

Abstract
Macrophages are specialized cells of the immune system that exhibit a prodigious capacity for phagocytosis. The ability of macrophages to internalize a substantial proportion of their plasma membrane during phagocytosis indicates that they possess a mechanism for the rapid renewal of plasma membrane. We examined the role of endocytic membrane recycling in promoting phagocytosis. In contrast to many other cell types, macrophages lack a morphologically distinct peri-centriolar recycling compartment but instead demonstrate an extensive network of transferrin receptor-positive tubules and vesicles that participated in recycling. The rate of transferrin recycling in thioglycollate-elicited murine peritoneal macrophages (thio-macrophages) was exceedingly rapid, with exocytic rate constants that were 2- to 3-fold higher than those of most other cells. Because the GTPase Rab11 has been implicated in transferrin recycling in other cells, we determined its role in transferrin recycling and phagocytosis in macrophages. Macrophages expressing epitope-tagged Rab11 demonstrated the presence of Rab11 in several intracellular membrane compartments, including endosomes and nascent phagosomes. Expression of Rab11 25N, a GTP binding-deficient allele of Rab11, led to a decreased rate of transferrin efflux and impaired FcγR-mediated phagocytosis, where FcγR is the receptor for the Fc portion of IgG. In contrast, expression of Rab11 70L, a GTPase-deficient allele of Rab11, led to an increased rate of transferrin efflux and enhanced phagocytosis. We conclude that macrophages have adapted a rapidly mobilizable, endocytic compartment to enhance phagocytosis. Rab11 participates in the recruitment of this compartment to the macrophage cell surface.
Although many cells have the capacity to ingest a wide variety of phagocytic targets, macrophages are particularly adept at particle engulfment. Macrophages internalize the equivalent of a significant percentage of their membrane surface area during phagocytosis, implying that they must replenish cell surface membrane from intracellular pools. For example, the average surface area of adherent thioglycollate-elicited murine peritoneal macrophages (thio-macrophages) has been estimated to be 1,367 μm2 (1). Each thio-macrophage is capable of ingesting an average of 10–30 sheep erythrocytes opsonized with rabbit anti-sheep IgG (EIgG) within 30 min (2). The surface area of a single sheep erythrocyte is approximately 66 μm2 (3, 4). Therefore, thio-macrophages are capable of ingesting the equivalent of 48–145% of their surface area during 30 min of FcγR-mediated phagocytosis, where FcγR is the receptor for the Fc portion of IgG. Potential sources of intracellular membrane required for phagocytosis include endosomes, lysosomes, and components of the Golgi apparatus. A recent study using bacterial toxins that cleave vesicle-soluble N-ethylmaleimide-sensitive factor attachment protein receptors (v-SNAREs) indicates that a v-SNARE-containing intracellular membrane compartment is a likely source of membrane required for phagocytosis (5). Recruitment of lysosomes, while necessary for trypanosome invasion (6), is not required for FcγR-mediated phagocytosis (7) and microbial pathogens that evade lysosomal fusion, such as Mycobacterium tuberculosis (8) and Legionella pneumophila (9), are avidly ingested by macrophages. Brefeldin A does not block FcγR-mediated phagocytosis (10), suggesting that the Golgi apparatus is not an immediate source of membrane required for phagocytosis and pseudopod extension. Several lines of evidence suggest that endosomes may contribute to membrane renewal during phagocytosis. The macrophage capacity for endocytosis is high compared with other cells (11), implying that macrophages contain an abundance of endosomal membrane that may participate in recycling. Endosomes accumulate beneath nascent pseudopods during phagocytosis (12), and endosome-phagosome fusion has been documented during early stages of FcγR-mediated phagocytosis (13).
