

Abstract
Apurinic/apyrimidinic (AP) sites are common DNA lesions that arise from spontaneous depurination or by base excision repair (BER) of modified bases. A biotin-containing aldehyde-reactive probe (ARP) [Kubo, K., Ide, H., Wallace, S. S. & Kow, Y. W. (1992) Biochemistry 31, 3703–3708] is used to measure AP sites in living cells. ARP penetrates the plasma membrane of cells and reacts with AP sites in DNA to form a stable ARP–DNA adduct. The DNA is isolated and treated with avidin-horseradish peroxidase (HRP), forming a DNA–HRP complex at each biotin residue, which is rapidly separated from free avidin–HRP by selective precipitation with a DNA precipitating dye (DAPER). The number of AP sites is estimated by HRP activity toward chromogenic substrate in an ELISA assay. The assay integrates the AP sites formed by the different glycosylases of BER during a 1-h incubation and eliminates artifactual depurination or loss of AP sites during DNA isolation. The assay was applied to living cells and nuclei. The number of AP sites after a 1-h incubation in old IMR90 cells was about two to three times higher than that in young cells, and the number in human leukocytes from old donors was about seven times that in young donors. The repair of AP sites was slower in senescent compared with young IMR90 cells. An age-dependent decline is shown in the activity of the glycosylase that removes methylated bases in IMR90 cells and in human leukocytes. The decline in excision of methylated bases from DNA suggests an age-dependent decline in 3-methyladenine DNA glycosylase, a BER enzyme responsible for removing alkylated bases.
Keywords: DNA damage, aging, glycosylase, AP site
Apurinic and apyrimidinic (AP or abasic) sites are common lesions in DNA and are formed either spontaneously or as intermediates during the course of base excision repair (BER) of oxidized, deaminated, or alkylated bases (1, 2). N-glycosylases of BER recognize and excise the aberrant bases from DNA, resulting in formation of an AP site. The AP site is then incised by an AP endonuclease to generate a 5′-terminal base-free deoxyribose phosphate at a single-strand break. The repair of the single-strand break (AP site) is accomplished by a DNA polymerase β (β-pol) and DNA ligase (ligase I or III/XRCC1) to restore the normal and correct base (3, 4). The rate-limiting step of BER is the lyase activity associated with the 8-kDa domain of β-pol, which removes the AP site from DNA (4). AP sites can be mutagenic (5–7) or can cause cell death (8–10).
The methods (11–14) that detect AP sites after DNA isolation, including those with ARP (15, 16), may be inaccurate as a result of several factors: (i) artifactual AP sites resulting from base loss and formation of AP sites in DNA by high temperature at neutral pH (12, 17, 18); (ii) AP sites in DNA are subject to loss by β-elimination, followed by δ-elimination catalyzed by high temperatures (19), primary amines in histones, polyamines (19–21), and thiols (22), causing underestimation of AP levels in DNA. Nakamura et al. (23) studied the endogenous level of AP sites in different tissues of the rat using aldehyde-reactive probe (ARP), isolating the DNA before trapping AP sites. They estimate that there are 50,000–200,000 AP sites/mammalian cell, that the brain of rat contained the most AP sites, and that most of the AP sites were cleaved 5′ to the AP sites. The first two results are different from the results reported here.
In this paper, we describe a method for biotinylating AP sites in live cells and then quantifying the biotin in the isolated DNA. This method is used to determine the age-dependent changes in BER in human fibroblasts (IMR90), leukocytes isolated from human blood, and nuclei isolated from rat tissues.
Materials and Methods
Materials.
A QIAamp blood kit was purchased from Qiagen (Chatsworth, CA). The DNA-precipitating agent, DAPER [N,N′-bis(3,3′-(dimethyl-amino)propylamine)-3,4,9,10-peryl-enetetra-carboxylicdiimide] and ImmunoPure 3,5,3′,5′-tetra-methylbenzidine horseradish peroxidase (HRP) substrate kits were from Pierce. Vectastain ABC kit was from Vector Laboratories. O-Methoxyamine, EGTA, and Histopaque 1077 were from Sigma. HK-UNG thermolabile uracil N-glycosylase (UNG) was from Epicentre Technologies (Madison, WI). Hydrogen peroxide (30%) and Tween 20 were from Fisher. Fatty acid-free BSA was from Calbiochem. Rnase A was from Roche Molecular Biochemicals. Calf thymus DNA was from Worthington. ARP N′-aminooxy-methylcarbonyl hydrazino-d-biotin, was from Dujindo Laboratories (Kumamoto, Japan). Methyl methanesulfonate (MMS) and 1-octane sulfonic acid were from Fluka.
Preparation of Nuclear Fraction from Rat Liver and Brain.
