid) incubation under in vitro assay conditions. BioB (100 μM) initially contained ∼1.5 [2Fe-2S]2+ clusters per monomer. Addition of DTT (10 mM), FeCl3 (400 μM), Na2S (400 μM), AdoMet (500 μM), flavodoxin (1 μM), flavodoxin reductase (1 μM), and NADPH (1 mM) under argon resulted in an increase in absorbance at ca. 410 nm, consistent with the formation of a [4Fe-4S]2+ cluster. Following anaerobic repurification, the spectrum (s) shows bands at 320 and 452 nm that are characteristic of a remaining [2Fe-2S]2+ cluster and at 410 nm characteristic of a [4Fe-4S]2+ cluster. The spectrum is fit to a 1:1 mixture of [2Fe-2S]2+ and [4Fe-4S]2+ clusters (dotted); a scaled spectrum of BioB containing one [4Fe-4S]2+ cluster (long/short dash) is shown for comparison.
We have found that the minimal requirements for formation of this mixed cluster state are incubation with FeCl3, Na2S, and DTT for 30-60 min at 25 °C. The protein can then be repurified by anaerobic gel filtration chromatography and is then resistant to degradation under sustained anaerobic conditions. When BioB containing one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer is titrated with dithionite and the electrochemical potential monitored, we observe that both clusters are stable from -100 to -400 mV but are eventually reduced with indistinguishable midpoint potentials of -430 ± 20 mV (Figure 6). Although the [2Fe-2S]2+ and [4Fe-4S]2+ clusters are apparently reduced in parallel, the data are best fit assuming an independent noncooperative reduction of each cluster (n = 1 in the Nernst equation). The relatively low reduction potentials for both clusters in this mixed cluster state suggest that BioB containing one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer would be the predominant stable enzyme form in both in vitro assays and potentially in vivo. We propose that BioB containing one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer is the native enzyme form but that aerobic purification results in degradation of the [4Fe-4S]2+ cluster to a partially saturated [2Fe-2S]2+ cluster. This interpretation is consistent with activity data presented in the accompanying paper (37).
Figure 6:
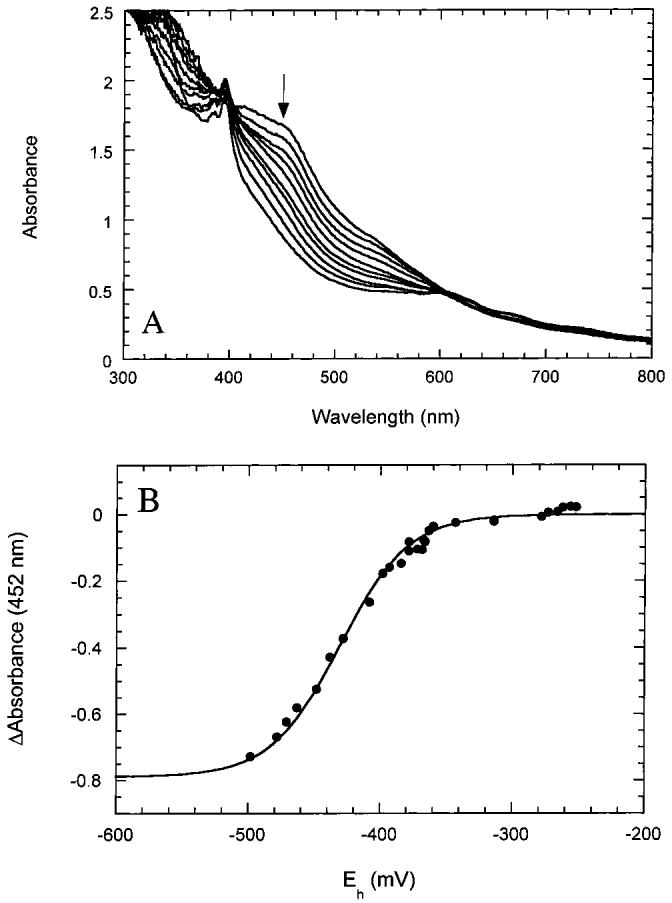
Titration of BioB containing one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer. BioB containing a 1:1 mixture of clusters was prepared and repurified as described in Materials and Methods. (A) UV/visible spectra were recorded as the cell potential was lowered by the addition of dithionite. The absorbance from 420 to 550 nm decreased in parallel, suggesting simultaneous reduction of [2Fe-2S]2+ and [4Fe-4S]2+ clusters. The sharp absorbance at 395 nm is due to reduced methyl viologen. (B) The spectral changes at 452 nm were plotted as a function of the measured cell potential and are fit to a single Nernst curve with Em ± -430 ± 20 mV. The uncertainty represents variability between experiments. Analysis at several other wavelengths gives similar results, suggesting that the [2Fe-2S]2+ and [4Fe-4S]2+ clusters are reduced in parallel.