The mechanisms governing endocytic trafficking have been intensively studied by several groups using labeled transferrin as a marker of bulk membrane flow (reviewed in refs. 14 and 15). After binding to cell surface receptors, transferrin undergoes endocytosis, entering a compartment (the sorting endosome) that either directly recycles back to the cell surface (16, 17), or merges with a peri-centriolar compartment that also recycles to the cell surface (14, 15). At steady state, approximately 80% of the interiorized transferrin is localized to the peri-centriolar recycling compartment (18). The kinetics of transferrin recycling consists of fast and slow components (15, 19, 20); depending on the cell type, the t1/2 for recycling of labeled transferrin is typically between 4 and 12.5 min (17, 21–24). A recent analysis suggests that direct recycling from early/sorting endosomes is more rapid than that of recycling endosomes in MDCK cells (25). The molecular regulation of recycling from either compartment is poorly understood, but apparently involves Rab11, a member of a large family of GTPases that are thought to promote tethering of vesicles before fusion (26). Rab11 originally was identified in post-Golgi membranes in secretory cells (27, 28) but also localizes to transferrin receptor-containing recycling compartments in a variety of cells (29–32). Expression of a GTP binding-deficient allele of Rab11 leads to a reduced rate of efflux of labeled transferrin in Chinese hamster ovary cells (17, 29).
In this study we examined the role of Rab11 in FcγR-mediated phagocytosis in macrophages. We used primary inflammatory macrophages (thio-macrophages) and a mouse macrophage cell line expressing various alleles of Rab11 to test the hypothesis that Rab11 is required for optimal FcγR-mediated phagocytosis.
Experimental Procedures
Cells and Reagents.
RAW LacR/FMLP.2 cells were derived from RAW 264.7 cells (33). Thio-macrophages were isolated as described (34). Adherent macrophages and NIH 3T3 cells were maintained in RPMI medium 1640 containing 10% (vol/vol) FCS, 100 units/ml penicillin G, and 100 μg/ml streptomycin. cDNA encoding hemagglutinin (HA)-tagged human Rab11 was kindly provided by Hsiao-Ping H. Moore (University of California, Berkeley). HA-Rab11 was subcloned into pSFFV (35). Site-directed mutagenesis was performed by using a QuikChange site-directed mutagenesis kit (Stratagene). All constructs were verified by DNA sequencing. Mouse mAb HA.11 against HA was from Berkeley Antibody, Richmond, CA. mAb C2 against mouse transferrin receptor and unconjugated and biotin-conjugated mAb 2.4G2 against FcγRII/III were from PharMingin. A mouse mAb against Rab11 was from Transduction Laboratories (San Diego, CA). Fluorescein-, Cy5-, and rhodamine-conjugated secondary antibodies and mouse transferrin were from Jackson ImmunoResearch. Streptavidin conjugates and Alexa 488 anti-mouse IgG were from Molecular Probes. Gold-conjugated goat anti-rabbit and anti-mouse IgG were from Amersham Nederland ('s-Hertogenbosch, The Netherlands).
Measurements of Transferrin Efflux.
Cells adherent to round coverslips were preincubated for 1–2 hr at 37°C in RPMI 1640 followed by 1 hr in buffer A (125 mM NaCl/5 mM KCl/1 mM KH2PO4/5 mM glucose/10 mM NaHCO3/1 mM MgCl2/1 mM CaCl2/20 mM Hepes, pH 7.4) containing 50 μg/ml iron-saturated rhodamine-transferrin and 2 mg/ml ovalbumin. Cells were washed twice in ice-cold buffer A, twice (1 min each) in ice-cold 200 mM NaCl, 50 μM (2[N-morphilino]ethanesulfonic acid, pH 5.0, to remove rhodamine-transferrin bound to the cells' surfaces and twice in ice-cold buffer A. Cells were incubated in buffer A supplemented with 2 mg/ml ovalbumin, 3 μg/ml unlabeled transferrin, and 100 μM desferrioxamine for varying times at 37°C followed by fixation in 3.7% formaldehyde. Quantitation of transferrin efflux was done by measuring cell-associated rhodamine-transferrin fluorescence using single-cell microspectrofluorometry (36). All fluorescence values were corrected for autofluorescence of unlabeled cells. A total of 30–50 cells per coverslip were selected for fluorescence quantitation without prior knowledge of rhodamine intensity. For experiments using transfected RAW LR/FMLPR.2 cells, cells expressing HA-tagged Rab11 constructs were identified by staining with mAb HA.11 followed by fluorescein-conjugated anti-mouse IgG. Fluorescein-positive cells were visualized without knowledge of rhodamine fluorescence, and rhodamine fluorescence intensities were measured as described above. Semilog plots of rhodamine-transferrin efflux were biphasic, suggesting that exocytic release of rhodamine-transferrin occurred by at least two separate processes with distinct kinetics. For thio-macrophages and NIH 3T3 cells, curve fitting was performed by using a double exponential decay model (deltagraph 4.0, SPSS, Chicago) according to the following equation: y = afe-kfastt + ase-kslowt, where y is the cell-associated rhodamine-transferrin, as and af are the relative amounts of ligand at t = 0, and kfast and kslow are the first-order rate constants for fast and slow exocytosis, respectively. A separate first-order rate constant for recycling, kr, was calculated for rhodamine-transferrin efflux in RAW LR/FMLPR.2 cells and thio-macrophages. For times between t = 0 and 8 min (RAW LR/FMLPR.2 cells) and 0 and 4 min (thio-macrophages), curve-fitting of rhodamine-transferrin efflux data using a single exponential decay model produced a straight line.