Young (2–4 mo) or old (24 mo) Sprague–Dawley male rats were sacrificed after anesthesia under ether, and liver and brain were removed and placed in ice-cold buffer consisting of 210 mM mannitol, 70 mM sucrose, 1 mM EDTA, 5 mM Hepes (pH 7.4) (MSH) supplemented with 0.1% fatty acid-free BSA. The tissues were weighed and diluted 1/5 by the buffer and homogenized with a Potter–Elvehjem homogenizer (Wheaton). Briefly, the homogenate was centrifuged twice in the cold at 500 × g for 10 min, and the pellet was collected and suspended into 5 ml ice-cold 0.25 M sucrose/50 mM Tris⋅HCl (pH 7.5)/25 mM KCl/5 mM MgCl2 (SKTM)/1 mM diethylenetriamine pentaacetic acid and filtered through four layers of cheese cloth into ice-cold 50-ml tubes. The nuclear fraction was washed twice in the cold and centrifuged at 500 × g in the same buffer and suspended at a concentration of 50 mg protein/ml. For the assay of AP sites, 2 mg of protein was used. Nuclear DNA was isolated by QIAgen or by phenol/chloroform extraction.
Isolation of Leukocytes from Human Blood.
Human subjects who participated in our study were not smokers and generally were in good health. The range of the ages of each group was 21–33 yr (n = 9) for young and 58–75 yr (n = 8) for old. Blood donations were collected by venipuncture in the morning and during fasting into 15-ml heparinized vacutainers (Becton Dickinson). Leukocytes were isolated from whole blood by Histopaque 1077, as described by the manufacturer. Contaminating erythrocytes were removed by suspending the pellet in 50 ml of ice-cold ammonium chloride lysis buffer (150 mM NH4Cl/1 mM EDTA/10 mM NaHCO3) and gently inverting for 10 min at room temperature. Afterward, the leukocytes were collected and resuspended into serum-free DMEM supplemented with 25 mM Hepes. The viability of the leukocytes was generally ≥90%. IMR90 cells were cultured as described in ref. 24.
Preparing DNA with Uracil as a Standard for AP Site Assay.
The Escherichia coli mutant CJ236 (dut−/ung−) deficient for the activities of uracil glycosylase (ung−) and dUTPase (dut−) were used for preparing DNA containing a high level of uracil (25). The purity of the DNA was determined by the ratio A260/A280, and the uracil content of the DNA was quantified by GC-MS as described (26). The DNA was stored in aliquots at −20°C.
Assay for AP Sites in Cultivated Cells, Blood Leukocytes, and Nuclei.
IMR90 (2 × 106), leukocytes (5–10 × 106) in 1 ml PBS/5 mM glucose, or nuclei (2–5 mg protein) in 1 ml isolation buffer were incubated with 3 mM ARP for 60 min at 37°C. The cells were then collected by centrifugation at high speed at room temperature and washed twice with 1 ml PBS. The DNA was isolated by the QIAamp blood kit as described by the manufacturer (or by chloroform/phenol extraction). The DNA was quantitated, and, in general, 1 μg was transferred into 200 μl of 10 mM Tris (pH 9) and mixed with 15 μl of 5 M NaCl (mole salt/mole DNA ≈25,000) and incubated for 60 min at room temperature with 30 μl of avidin-HRP (ABC kit, prepared as described by the manufacturer but scaled down to 1 ml and diluted 1:3). The remaining DNA–ARP adduct can be frozen at −20°C for at least a year and reanalyzed. The DNA-avidin-HRP complex (DNA–HRP) was separated from unbound avidin-HRP by gently mixing 65 μl of 1 mM DAPER (mole DAPER/mole DNA ≈66) and incubated at room temperature for 5 min (DAPER forms aggregates that are easily removed by centrifugation). The DNA–DAPER precipitate was then collected by centrifugation for 5 min at 12,500 × g at 4°C. The pellet was resuspended by pipette into 1.4 ml of wash buffer [0.17 M NaCl/20 mM Tris/0.25% Tween 20/1% BSA (pH 8)] followed by centrifugation for 5 min at 12,500 × g at 4°C. The wash is repeated once more and the residual washing buffer is removed carefully. The DNA–HRP precipitate was suspended in 500 μl ice-cold 50 mM Na-citrate (pH 5.3) and sonicated at output 1–2 watts for 2–3 s (Sonifier Cell Disrupter, model w185D; Branson) and cooled immediately. Sonication can be repeated as long as the sample is kept cold. We measured HRP activity, as an indicator of AP sites in DNA–HRP by using the chromogenic ImmunoPure TMB or the fluorogenic QuantaBlu substrate kits (Pierce).
A standard curve for AP sites was prepared with 100 ng of DNA containing a known amount of uracil. The DNA was suspended into 50 μl of 10 mM Na2HPO4 (pH 7.5) and incubated with 25 μM spermine for 5 min at room temperature. Then, the DNA was incubated at 37°C for 60 min with 0.1–1 unit of uracil-DNA N-glycosylase (UNG) to catalyze the removal of uracil residues and form AP sites, together with 3 mM ARP that will trap the AP sites. The resulting “DNA–ARP” adduct was isolated from unbound ARP and UNG by QIAamp columns (minus the protease step) and quantitated. The amount of AP sites was corrected for losses of DNA during isolation (10–30% loss). The biotinylated DNA was incubated with avidin–HRP and processed as above. Sodium chloride was not added because the amount of salt in PBS is enough to establish the ratio (mole salt/mole DNA ≈25,000), and the volume of DAPER used was 5–6 μl. The background for standard AP-DNA and unknown samples was established by running in parallel 1 μg of calf thymus DNA. Equivalent amounts of DNA were run for each enzymatic assay of HRP; generally, 20–100 ng was run for an unknown sample and 0.03–1.2 ng for standard AP DNA.