DISCUSSION
Biotin synthase catalyzes the reductive cleavage of AdoMet, generating methionine and 5′-deoxyadenosine (38). Similar chemistry is catalyzed by several AdoMet-dependent radical enzymes, including pyruvate formate-lyase (39), anaerobic ribonucleotide reductase (40), lysine 2,3-aminomutase (17, 41), and lipoyl synthase (12), and may be catalyzed by benzylsuccinate synthase (13). All of these enzymes contain air-sensitive iron-sulfur clusters that are thought to be intimately involved in the AdoMet cleavage chemistry. The iron-sulfur cluster may serve two mechanistic roles: first, it probably serves as the immediate acceptor of an electron from flavodoxin in E. coli (or a similar native electron donor in other species), and second, interaction of the cluster with AdoMet may assist in labilizing the relatively stable sulfonium center toward one-electron reductive decomposition (15, 42). The product of C-S bond cleavage, the 5′-deoxyadenosyl radical, is a very strong oxidant that is essential for generating protein or substrate radicals in each of these enzymes.
Unlike other AdoMet-dependent radical enzymes, the iron-sulfur clusters in biotin synthase may fulfill an additional role as the sulfur-donating substrate for formation of the thioether ring in the product. Consistent with this hypothesis, BioB that is reconstituted with [34S]sulfide (23) or expressed in media containing [35S]cysteine (24) results in enzyme that incorporates high levels of isotopic label into biotin in the absence of added exogenous sulfur donors. Further, addition to in vitro assays of NifS, a protein that catalyzes sulfide incorporation into iron-sulfur clusters, allows high-level transfer of 35S from cysteine to biotin (43). However, addition of NifS or NifS/NifU fails to result in multiple turnovers (43, 44). Thus, although the iron-sulfur clusters may serve as a source of sulfide for biotin formation, it is not clear how the enzyme is regenerated for multiple turnovers.
The behavior of the iron-sulfur clusters in biotin synthase is certainly not typical of other cluster-containing proteins. For example, iron-sulfur proteins involved in pure electron transfer such as ferredoxins tend to have a single stable cluster configuration that is not altered upon reduction. In contrast, AdoMet-dependent radical enzymes apparently share a flexible cluster binding site that can contain [2Fe-2S], [3Fe-4S], and [4Fe-4S] clusters, depending upon the availability of iron and sulfide and the redox potential of the system. One of the unique challenges in working with these enzymes is identifying from among these possibilities the active cluster configuration.