Cryoimmunogold Electron Microscopy.
Sixteen hours after transfection, RAW LacR/FMLP.2 cells were incubated in the presence or absence of EIgG for 5 min at 37°C. Transfected cells were fixed, and ultra-thin frozen sections were incubated at room temperature with the indicated antibodies and 5 and/or 10 nm gold conjugates as described (37). After immunolabeling, cryosections were embedded in a mixture of methylcellulose and uranyl acetate and examined with a Philips CM10 electron microscope (Eindhoven, The Netherlands).
Phagocytosis Assays.
Phagocytosis assays were performed in buffer A. To determine the rate of phagocytosis, a synchronous wave of phagocytosis at 37°C was initiated after adherence of EIgG at 4°C (36). Bound, uningested EIgG were detected by staining with fluorescein-conjugated anti-rabbit IgG for 30 min at 4°C. Cells were fixed in 3.7% formaldehyde, permeabilized with 0.2% Triton X-100, and stained for expression of HA-tagged Rab11 constructs by using a mAb HA.11 followed by aminoethylcoumarin-anti-mouse IgG and for total EIgG by using rhodamine anti-rabbit IgG. Ingested EIgG were identified as fluorescein-negative rhodamine-positive phagocytic vacuoles. A total of 50 cells expressing the indicated Rab constructs and 50 nonexpressing control cells on the same coverslip were scored for internalized or bound EIgG. The phagocytic index was calculated as the average number of EIgG ingested per cell. To determine the extent of phagocytosis (i.e., unsynchronized phagocytosis), transfected RAW LacR/FMLPR.2 cells were incubated with 5 × 106 EIgG at 37°C for 30 min, and phagocytosis was quantitated as described above.
Results
Localization and Dynamics of Transferrin in Thio-Macrophages.
We examined the distribution of rhodamine-transferrin and transferrin receptors in thio-macrophages and NIH 3T3 cells. In both cell types, endocytosed rhodamine-transferrin and transferrin receptors showed extensive overlap. In NIH 3T3 cells, both markers localized to a peri-centriolar location, similar to results obtained in other cells. However, in thio-macrophages the bulk of internalized rhodamine-transferrin was present in peripheral vesicles. There was a conspicuous lack of accumulation of rhodamine-transferrin in a peri-centriolar location (Fig. 1A). Even during various chase times after loading, no appreciable rhodamine-transferrin accumulated in a peri-centriolar location (data not shown). Labeling of rhodamine-transferrin in thio-macrophages was specific because, using microspectrofluorometry, the rhodamine fluorescence intensity was reduced by 80% in the presence of 100-fold excess unlabeled transferrin. Thio-macrophages and NIH 3T3 cells were loaded with rhodamine-transferrin to steady state, and efflux was monitored over time. Both cell types showed a biphasic efflux of rhodamine-transferrin, but the efflux from thio-macrophages was more rapid (Fig. 1B). Values for kfast in thio-macrophages and NIH 3T3 cells were 0.652 and 0.239, respectively, and the t1/2 of rhodamine-transferrin efflux for thio-macrophages was 1.8 min, as compared with 6 min for NIH 3T3 cells. Thus, trafficking of transferrin in macrophages was notable in two respects: the absence of a distinct peri-centriolar recycling compartment and the extremely rapid rate of transferrin efflux.
Figure 1.