Measuring BER in Living Cells.
The activity of glycosylases was determined by the capability of hydrogen peroxide or MMS to induce AP sites above their basal level. Leukocytes from human blood were suspended at 10 × 106/ml in serum-free DMEM supplemented with 25 mM Hepes (pH 7.3) and pulse-treated for 5 min with 20 mM MMS at 37°C. The cells were washed once with this supplemented media and incubated immediately with ARP to assess the induction of AP sites. The viability of the cells was determined by trypan blue exclusion. Cultivated primary human lung fibroblasts (IMR90) were harvested and incubated in PBS/5 mM glucose at concentration of 2 × 106/ml. ARP was added to the cells and split in aliquots of 1 ml. Hydrogen peroxide or MMS were added to the cells at final concentration of 10 mM or 20 mM, respectively, and the cells were incubated for 1 h at 37°C. Then, the cells were washed twice with 1 ml PBS, and the assay for AP sites was performed as described above.
Repair of AP sites was measured after inducing them by treating 2 × 106/ml IMR90 cells for 5 min with a pulse of 10 mM H2O2 at 37°C in PBS/5 mM glucose. Then, cells were incubated in fresh DMEM (supplemented only with 25 mM Hepes) for recovery. Aliquots were collected just before (control) and immediately after the end of the H2O2 pulse and at intervals of 15 min and 45 min during the recovery. Samples were treated immediately with ARP after aliquots were withdrawn and AP sites were determined. The viability of the cells was determined by trypan blue exclusion.
Permeability of ARP into IMR90 Cells.
IMR90 cells (3 × 106/1 ml) were incubated in PBS/5 mM glucose with 3 mM ARP for different intervals (1, 5, 10, 15, and 30 min). At each time point, 1 ml was sampled and the cells were collected by centrifugation and washed in cold PBS. The cells were suspended into 150 μl of lysis solution (0.2 M methanesulfonic acid) and incubated at room temperature for 5 min. Denatured macromolecules were removed by spinning at high speed and used for protein quantitation. The supernatant was filtered through a 30,000 cut-off, ultra-free filter, and 50 μl were injected on an HPLC column (3-μm, 0.46 × 15-cm LC18-DB). ARP was eluted after 12 min with a mobile phase of 25 mM NaH2PO4, 5 mM 1-octane sulfonic acid, and 2% acetonitrile adjusted to pH 2.7 by phosphoric acid with a flow rate of 1 ml/min. An ESA coulochem detector 5100A using a model 5010 analytical cell and 5020 guard cell was used for analysis. The guard cell was set at +800 mV, electrode 1 at +100 mV, and electrode 2 at +880 mV. Full-scale output was 10 μA, and peak area was analyzed and compared with peak area of standard ARP.
Statistical Analysis.
Statistical analysis, using the student's two-tailed t test or nonparametric Mann–Whitney test, was performed with an Instat (San Diego, CA) statistical analysis program.
Results
The Assay for AP Sites in Standard DNA.
A standard DNA with AP sites was made with uracil-enriched DNA treated with UNG to generate AP sites. (i) DNA is incubated with UNG and 3 mM ARP for 60 min to generate the biotinylated-DNA (ARP-DNA). (ii) Free ARP and ARP-DNA are separated during isolation of DNA. ARP-DNA is prepared in 10 mM Tris (pH 9), which is compatible with the next step of the assay. The DNA is quantitated, and an aliquot is put into new buffer followed by 306 mM NaCl (75 μmol NaCl/1 μg DNA). The rest of the ARP-DNA is saved at −20°C for further analysis. (iii) The DNA-ARP is treated with avidin-HRP at room temperature for 60 min. The DNA–HRP complex and unbound avidin-HRP are separated by selective precipitation of the DNA–HRP complex by the DNA precipitating dye DAPER (27). This method of washing unbound avidin-HRP overcomes the necessity for immobilizing DNA to wash unbound avidin-HRP. DNA precipitation by DAPER is rapid and does not affect the activity of HRP, even after a 1-h incubation at room temperature (1.12 ± 0.05 vs. 1.164 ± 0.05 absorbance units). The precipitate was dissolved by gentle sonication in the cold. (iv) The HRP activity in the DNA-HRP was measured. A typical standard curve using the chromogenic substrate and the contribution of the DNA to the background is shown in Fig. 1.
Figure 1.

Standard curve and background at 450 nm from ARP-derivatized DNA. DNA with AP sites was prepared by incubating uracil-DNA glycosylase with 100 ng of genomic DNA of E. coli that contains a known amount of uracil. The DNA was then treated with ARP to trap AP sites, as described. A DNA sample not treated with ARP was run in parallel as background. Both samples of DNA were incubated with HRP-avidin and prepared as described. (A) Typical standard curve. (B) Shows signal and background.