In our hands, biotin synthase is purified containing ca. 2.5-3 Fe and 3-4 S2- atoms per monomer, and one might assume experimental error and estimate one [2Fe-2S]2+ cluster per monomer. However, several experimental observations suggest this is not the case: first, electrochemical analysis of [2Fe-2S]2+ cluster reduction indicates that two clusters with different redox properties are present in ca. 2:1 ratio (Figure 1). Second, incubation of the oxidized protein with FeCl3 followed by anaerobic gel filtration chromatography results in an increase to ∼3.7 Fe atoms per monomer, consistent with an increase to ∼1.9 [2Fe-2S]2+ clusters per monomer and an increase in ε452 to 11 300 M-1 cm-1. Finally, reconstitution under reducing conditions in the presence of excess Fe3+ and S2- results in an increase to ∼6-8 Fe and 7-9 S2- atoms per monomer, again consistent with two clusters per monomer. Previous analyses of the aerobically purified protein have resulted in Fe contents that range from 0.96 to 2.1 Fe atoms per monomer (23, 29, 30), and S2- content of 1.4-2.0 per monomer (23, 29). Reconstitution of apoprotein under anaerobic conditions followed by desalting resulted in ∼3.8 Fe atoms per monomer (22). Although we can only speculate about potential reasons for the discrepancy between these data and the present study, we would suggest that the [4Fe-4S]2+ cluster in biotin synthase is oxidatively degraded during purification, resulting in initial formation of a partially saturated [2Fe-2S]2+ cluster, but that even this cluster is lost upon prolonged exposure to air. However, a second [2Fe-2S]2+ cluster bound at a different site is air-stable and is preserved throughout the purification.
The electrochemical behavior of BioB upon exposure to chemical reductants is complex, with conversions between different cluster forms, and we find it useful to think about this property in terms of cluster “phase” transitions (Figure 7). One of the [2Fe-2S]2+ clusters within BioB has a high reduction potential (-140 mV) that is within range of numerous in vivo reducing systems and upon reduction undergoes a transition to an EPR-silent cluster that we suspect is a [4Fe-4S]2+ cluster. The midpoint potential for reduction of the remaining [2Fe-2S]2+ cluster is much lower (-430 mV) and upon reduction also undergoes a transition to a [4Fe-4S]2+/+ cluster. The midpoint potentials for reduction of the [4Fe-4S]2+ clusters are also low (-440 and -505 mV) and appear to be associated with reversible reduction to [4Fe-4S]+ clusters. The relatively easy reduction of one [2Fe-2S]2+ cluster suggests, assuming sufficient Fe and S2- are available, that as the electrochemical potential experienced by BioB is lowered (Figure 7), one of the [2Fe-2S]2+ clusters in the dimer will undergo cluster reassembly to a [4Fe-4S] cluster at high potential (-140 to -250 mV), while the second [2Fe-2S]2+ cluster in the dimer will remain stable until a much lower potential (-430 mV). Conversely, [4Fe-4S]2+ clusters in BioB are not reduced until even lower potentials of -440 and -505 mV. The effective redox potential experienced by BioB in aerobic E. coli in exponential growth is probably defined by the NADP+/NADPH ratio ≈ 1 (45), giving an approximate cell potential of -330 ± 30 mV, similar to the potential measured in our assays (-330 ± 20 mV). Under these electrochemical conditions, and provided that sufficient iron and sulfide are available, we predict that BioB should exist predominantly containing one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer (Figure 7). Consistent with this proposal, protein incubated under in vitro assay conditions in the absence of dethiobiotin exhibits a UV/visible spectrum that indicates an approximate 1:1 mixture of [2Fe-2S]2+ and [4Fe-4S]2+ clusters (Figure 6). These clusters are only reduced at very low potentials of ca. -450 mV, suggesting that this mixed cluster form of the enzyme would be resistant to further reductive cluster conversion. Clearly UV/visible spectroscopy is a fairly crude measure of cluster states, and it is certainly possible that other cluster forms are present in minor amounts. Recent studies of biotin synthase using Mossbauer spectroscopy have demonstrated varying mixtures of [2Fe-2S]2+, [4Fe-4S]2+, and [4Fe-4S]+ clusters, depending upon the sample preparation method and extent of reduction (20, 22). Trautwein and co-workers report that one [2Fe-2S]2+ cluster in biotin synthase is resistant to dithionite reduction (20), consistent with our electrochemical results that indicate the stabilization of one [2Fe-2S]2+ cluster.