Macrophages lack a morphologically distinct peri-centriolar recycling compartment and efflux rhodamine-transferrin at a rapid rate. (A) Confocal fluorescence micrographs of the indicated cells incubated with biotin-transferrin for 1 hr at 37°C, followed by fixation and staining for biotin-transferrin (Right) using rhodamine-streptavidin, or transferrin receptor (Left) using mAb C2 followed by fluorescein-conjugated anti-rat IgG. Data are representative of three similar experiments. (Bar = 10 μm.) (B) Recycling of rhodamine-transferrin (Tfn). After acid washing to remove surface-bound rhodamine-transferrin, cell-associated rhodamine-transferrin is expressed as percent of initial rhodamine-transferrin present at time t = 0. Data points represent mean ± SEM (n = 3–4).
Rab11 Regulates Transferrin Recycling in Macrophages.
Rab11 plays an important role in the regulation of transferrin recycling in epithelial cells (17, 29). By immunoblotting with a mAb against Rab11, this GTPase was readily detectable in lysates derived from thio-macrophages and RAW LR/FMLPR.2 cells; however, this mAb was not useful for the detection of Rab11 in glutaraldehyde-fixed cells, thus precluding its use in cryoimmunogold electron microscopy (not shown). Therefore, we performed cryoimmunogold electron microscopy on RAW LR/FMLPR.2 cells transfected with HA-Rab11 WT. Rab11 was associated prominently with a peripheral population of intracellular tubules and vesicles (Fig. 2) similar to the localization of internalized rhodamine-transferrin in thio-macrophages. In other sections, Rab11 also appeared in Golgi stacks (not shown). Confocal fluorescence microscopy demonstrated significant colocalization between HA-Rab11 WT and transferrin receptors (Fig. 3) and rhodamine-transferrin (not shown). In contrast, a GTP binding-deficient allele of Rab11 (Rab11 25N) had a slightly more diffuse appearance (Fig. 3), although cryo-immunogold electron micrographs of RAW LR/FMLPR.2 expressing Rab11 25N showed extensive membrane association of this construct as well (data not shown). Localization of Rab11 25N was not prominent in filopodia and overlap with transferrin receptor was not obvious (Fig. 3). A GTPase-deficient version of Rab 11 (Rab11 70L) appeared in a punctate distribution, was present in filopodia, and demonstrated partial colocalization with transferrin receptors, similar to Rab11 WT. As compared with mock-transfected cells, cells expressing Rab11 25N demonstrated a decreased rate of rhodamine-transferrin efflux whereas cells expressing Rab11 70L demonstrated an accelerated rate of rhodamine-transferrin efflux (Table 1).
Figure 2.

Rab11 localizes to endocytic tubules and vesicles in macrophages. Adherent RAW LR/FMLPR.2 cells were transfected with HA-Rab11 WT. After fixation, ultrathin cryosections were labeled with monoclonal anti-HA followed by rabbit anti-mouse IgG and anti-rabbit-10 nm gold. An area of a cell showing Rab11-labeled tubular cisternae and vesicles (large arrows) underneath the cell membrane; a short ruffle also is labeled (small arrows) on the membrane and in the cytosol. (Bar = 200 nm.)
Figure 3.
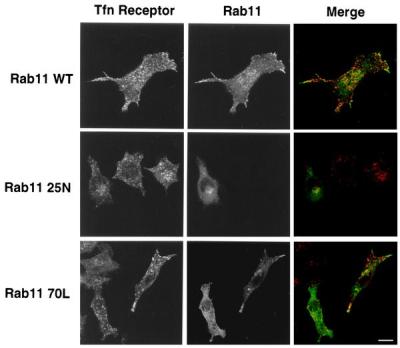
Localization of transfected Rab11 alleles in RAW LR/FMLPR.2 cells. Adherent macrophages transfected with HA-tagged constructs as indicated were fixed and stained for transferrin (Tfn) receptor with mAb C2 followed by biotin anti-rat IgG and Alexa 532-streptavidin, and for HA with mAb HA.11 followed by Alexa 488-anti-mouse IgG. Confocal fluorescence micrographs are representative of five similar experiments. (Bar = 10 μm.)
Table 1.
Kinetic parameters for rhodamine-transferrin recycling in RAW LR/FMLPR.2 cells expressing various Rab11 alleles
| Transfected plasmid | Relative expression, % | kr (min−1)* | t1/2 (min)† |
|---|---|---|---|
| Mock (vector) | 0.130 ± 0.011 | 4.9 ± 0.21 | |
| Rab11 WT | 100 ± 8.6 | 0.120 ± 0.007 | 5.3 ± 0.68 |
| Rab11 25N | 102 ± 8.2 | 0.077 ± 0.010‡ | 9.1 ± 1.2‡ |
| Rab11 70L | 106 ± 7.7 | 0.231 ± 0.034‡ | 2.2 ± 0.70‡ |
Based on single-cell microspectrofluorometry. Each value represents mean ± SEM, n = 4.