For optimal signal/noise ratio, the pH during the interaction of avidin-HRP with the ARP-DNA should be 9, the mole salt/mole DNA ratio ≈25,000, and the mole DAPER/mole DNA ratio 66. These conditions, together with the suggested washing buffer, create a low background and increase the sensitivity of the assay (Fig. 1). The optimal concentration of ARP needed to maximize the signal was 1 mM, similar to previous reports (15). A similar concentration was essential for treating the whole cells. We used 3 mM of ARP in all of our experiments.
To establish the validity of this assay, the standard DNA was used to compare the amount of uracil residues liberated to the amount of AP sites generated by treatment with UNG. Uracil was determined by GC-MS (26) and AP sites by the described assay. The values were essentially the same (0.28% vs. 0.3%), thereby validating the accuracy of the assay.
Permeability of ARP into Living Cells.
Cells from early or late passages were incubated with 3 mM ARP for different intervals, and the level of ARP in the cells was determined by HPLC electrochemical detection. The kinetics of ARP accumulation in both types of cells was similar and is shown in Fig. 2. ARP accumulates in the cells and reaches a plateau within 10–15 min at an intracellular concentration of 5.46 nmol/3 × 106 cells.
Figure 2.

Kinetics of ARP accumulation into intact IMR90 cells. IMR90 cells (3 × 106/ml) in PBS/5 mM glucose were incubated with 3 mM ARP at 37°C for different intervals. At each time point, cells were sampled and processed as described in Materials and Methods, and the intracellular ARP was measured by HPLC-EC. The data is an average of three independent experiments (mean ± SD).
Treating Intact Cells with ARP.
AP sites in DNA are trapped while the cells are still viable. We find that ARP adducts persist in the DNA. The number of AP sites was not significantly different in young IMR90 cells immediately after trapping by ARP and over 2 h of recovery after ARP, 8.4 ± 3 vs. 9.1 ± 1.8 AP site/106 nucleotides, respectively. In addition, when cells were treated with 3 mM ARP for 60 min, then ARP washed out and seeded under normal culture conditions for a week, they grew as the vehicle-treated control (2.86 ± 0.11 × 106 vs. 2.68 ± 0.035 × 106 cells/dish, respectively). These results suggest that ARP is tolerated by the cells for at least the first 60 min of treatment, that ARP-DNA adducts are removed at a very slow rate from the DNA, and that a 60-min treatment with ARP does not affect cellular viability. The specificity of ARP for AP sites was shown by blocking AP sites in DNA with O-methoxyamine (13). When IMR90 cells were treated with O-methoxyamine for 10 min before adding ARP and then for 1 h in the presence of ARP, the number of AP sites decreased by 70% compared with the control (6.2 ± 0.45 vs. 1.8 ± 0.77 AP sites/106 nucleotides).
Rate of Formation and Estimation of the Steady-State Level of AP Sites in Living Cells.
Young (or old) IMR90 cells were incubated with or without 10 mM methoxyamine for 10 min before adding ARP. Then, the cells were washed three times with PBS at 37°C, and AP sites were assayed. In cells treated with methoxyamine, compared with control cells, the number of AP sites decreased from 4.28 ± 0.55 to 3.61 ± 0.39 AP sites/106 nucleotides/1 h (n = 3, P = 0.16). The difference in the number of AP sites between the two treatments gives an estimation of the steady-state level of AP sites, which is <0.67 AP sites/106 nucleotides.
Formation of AP sites in the cellular DNA is linear over 90 min for young and senescent fibroblasts. However, the rate of AP sites formed in young cells is one-third the rate in senescent cells (Fig. 3).
Figure 3.

Endogenous rate of AP sites formation in intact IMR90 cells. Senescent and young IMR90s (2 × 106/ml) in PBS/5 mM glucose were treated for different intervals with 5 mM ARP at 37°C. Then, free ARP was washed out followed by isolation of DNA, and the level of AP sites was determined as described in Materials and Methods. The values are mean ± SE. *, P < 0.05; **, P < 0.01.
Changes in the Activity of BER During in Vitro Senescence of Cultivated Human Lung Fibroblasts (IMR90).
To measure the ability of AP endonuclease and DNA pol β of BER to repair AP sites, we challenged IMR90 cells with 10 mM hydrogen peroxide for 5 min at 37°C. The level of AP sites was determined immediately after the pulse and at 15-min intervals (Table 1). The repair of AP sites was slower in senescent cells than young cells. In senescent cells, after 15 min of recovery, the induced AP sites remain significantly higher than the basal level (P < 0.04), whereas, in young cells, they were completely repaired.
Table 1.
Repair of AP sites in old and young IMR90 cells
 |
Senescent [population-doubling levels (PDLs) 46-54] and young (PDLs 25–30) IMR90 cells (2 × 106/ml in DMEM supplemented only with 25 mM Hepes) were treated with a pulse of 10 mM hydrogen peroxide for 5 min at 37°C and incubated for recovery. Immediately after, and at 15-min intervals, aliquots were removed and AP sites were determined as described in Materials and Methods. The basal level of AP sites was determined in nontreated cells. *, P < 0.05; **, P < 0.01.
Number of trapped AP sites/106 nucleotides. The values are mean ± SE of at least five experiments.