Figure 7:
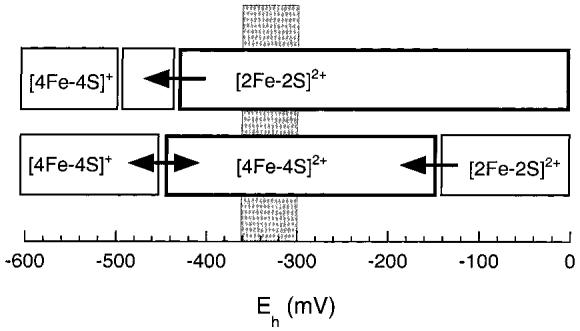
Diagram that shows the cluster transitions in BioB associated with decreasing redox potentials. The upper bar represents the “stable” [2Fe-2S]2+ cluster that is reductively reassembled to a [4Fe-4S] cluster only at very low potential. The lower bar represents the “labile” [2Fe-2S]2+ cluster that is converted to a [4Fe-4S]2+ cluster at a potential of -140 mV. Further reduction to [4Fe-4S]+ clusters is predicted at -450 and -505 mV. The electrochemical potential experienced by BioB in anaerobic assays and in aerobic E. coli (-330 ± 30 mV) is represented by the gray bar, showing that the stable enzyme form in this region is predicted to contain one [2Fe-2S]2+ and one [4Fe-4S]2+ cluster.
The nature of the in vivo active iron-sulfur clusters in biotin synthase remains unknown. The overexpressed enzyme is inactive in the absence of added iron and reductants, and therefore we conclude that the active state of the enzyme contains clusters that are assembled by the addition of iron and sulfide to the initial [2Fe-2S]2+ clusters. In vitro, the dimeric enzyme stabilizes a 1:1 mixture of [2Fe-2S]2+ and [4Fe-4S]2+ clusters over a wide potential range. The case in vivo would be similar if access to iron, sulfide, and reductants were unrestricted; however, it is becoming increasingly evident that in many instances iron-sulfur cluster assembly is mediated by specific enzymes [e.g., IscS and IscU or homologues in E. coli (32, 46)]. In the following paper, we show that biotin synthase containing a 1:1 mixture of [2Fe-2S]2+ and [4Fe-4S]2+ clusters is the active enzyme form and that turnover results in the selective destruction of the [2Fe-2S]2+ cluster (37). Clearly, a better understanding of the mechanisms of in vivo iron-sulfur cluster assembly in BioB is essential to furthering our understanding of the radical-mediated sulfur insertion catalyzed by biotin synthase.
ACKNOWLEDGMENT
We thank Dr. Takahiro Yano and Dr. Tomoko Ohnishi (University of Pennsylvania) for advice and assistance with EPR experiments.
Footnotes
This research has been supported by NIH Research Grant GM59175 and the Thomas B. and Jeanette E. Laws McCabe Fund.
- AdoMet
- S-adenosyl-l-methionine
- anRR
- anaerobic (class III) ribonucleotide reductase
- BioB
- biotin synthase
- Bis-Tris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- DTT
- dithiothreitol
- EPR
- electron paramagnetic resonance
- LAM
- lysine 2,3-aminomutase
- NADPH
- nicotinamide adenine dinucleotide phosphate, reduced form
- PFL AE
- pyruvate formate-lyase activating enzyme
- Tris-HCl
- tris(hydroxymethyl)aminomethane hydrochloride
REFERENCES
- 1.Cleary PP, Campbell A. J. Bacteriol. 1972;112:830–839. doi: 10.1128/jb.112.2.830-839.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rolfe B, Eisenberg MA. J. Bacteriol. 1968;96:515–524. doi: 10.1128/jb.96.2.515-524.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mejean A, Bui BT, Florentin D, Ploux O, Izumi Y, Marquet A. Biochem. Biophys. Res. Commun. 1995;217:1231–1237. doi: 10.1006/bbrc.1995.2900. [DOI] [PubMed] [Google Scholar]