Rate constant for efflux of rhodamine-transferrin during 8 min of chase after 1 hr of loading.
† Amount of time required for efflux of 50% of total rhodamine-transferrin after 1 hr of loading.
‡P < 0.05 (Rab11 25N or Rab11 70L vs. mock-transfected).
Rab11 Regulates Phagocytosis in Macrophages.
Transferrin and transferrin receptors have been shown to be closely associated with leading lamella in fibroblasts (16) and in membrane ruffles produced in response to epidermal growth factor in KB cells (38), cytoskeletal alterations that resemble those accompanying phagocytosis. Therefore, we determined the distribution of transferrin receptors in macrophages undergoing phagocytosis. Transferrin receptors appeared adjacent to nascent phagosomes. This was most apparent in cells in which the majority of phagocytic targets localized to one pole of the cell (Fig. 4), although this pattern also was seen in cells engaging in phagocytosis in an unlocalized fashion (not shown). Rab11 also labeled structures that were intimately associated with phagocytic targets. Cryoimmunogold electron micrographs of macrophages transfected with HA-Rab11 WT demonstrated labeling of membranes associated with the base of pseudopods (Fig. 5) and with filopodia not engaging in phagocytosis (Fig. 3 and data not shown). Cells expressing various Rab alleles were assessed for both rate and extent of phagocytosis. Expression of Rab11 25N led to an inhibition in both the rate and extent of phagocytosis of EIgG compared with nonexpressing controls. In contrast, expression of Rab11 70L led to enhanced phagocytosis (Fig. 6). The effect of Rab11 25N and Rab11 70L expression on phagocytosis was not caused by an alteration in the binding of EIgG (Fig. 6), indicating that Rab11 does not appreciably affect early events after engagement of FcγRs, such as receptor affinity or surface expression.
Figure 4.

Transferrin receptors are associated with nascent phagosomes. Adherent RAW LR/FMLPR.2 cells were incubated with EIgG for 15 min at 4°C to allow binding, but not ingestion, then incubated at 37°C for 2 min before fixation and staining for EIgG with Cy5-conjugated anti-rabbit IgG and for transferrin receptor with mAb C2 followed by biotin anti-rat IgG and Alexa 532-streptavidin. (A) Phase-contrast. (B) EIgG. (C) Transferrin receptor. Micrographs are representative of four similar experiments. (Bar = 10 μm.)
Figure 5.

Rab11 localizes to regions of the cell engaging in phagocytosis. Adherent RAW LR/FMLPR.2 cells transfected with HA-Rab11 WT were incubated with EIgG for 4 min. After fixation, ultrathin cryosections were labeled with monoclonal anti-HA followed by rabbit anti-mouse IgG and anti-rabbit-10 nm gold. An area of a macrophage in contact with two erythrocytes (E) showing labeling on filopodia (f). Vacuoles (v) and an invagination of the plasma membrane (i) are labeled on the membrane. (Bar = 300 nm.)
Figure 6.

Expression of Rab11 mutants alters the rate and extent of phagocytosis of EIgG. (A) Adherent RAW LR/FMLPR.2 cells transfected with the indicated constructs were incubated with EIgG at 4°C and the extent of binding was determined. (B) In parallel cultures, macrophages preincubated with EIgG at 4°C were incubated for varying times at 37°C to monitor the rate of phagocytosis in cells expressing either HA-Rab11 25N (■), HA-Rab11 70L (▴), or nonexpressing controls (●). Numbers in parentheses refer to percent of cell-associated EIgG ingested by 30 min. Data points represent mean ± SEM (n = 3). (C) Unsynchronized phagocytosis of EIgG was monitored in RAW LR/FMLPR.2 cells transfected with the indicated constructs. Percent phagocytosis of expressing cells compared with nonexpressing controls represents mean ± SEM (n = 3).