Simultaneously, IMR90 cells were challenged with hydrogen peroxide or MMS to measure the activity of different glycosylases that form AP sites. This approach revealed a 5-fold (P < 0.008) and 2-fold (P < 0.002) increase in AP sites by H2O2 in young and senescent cells, respectively (Table 1). A similar but more marked difference in the extent of AP site induction was seen in senescent vs. young cells when the alkylating agent MMS was used. MMS induces a 234-fold (P < 0.0001) vs. 24-fold (P < 0.0001) increase in AP sites in young vs. senescent cells, respectively (Table 2). These results show a decline with age in the activity of the 3-methyladenine-DNA glycosylase that excises methylated bases. A similar but milder decline is also seen with glycosylases that remove oxidized bases.
Table 2.
Assessment of the activity of 3-methyl adenine DNA glycosylase in senescent and young IMR90 cells
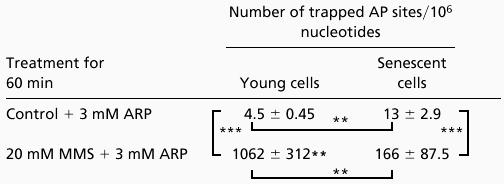 |
Young or senescent IMR90 cells (2 × 106/ml) in PBS/5 mM glucose were treated with 3 mM ARP (Control), 3 mM ARP + 20 mM MMS for 1 h at 37°C. The cells were then washed in PBS, the DNA was isolated, and 1 μg of DNA was used for detection of AP sites as described. The values are mean ± SD. **, P < 0.01; ***, P < 0.001.
AP Sites in Leukocytes from Young vs. Old Humans.
The number of AP sites was determined in leukocytes from young and old humans (young, n = 9; and old, n = 8; all males). The AP sites observed in old donors (ages 58–75 yr) is 7-fold higher than the AP sites observed in leukocytes from young donors (ages 22–33 yr; Table 3). We used MMS as a tool to measure 3-methyl-adenine DNA-glycosylase in leukocytes from old (n = 4) and young (n = 5) donors (Table 3). Interestingly, the level of AP sites induced by MMS depends on the age of the donor, similar to old and young IMR90 cells. MMS was very efficient in inducing AP sites in leukocytes from young donors (50-fold), but essentially no induction was observed in leukocytes from old donors.
Table 3.
The basal and the induced level of AP sites in human leukocytes isolated from whole blood of old and young donors
 |
Human leukocytes were isolated from young (n = 9) and old (n = 8) donors immediately after drawing blood, as described in Materials and Methods. The isolated leukocytes were incubated for 20 min at 37°C for recovery at a density of 5–10 × 106/ml DMEM supplemented only with 25 mM Hepes. AP sites were induced by treating the leukocytes from old and (n = 4) and young (n = 5) donors with a pulse of 20 mM MMS for 5 min at 37°C. The basal level and induced level of AP sites in DNA from leukocytes were measured as described in Materials and Methods. The viability of the leukocytes as estimated by trypan blue exclusion is over 95%. The values are mean ± SE. **, P < 0.01; ***, P < 0.001.
Age-Dependent Changes in the Level of AP Sites in nuclear DNA (nDNA) in Brain and Liver from Rats.
The number of AP sites in nDNA of liver increased above control when intact nuclei were treated with MMS (data not shown). The level of AP sites in liver nDNA of old rats is 69% higher than liver nDNA of young rats (Table 4). On the other hand, no age-dependent difference was seen in the level of AP sites in nDNA from brain (Table 4).
Table 4.
The level of AP sites in nuclear DNA from brain and liver of old and young rats
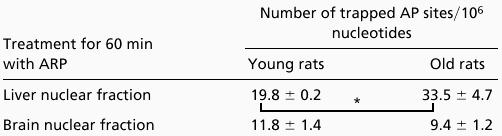 |
The number of AP sites in isolated nuclei was determined in liver and brain from five young rats (4 mo) and six old rats (24 mo). Both tissues were collected into iced buffer and homogenized; all the steps were performed at 4°C. The number of AP sites was determined as described. The data is a mean ± SE. *, P < 0.05.
Discussion
In this study, we report an approach for measuring AP sites and BER in live cells. The method is used to study the age-dependent changes in the level of AP sites and activity of BER in cultivated human lung fibroblasts, leukocytes, and nuclei from rat tissues. The approach traps AP sites by ARP before isolating the DNA, which reduces the possibility of misrepresenting the level of AP sites in vivo (19–22). We measure the AP sites trapped during a 1-h incubation, during which AP sites are formed and removed. This is different from the standard assay where DNA is isolated and the AP sites measured (11–14). By comparison, higher levels of AP sites were found when DNA was isolated from rat liver before treating with ARP (17.5 ± 3.1 AP sites/106 nucleotides), similar to ref. 23. The number of AP sites from the current method is 12.3 ± 0.65 AP sites/106 nucleotides/1 h, and the steady-state level is estimated as <1 AP site/106 nucleotides. The elevated steady-state level of AP sites in the traditional methods could be the result of the depurination of the DNA during the proteolytic digestion at 50°C (17), possibly catalyzed by different nucleophiles in the cellular homogenate. Thus, the approach for detection of AP sites described here appears to avoid the aforementioned limitations.