- 4.Birch OM, Fuhrmann M, Shaw NM. J. Biol. Chem. 1995;270:19158–19165. doi: 10.1074/jbc.270.32.19158. [DOI] [PubMed] [Google Scholar]
- 5.Sanyal I, Gibson KJ, Flint DH. Arch. Biochem. Biophys. 1996;326:48–56. doi: 10.1006/abbi.1996.0045. [DOI] [PubMed] [Google Scholar]
- 6.Ifuku O, Kishimoto J, Haze S, Yanagi M, Fukushima S. Biosci. Biotechnol. Biochem. 1992;56:1780–1785. doi: 10.1271/bbb.56.1780. [DOI] [PubMed] [Google Scholar]
- 7.Ifuku O, Koga N, Haze S, Kishimoto J, Wachi Y. Eur. J. Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 8.Ohshiro T, Yamamoto M, Bui BT, Florentin D, Marquet A, Izumi Y. Biosci. Biotechnol. Biochem. 1995;59:943–944. doi: 10.1271/bbb.59.943. [DOI] [PubMed] [Google Scholar]
- 9.Rodel W, Plaga W, Frank R, Knappe J. Eur. J. Biochem. 1988;177:153–158. doi: 10.1111/j.1432-1033.1988.tb14356.x. [DOI] [PubMed] [Google Scholar]
- 10.Sun X, Eliasson R, Pontis E, Andersson J, Buist G, Sjoberg BM, Reichard P. J. Biol. Chem. 1995;270:2443–2446. doi: 10.1074/jbc.270.6.2443. [DOI] [PubMed] [Google Scholar]
- 11.Ruzicka FJ, Lieder KW, Frey PA. J. Bacteriol. 2000;182:469–476. doi: 10.1128/jb.182.2.469-476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Marletta MA. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 13.Leuthner B, Leutwein C, Schulz H, Horth P, Haehnel W, Schiltz E, Schagger H, Heider J. Mol. Microbiol. 1998;28:615–628. doi: 10.1046/j.1365-2958.1998.00826.x. [DOI] [PubMed] [Google Scholar]
- 14.Frey PA. FASEB J. 1993;7:662–670. doi: 10.1096/fasebj.7.8.8500691. [DOI] [PubMed] [Google Scholar]
- 15.Cosper NJ, Booker SJ, Ruzicka F, Frey PA, Scott RA. Biochemistry. 2000;39:15668–15673. doi: 10.1021/bi0022184. [DOI] [PubMed] [Google Scholar]
- 16.Magnusson OT, Reed GH, Frey PA. J. Am. Chem. Soc. 1999;121:9764–9765. [Google Scholar]
- 17.Baraniak J, Moss ML, Frey PA. J. Biol. Chem. 1989;264:1357–1360. [PubMed] [Google Scholar]
- 18.Escalettes F, Florentin D, Tse Sem Bui B, Lesage D, Marquet A. J. Am. Chem. Soc. 1999;121:3571–3578. [Google Scholar]
- 19.Duin EC, Lafferty ME, Crouse BR, Allen RM, Sanyal I, Flint DH, Johnson MK. Biochemistry. 1997;36:11811–11820. doi: 10.1021/bi9706430. [DOI] [PubMed] [Google Scholar]
- 20.Tse Sum Bui B, Florentin D, Marquet A, Benda R, Trautwein AX. FEBS Lett. 1999;459:411–414. doi: 10.1016/s0014-5793(99)01300-9. [DOI] [PubMed] [Google Scholar]
- 21.Ugulava NB, Gibney BR, Jarrett JT. Biochemistry. 2000;39:5206–5214. doi: 10.1021/bi9926227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ollagnier-De Choudens S, Sanakis Y, Hewitson KS, Roach P, Baldwin JE, Munck E, Fontecave M. Biochemistry. 2000;39:4165–4173. doi: 10.1021/bi992090u. [DOI] [PubMed] [Google Scholar]
- 23.Tse Sum Bui B, Florentin D, Fournier F, Ploux O, Mejean A, Marquet A. FEBS Lett. 1998;440:226–230. doi: 10.1016/s0014-5793(98)01464-1. [DOI] [PubMed] [Google Scholar]
- 24.Gibson KJ, Pelletier DA, Turner IM., Sr. Biochem. Biophys. Res. Commun. 1999;254:632–635. doi: 10.1006/bbrc.1998.9991. [DOI] [PubMed] [Google Scholar]