Discussion
The data presented here indicate that Rab11 regulates membrane recycling from an endosomal compartment and that intact Rab11 function is required for optimal FcγR-mediated phagocytosis. The finding that cells expressing Rab11 70L demonstrated enhanced transferrin recycling differ from those of an earlier study in BHK cells (29) in which expression of Rab11 70L led to impaired transferrin recycling. The presence of a prominent peri-centriolar recycling compartment in other cells may complicate the analysis of transferrin recycling from an early endosomal compartment. Our data are consistent with a recent study of TRVb cells in which the authors examined transferrin recycling after preincubating the cells at 16°C (17), a temperature that prevented entry of transferrin into the peri-centriolar recycling compartment. In this study, expression of Rab11 70L led to a slightly enhanced rate of transferrin recycling at the reduced temperature. Because macrophages lack a morphologically distinct transferrin receptor-positive peri-centriolar recycling compartment, the fate of internalized transferrin in macrophages is more restricted, and the effects of expression of different Rab11 alleles on transferrin recycling are easier to interpret. Our findings indicate that GTP hydrolysis is not only dispensable for transferrin receptor recycling, but actually may inhibit recycling. In this respect, Rab11 in macrophages resembles GTPases of the Ras and Rho family, whose biological activities are enhanced when GTP hydrolysis is inhibited.
The enhanced endocytic capacity of macrophages relative to most other cells indicates that macrophages must have evolved mechanisms of rapid replenishment of plasma membrane from intracellular sources. With the exception of rabbit reticulocytes (39), the rate constant for transferrin recycling in thio-macrophages is greater than that of any other cell type measured to date. For example, kfast in TRVb-1 and K562 cells are 0.249 and 0.202, respectively (40, 41), as compared with 0.652 for thio-macrophages (this study). The t1/2 of transferrin recycling is 1.8 min for thio-macrophages as compared with 5 min for Chinese hamster ovary cells (42), 6.5 min for A431 cells (43), and 11.5 min for a choriocarcinoma cell line (23). Interestingly, transferrin recycling in RAW LR/FMLPR.2 cells is not as rapid as that in thio-macrophages, which mirrors differences in their phagocytic efficacy.
Our data suggest that the accelerated rate of transferrin recycling in macrophages may be partly because of the presence of an extensive peripherally based endosomal network that contributes to rapid recycling. Despite the absence of a distinct peri-centriolar recycling compartment, the rate of transferrin recycling in macrophages was biphasic, with fast and slow components (Fig. 1). In this respect, transferrin recycling in macrophages resembles that of other cells, indicating the presence of at least two kinetically distinguishable mechanisms of recycling. This finding implies that early endosomes themselves are heterogeneous with respect to recycling and that the peri-centriolar recycling compartment that is characteristic of other cells may not be the only source of heterogeneity in endosomal recycling. By cryoimmunogold electron microscopy, Rab11-positive compartments in macrophages appeared highly pleiomorphic, consisting of tubules and vesicles. Perhaps the complex appearance of this compartment reflects functional differences as well.
We do not know how this compartment is regulated. In particular, we have not yet determined whether transferrin recycling in macrophages is purely constitutive or is regulated by external stimuli, including phagocytic particles. Preliminary experiments have not demonstrated that FcγR clustering triggers a further enhancement in the normally rapid rate of transferrin efflux in macrophages, findings that are reminiscent of an earlier study using J774 macrophages and zymosan targets (44). We also do not know whether Rab11 promotes recycling and tethering of membrane vesicles or tubules in a localized fashion (e.g., intraphagosomally). Electron micrographs demonstrated staining for Rab11 in multiple compartments, including phagosomes. Because endocytosis and pinocytosis occur throughout the plasma membrane in a constitutive fashion, it may be unrealistic to expect the demonstration of Rab11 accumulation at sites of active membrane turnover, such as phagosomes. Furthermore, Rab11 may diffuse rapidly within the plane of the membrane after membrane fusion or may immediately dissociate from the membrane after the initial tethering event, thus making it difficult to appreciate rapid subcellular dynamics of Rab11.