ARP penetrates living cells and equilibrates with extracellular ARP within 10–15 min at 37°C. A fibroblast (IMR90) has an approximate cell volume of ≈500 fL (r = 50 μm), assuming a spherical shape and a fraction of water of 1. We estimated the concentration of ARP at the saturation (10–15 min) to be ≈3.5 mM. Thus, intracellular saturating concentrations of ARP toward AP sites (1 mM) are achieved after 1 min (Fig. 2). Inside the cells, ARP reacts with AP sites in the chromosome. The increasing signal for AP sites from cells treated with hydrogen peroxide or MMS attests to the ability of ARP to penetrate cells and react with AP sites in the DNA. O-Methoxyamine, which reacts with AP sites, competed for AP sites in DNA of living cells and decreases the signal from ARP. Also, in broken nuclei (prepared by cycles of freezing/thawing), where glycosylases were released and thus have less access to DNA and therefore fewer AP sites will be formed, only 30% of the AP sites were detected relative to control. To estimate the steady-state level of AP sites, we used methoxyamine to bind AP sites for 10 min and then washed it out. Methoxyamine binds to existing AP sites and those that have been generated during the incubation. When the AP assay was applied to these cells, the level of AP sites was lower than their level in control cells by 0.67 AP site/106 nucleotides (or ≈20%). This suggests that the level of AP sites at steady state is lower than 1 AP site/106 nucleotides. Since ARP is readily permeable to cells and reacts quickly with AP sites (12), we conclude that ARP measures the AP sites that formed spontaneously and those formed by the activity of glycosylases. Consequently, we suggest that ARP competes for AP sites (or the 5′-deoxyribose phosphate flap in DNA) with the lyase activity of β-pol. Thus, the number of AP sites measured by this approach is an integrated value over a defined interval.
To assess the status of AP sites with aging, we have used human lung fibroblasts (IMR90 cells), an in vitro model for studying aging, and human leukocytes isolated from old and young donors. IMR90 cells are young at low population doubling level (PDL) and old (or senescent) at high population doubling. A 2- to 3-fold increase in the number of AP sites is observed in senescent IMR90 cells compared with young cells (Table 2). In addition, the rate of generation of AP sites in senescence IMR90 cells is found to be 3-fold higher than the rate in young cells (Fig. 3). Similarly, in human leukocytes from old donors, the number of AP sites significantly increases (7-fold) compared with those from young donors. The high number of AP sites in old cells may reflect (i) an increase in endogenous oxidative insult with age that increases the damage to DNA (28, 29), repair by BER, and formation of AP sites, and/or (ii) an age-dependent decline in repair of AP sites. To test these possibilities, we used IMR90 cells to induce AP sites by pulse-treatment of old and young cells with H2O2, and then followed their repair during recovery. The induced AP sites were repaired in young IMR90 cells within ≈15 min posttreatment, which also indicates normal performance of AP endonuclease and β-pol. A similar time of repair of oxidized bases was also seen in human lymphoblasts (30), in Hela cells (31), and in Hela cells for oxo8Gua (K. Beckman and B.N.A., unpublished data). Senescent IMR90 cells, on the other hand, were slower in repairing AP sites with concomitant loss of 39% of viability after only 45 min, presumably because of the elevated levels of general oxidative damage. The limitation of repairing AP sites, together with an elevated oxidative stress with age (32–35) also (36) may explain the age-dependent increase in the level of AP sites in the genome of senescent cells. A decline in repair factors, such as AP endonuclease or β-pol, which function downstream to glycosylases could also increase the duration of AP sites in the genome. Unbalanced activity of the repair enzymes has been suggested previously to induce a high level of AP sites in DNA, and, as a result, increase the rate of mutation (37) and cell death (38, 39). Furthermore, the fact that ARP binds to AP sites in the genome of living cells suggests that the AP site in vivo is available to react with exogenous factors (i.e., ARP) and probably intracellular components (polyamines and basic proteins). In vitro polyamines (or basic proteins) react with AP sites in chromatin and convert them to single-strand breaks (40), a reaction that may occur in vivo as well. A high level of AP sites in both strands of DNA presumably will increase the formation of DNA double-strand breaks (39, 41, 42).