- 25.Fish WW. Methods Enzymol. 1988;158:357–364. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 26.Beinert H. Anal. Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 27.Broderick JB, Henshaw TF, Cheek J, Wojtuszewski K, Smith SR, Trojan MR, McGhan RM, Kopf A, Kibbey M, Broderick WE. Biochem. Biophys. Res. Commun. 2000;269:451–456. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 28.Dutton PL. Methods Enzymol. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 29.Sanyal I, Cohen G, Flint DH. Biochemistry. 1994;33:3625–3631. doi: 10.1021/bi00178a020. [DOI] [PubMed] [Google Scholar]
- 30.McIver L, Baxter RL, Campopiano DJ. J. Biol. Chem. 2000;275:13888–13894. doi: 10.1074/jbc.275.18.13888. [DOI] [PubMed] [Google Scholar]
- 31.Orme-Johnson WH, Orme-Johnson NR. In: Iron-Sulfur Proteins. Spiro TG, editor. John Wiley & Sons; New York: 1982. pp. 67–96. [Google Scholar]
- 32.Agar JN, Krebs C, Frazzon J, Huynh BH, Dean DR, Johnson MK. Biochemistry. 2000;39:7856–7862. doi: 10.1021/bi000931n. [DOI] [PubMed] [Google Scholar]
- 33.Hurley JK, Weber-Main AM, Hodges AE, Stankovich MT, Benning MM, Holden HM, Cheng H, Xia B, Markley JL, Genzor C, Gomez-Moreno C, Hafezi R, Tollin G. Biochemistry. 1997;36:15109–15117. doi: 10.1021/bi972001i. [DOI] [PubMed] [Google Scholar]
- 34.Ohnishi T. Biochim. Biophys. Acta. 1998;1364:186–206. doi: 10.1016/s0005-2728(98)00027-9. [DOI] [PubMed] [Google Scholar]
- 35.Ugulava NB, Crofts AR. FEBS Lett. 1998;440:409–413. doi: 10.1016/s0014-5793(98)01493-8. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee RV, Harder SR, Ragsdale SW, Matthews RG. Biochemistry. 1990;29:1129–1135. doi: 10.1021/bi00457a005. [DOI] [PubMed] [Google Scholar]
- 37.Ugulava NB, Sacanell CJ, Jarrett JT. Biochemistry. 2001;40:8352–8358. doi: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guianvarc'h D, Florentin D, Tse Sum Bui B, Nunzi F, Marquet A. Biochem. Biophys. Res. Commun. 1997;236:402–406. doi: 10.1006/bbrc.1997.6952. [DOI] [PubMed] [Google Scholar]
- 39.Knappe J, Schmitt T. Biochem. Biophys. Res. Commun. 1976;71:1110–1117. doi: 10.1016/0006-291x(76)90768-3. [DOI] [PubMed] [Google Scholar]
- 40.Harder J, Eliasson R, Pontis E, Ballinger MD, Reichard P. J. Biol. Chem. 1992;267:25548–25552. [PubMed] [Google Scholar]
- 41.Moss M, Frey PA. J. Biol. Chem. 1987;262:14859–14862. [PubMed] [Google Scholar]
- 42.Lieder KW, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. Biochemistry. 1998;37:2578–2585. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- 43.Tse Sum Bui B, Escalettes F, Chottard G, Florentin D, Marquet A. Eur. J. Biochem. 2000;267:2688–2694. doi: 10.1046/j.1432-1327.2000.01284.x. [DOI] [PubMed] [Google Scholar]
- 44.Kiyasu T, Asakura A, Nagahashi Y, Hoshino T. J. Bacteriol. 2000;182:2879–2885. doi: 10.1128/jb.182.10.2879-2885.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunlap RB, Harding NGL, Huennekens FM. Biochemistry. 1971;10:88–97. doi: 10.1021/bi00777a014. [DOI] [PubMed] [Google Scholar]
- 46.Flint DH. J. Biol. Chem. 1996;271:16068–16074. [PubMed] [Google Scholar]