There are likely to be multiple sources of intracellular membrane in phagocytic leukocytes that contribute to phagocytosis. The ability of early and late endosomes to fuse with phagosomes and the apposition of components of the Golgi apparatus to regions beneath the plasma membrane of macrophages undergoing spreading on immune complexes (45) attest to the diversity of membrane compartments that are affected by phagocytosis. It is possible that Rab11-containing recycling endosomes receive input from multiple compartments besides the plasma membrane. It is also possible that Rab11 controls the pool size of plasma membrane available for phagocytosis, and that this pool originates from multiple intracellular compartments in addition to the recycling compartment described here. Several studies have documented an endosome-to-Golgi pathway, and proteins that accumulate in the trans-Golgi network (TGN), such as TGN-38, recycle between the plasma membrane and the Golgi apparatus (14, 15). Rab11 also has been implicated in a TGN-to-basolateral plasma membrane pathway in polarized cells (31). Although we currently do not favor a model for membrane trafficking in phagocytosis that involves the Golgi (10), we cannot eliminate the possibility that the TGN, directly or indirectly, contributes to phagocytosis, nor can we conclude that the sole effect of expression of mutant Rab11 alleles in macrophages is to alter trafficking in the recycling compartment. Thus, Rab11 may contribute to phagocytosis by promoting a generalized flow of membrane from several intracellular sites to the plasma membrane. These membrane sources may funnel through the transferrin receptor-positive recycling compartment or may traffic to the plasma membrane by an independent route. However, the efficient loading of transferrin via endocytosis and the rapid Rab11-dependent transferrin recycling argue that a substantial pool of membrane available for phagocytosis comes from this pool. In any case, the finding that expression of a GTP binding-deficient allele of Rab11 inhibits phagocytosis in macrophages indicates that Rab11 must play a nonredundant role in this macrophage function. Although the inhibition of phagocytosis by Rab11 25N was only partial (≈50%), the difference between rapid ingestion and efficient killing of pathogenic microbes and compromised, delayed killing of these organisms can have a profound influence on host survival.
Abbreviations
- EIgG
sheep erythrocytes opsonized with rabbit anti-sheep IgG
- FcγR
receptor for the Fc portion of IgG
- thio-macrophage
thioglycollate-elicited murine peritoneal macrophage
- HA
hemagglutinin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Norton J M, Munck A. J Immunol. 1980;125:259–266. [PubMed] [Google Scholar]
- 2.Michl J, Unkeless J C, Pieczonka M M, Silverstein S C. J Exp Med. 1983;157:1746–1757. doi: 10.1084/jem.157.6.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schalm O W, Jain N C, Carroll J E. Veterinary Hematology. Philadelphia: Lea and Febiger; 1975. [Google Scholar]
- 4.Norton J M. Biorheology. 1990;27:21–37. doi: 10.3233/bir-1990-27103. [DOI] [PubMed] [Google Scholar]
- 5.Hackam D J, Rotstein O D, Sjolin C, Schreiber A D, Trimble W S, Grinstein S. Proc Natl Acad Sci USA. 1998;95:11691–11696. doi: 10.1073/pnas.95.20.11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tardieux I, Webster P, Ravesloot J, Boron W, Lunn J A, Heuser J E, Andrews N W. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- 7.Capo C, Farnarier C, Benoliel A M, Bongrand P, Depieds R. J Reticuloendothel Soc. 1983;34:359–369. [PubMed] [Google Scholar]
- 8.Armstrong J A, D'Arcy Hart P. J Exp Med. 1971;134:713–740. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horwitz M A. J Exp Med. 1983;158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q, Cox D, Tseng C-C, Donaldson J G, Greenberg S. J Biol Chem. 1998;273:19977–19981. doi: 10.1074/jbc.273.32.19977. [DOI] [PubMed] [Google Scholar]
- 11.Steinman R M, Mellman I S, Muller W A, Cohn Z A. J Cell Biol. 1983;96:1–27. doi: 10.1083/jcb.96.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lennartz M R, Yuen A F C, Masi S M, Russell D G, Buttle K F, Smith J J. J Cell Sci. 1997;110:2041–2052. doi: 10.1242/jcs.110.17.2041. [DOI] [PubMed] [Google Scholar]