Using the same method, we assessed the activity of glycosylases for oxidized and alkylated bases in both age groups. For this purpose, we challenged IMR90 cells with: (i) hydrogen peroxide to generate oxidized bases or (ii) MMS to generate alkylated bases (43). Both agents induced AP sites considerably above the basal level. In IMR90 cells, MMS induced AP sites by 24- and 324-fold in senescent vs. young cells, respectively (Table 2). Hydrogen peroxide, on the other hand, induces a 2- and 5-fold increase in AP sites in senescent and young cells, respectively (Table 1). The difference between senescent and young cells in the fold of induction of AP sites suggests that the glycosylases that remove oxidized bases (8-oxoguanine DNA glycosylase and others) and methylated bases (3-methyladenine DNA glycosylase) decline in activity with age. The difference at the absolute levels of induction of AP sites between H2O2 and MMS (Tables 1 and 2) may be because of the fact that H2O2 is destroyed by catalase. This activity does not change with age (data not shown). Thus, the effective intracellular concentration of H2O2 will be very low; whereas one protective mechanism against the high concentration of MMS (20 mM) relies on glutathione, which does decline with age (28). Thus the decline with age in the induction of AP sites by MMS cannot be explained by efficient detoxification of MMS or H2O2 by senescent cells and suggests equal interaction with the DNA in both types of cells. In IMR90 cells, the changes with age in the glycosylases that remove oxidized bases from DNA were less dramatic (5-fold vs. 2-fold) compared with those that remove alkylated bases (i.e., 3-methyladenine-DNA glycosylase, 324-fold vs. 24-fold). In conclusion, it seems likely that the age-dependent increase at the basal level of AP sites originates, in large part, from endogenous oxidative damage to DNA by the activity of glycosylases that excise oxidized bases. Also, the decline in removal of AP sites from the DNA (Table 1) could contribute to the elevated level of AP sites in senescent cells. Similarly, on challenge with MMS, leukocytes from old and young donors respond differentially. A high level of AP sites was induced (50-fold) in leukocytes from young donors, whereas only low level of AP sites was induced in leukocytes from old donors. These findings suggest a decline with age in 3-methyladenine-DNA glycosylase that excises methylated bases (and other alkylated bases) from DNA in senescent IMR90 cells and leukocytes form old donors. This is consistent with the previously reported age-dependent decline in DNA repair in lymphocytes and primary skin fibroblasts of normal donors (44–46).
The level of AP sites in nDNA from old rat livers increased 69% compared with nDNA from young rat livers. These data agree with the increase in oxidative events that cause DNA damage and/or differential BER activity (as in IMR90 and leukocytes) and in previously described results for hepatocytes isolated from old and young livers (47). In contrast, we find no significant increase in AP sites in nDNA from brains of old rats. This could be because of a low rate of their formation by glycosylases, which agrees with the suggested age-dependent decline in BER in brain (48). This issue still needs to be studied further to assess the changes of BER with age in different tissues.
The discrepancy in the level of AP sites between brain and liver from young rats between our study and that presented in ref. 23 could possibly be because of: (i) differences in the anesthesia used, ether in the present study vs. metofane in ref. 23; (ii) nuclei were isolated in ref. 23 under conditions that permit oxidative stress, which can modify the level and the distribution of AP sites; and (iii) nuclei were isolated in ref. 23 at 2,000 × g instead of 500 × g in our study. Precipitation of nuclei with contaminating mitochondria may occur especially in brain and muscles. In homogenization of brain, synaptosomes are formed that may trap mitochondria, also mitochondria from heart muscle are connected to muscular filaments. These structures tend to precipitate at 2,000 × g. In our hands, isolated mitochondria possess a very high level of AP sites (data not shown).
We conclude that: (i) The assay for AP sites is fast, quantitative and can be applied to a wide range of sample types including living cells and isolated nuclei. (ii) Trapping AP sites in living cells helps overcome the possible artifacts introduced to the level of AP sites in DNA by isolation. (iii) The assay for AP sites can be used to demonstrate BER in living cells. (iv) Old IMR90 cells and human leukocytes isolated from old donors possess a reduced activity of glycosylases that remove methylated bases, suggesting a decline in the activity of BER.
Acknowledgments
We thank J. Nakamura and J. Swenberg for their help in learning the assay; M. Mack and R. Ingersoll for help with the GC-MS assay; and S. Linn, S. Wallace, J. Nakamura, J. Swenberg, and S. Wilson for criticisms. This work was supported by National Institutes of Health/National Institute on Aging Grant AG17140, National Cancer Institute Outstanding Investigator Grant CA39910, and National Institute of Environmental Health Sciences Center Grant ES1896 (to B.N.A.).