- 13.Mayorga L S, Bertini F, Stahl P D. J Biol Chem. 1991;266:6511–6517. [PubMed] [Google Scholar]
- 14.Mellman I. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 15.Mukherjee S, Ghosh R N, Maxfield F R. Physiol Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 16.Hopkins C R, Gibson A, Shipman M, Strickland D K, Trowbridge I S. J Cell Biol. 1994;125:1265–1274. doi: 10.1083/jcb.125.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini D D. Proc Natl Acad Sci USA. 1998;95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruenberg J, Maxfield F R. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- 19.Hopkins C R, Trowbridge I S. J Cell Biol. 1983;97:508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayor S, Presley J F, Maxfield F R. J Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein B S, Sussman H H. J Biol Chem. 1986;261:10319–10331. [PubMed] [Google Scholar]
- 22.McGraw T E, Dunn K W, Maxfield F R. J Cell Biol. 1988;106:1061–1066. doi: 10.1083/jcb.106.4.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Ende A, du Maine A, Schwartz A L, Strous G J. Biochem J. 1990;270:451–457. doi: 10.1042/bj2700451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beauchamp J R, Woodman P G. Biochem J. 1994;303:647–655. doi: 10.1042/bj3030647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheff D R, Daro E A, Hull M, Mellman I. J Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfeffer S R. Nat Cell Biol. 1999;1:E17–E22. doi: 10.1038/8967. [DOI] [PubMed] [Google Scholar]
- 27.Hori Y, Takeyama Y, Hiroyoshi M, Ueda T, Maeda A, Ohyanagi H, Saitoh Y, Kaibuchi K, Takai Y. Dig Dis Sci. 1996;41:133–138. doi: 10.1007/BF02208595. [DOI] [PubMed] [Google Scholar]
- 28.Urbe S, Huber L A, Zerial M, Tooze S A, Parton R G. FEBS Lett. 1993;334:175–182. doi: 10.1016/0014-5793(93)81707-7. [DOI] [PubMed] [Google Scholar]
- 29.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton R G. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green E G, Ramm E, Riley N M, Spiro D J, Goldenring J R, Wessling-Resnick M. Biochem Biophys Res Commun. 1997;239:612–616. doi: 10.1006/bbrc.1997.7520. [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Feng Y, Chen D, Wandinger-Ness A. Mol Biol Cell. 1998;9:3241–3257. doi: 10.1091/mbc.9.11.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casanova J E, Wang X Y, Kumar R, Bhartur S G, Navarre J, Woodrum J E, Altschuler Y, Ray G S, Goldenring J R. Mol Biol Cell. 1999;10:47–61. doi: 10.1091/mbc.10.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox D, Chang P, Zhang Q, Reddy P G, Bokoch G M, Greenberg S. J Exp Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Virgilio F, Meyer B C, Greenberg S, Silverstein S C. J Cell Biol. 1988;106:657–666. doi: 10.1083/jcb.106.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmer L E, Pancetti A R, Greenberg S, Bliska J B. Infect Immun. 1999;67:708–716. doi: 10.1128/iai.67.2.708-716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenberg S, El Khoury J, Di Virgilio F, Kaplan E M, Silverstein S C. J Cell Biol. 1991;113:757–767. doi: 10.1083/jcb.113.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calafat J, Janssen H, Stahle-Backdahl M, Zuurbier A E, Knol E F, Egesten A. Blood. 1997;90:1255–1266. [PubMed] [Google Scholar]
- 38.Bretscher M S, Aguado-Velasco C. Curr Biol. 1998;8:721–724. doi: 10.1016/s0960-9822(98)70281-7. [DOI] [PubMed] [Google Scholar]
- 39.Iacopetta B J, Morgan E H. J Biol Chem. 1983;258:9108–9115. [PubMed] [Google Scholar]
- 40.Spiro D J, Boll W, Kirchhausen T, Wessling-Resnick M. Mol Biol Cell. 1996;7:355–367. doi: 10.1091/mbc.7.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schonhorn J E, Wessling-Resnick M. Mol Cell Biochem. 1994;135:159–169. doi: 10.1007/BF00926519. [DOI] [PubMed] [Google Scholar]
- 42.Martys J L, Shevell T, McGraw T E. J Biol Chem. 1995;270:25976–25984. doi: 10.1074/jbc.270.43.25976. [DOI] [PubMed] [Google Scholar]
- 43.Davis R J, Faucher M, Racaniello L K, Carruthers A, Czech M P. J Biol Chem. 1987;262:13126–13134. [PubMed] [Google Scholar]
- 44.Buys S S, Kaplan J. J Cell Physiol. 1987;131:442–449. doi: 10.1002/jcp.1041310317. [DOI] [PubMed] [Google Scholar]
- 45.Bainton D F, Takemura R, Stenberg P E, Werb Z. Am J Pathol. 1987;134:15–26. [PMC free article] [PubMed] [Google Scholar]