Abbreviations
- AP
apurinic/apyrimidinic
- ARP
aldehyde-reactive probe
- UNG
uracil-DNA-N-glycosylase
- nDNA
nuclear DNA
- MMS
methyl methanesulfonate
- HRP
horseradish peroxidase
- BER
base excision repair
- DAPER
(N,N′-bis(3,3′-(dimethyl-amino)propylamine)-3,4,9,10-perylenetetra-carboxylicdiimide)
References
- 1.Croteau D L, Bohr V A. J Biol Chem. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T. Nature (London) 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 3.Kubota Y, Nash R A, Klungland A, Schar P, Barnes D E, Lindahl T. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava D K, Berg B J, Prasad R, Molina J T, Beard W A, Tomkinson A E, Wilson S H. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 5.Loeb L A. Cell. 1985;40:483–484. doi: 10.1016/0092-8674(85)90191-6. [DOI] [PubMed] [Google Scholar]
- 6.Strauss B S. BioEssays. 1991;13:79–84. doi: 10.1002/bies.950130206. [DOI] [PubMed] [Google Scholar]
- 7.Zhou W, Doetsch P W. Proc Natl Acad Sci USA. 1993;90:6601–6605. doi: 10.1073/pnas.90.14.6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pourquier P, Ueng L M, Kohlhagen G, Mazumder A, Gupta M, Kohn K W, Pommier Y. J Biol Chem. 1997;272:7792–7796. doi: 10.1074/jbc.272.12.7792. [DOI] [PubMed] [Google Scholar]
- 9.Kingma P S, Osheroff N. J Biol Chem. 1997;272:1148–1155. doi: 10.1074/jbc.272.2.1148. [DOI] [PubMed] [Google Scholar]
- 10.Kingma P S, Osheroff N. J Biol Chem. 1997;272:7488–7493. doi: 10.1074/jbc.272.11.7488. [DOI] [PubMed] [Google Scholar]
- 11.Bertrand J R, Malvy C, Paoletti C. Biochem Biophys Res Commun. 1987;143:768–774. doi: 10.1016/0006-291x(87)91420-3. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura J, Walker V E, Upton P B, Chiang S Y, Kow Y W, Swenberg J A. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 13.Liuzzi M, Talpaert-Borle M. J Biol Chem. 1985;260:5252–5258. [PubMed] [Google Scholar]
- 14.Weinfeld M, Liuzzi M, Paterson M C. Biochemistry. 1990;29:1737–1743. doi: 10.1021/bi00459a011. [DOI] [PubMed] [Google Scholar]
- 15.Ide H, Akamatsu K, Kimura Y, Michiue K, Makino K, Asaeda A, Takamori Y, Kubo K. Biochemistry. 1993;32:8276–8283. doi: 10.1021/bi00083a031. [DOI] [PubMed] [Google Scholar]
- 16.Kubo K, Ide H, Wallace S S, Kow Y W. Biochemistry. 1992;31:3703–3708. doi: 10.1021/bi00129a020. [DOI] [PubMed] [Google Scholar]
- 17.Lindahl T, Nyberg B. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 18.Maulik G, Botchway S, Chakrabarti S, Tetradis S, Price B, Makrigiorgos G M. Nucleic Acids Res. 1999;27:1316–1322. doi: 10.1093/nar/27.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strauss P R, Beard W A, Patterson T A, Wilson S H. J Biol Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 20.Bailly V, Verly W G. Biochem J. 1988;253:553–559. doi: 10.1042/bj2530553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailly V, Verly W G. Biochem J. 1989;259:761–768. doi: 10.1042/bj2590761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bricteux-Gregoire S, Verly W G. Biochem J. 1991;273:777–782. doi: 10.1042/bj2730777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura J, Swenberg J A. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 24.Chen Q, Fischer A, Reagan J D, Yan L J, Ames B N. Proc Natl Acad Sci USA. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warner H R, Duncan B K, Garrett C, Neuhard J. J Bacteriol. 1981;5:687–695. doi: 10.1128/jb.145.2.687-695.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blount B C, Ames B N. Anal Biochem. 1994;219:195–200. doi: 10.1006/abio.1994.1257. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z R, Rill R L. Anal Biochem. 1996;236:139–145. doi: 10.1006/abio.1996.0142. [DOI] [PubMed] [Google Scholar]
- 28.Ito Y, Kajkenova O, Feuers R J, Udupa K B, Desai V G, Epstein J, Hart R W, Lipschitz D A. J Gerontol A Biol Sci Med Sci. 1998;53:M169. doi: 10.1093/gerona/53a.3.m169. —M175. [DOI] [PubMed] [Google Scholar]
- 29.Chan S S, Monteiro H P, Deucher G P, Abud R L, Abuchalla D, Junqueira V B. Free Radical Biol Med. 1998;24:1411–1418. doi: 10.1016/s0891-5849(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 30.Jaruga P, Dizdaroglu M. Nucleic Acids Res. 1996;24:1389–1394. doi: 10.1093/nar/24.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moran M F, Ebisuzaki K. Carcinogenesis. 1987;8:607–609. doi: 10.1093/carcin/8.4.607. [DOI] [PubMed] [Google Scholar]
- 32.Ames B N, Shigenaga M K, Hagen T M. Biochim Biophys Acta. 1995;1271:165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- 33.Beckman K B, Ames B N. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 34.Hagen T M, Yowe D L, Bartholomew J C, Wehr C M, Do K L, Park J Y, Ames B N. Proc Natl Acad Sci USA. 1997;94:3064–3069. doi: 10.1073/pnas.94.7.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shigenaga M K, Hagen T M, Ames B N. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atamna, H., Paler-Martínez, A. & Ames, B. N. (2000) J. Biol. Chem., in press. [DOI] [PubMed]
- 37.Glassner B J, Rasmussen L J, Najarian M T, Posnick L M, Samson L D. Proc Natl Acad Sci USA. 1998;95:9997–10002. doi: 10.1073/pnas.95.17.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ochs K, Sobol R W, Wilson S H, Kaina B. Cancer Res. 1999;59:1544–1551. [PubMed] [Google Scholar]
- 39.Wallace S S. Radiat Res. 1998;150:S60–S79. [PubMed] [Google Scholar]
- 40.Male R, Fosse V M, Kleppe K. Nucleic Acids Res. 1982;10:6305–6318. doi: 10.1093/nar/10.20.6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blount B C, Mack M M, Wehr C M, MacGregor J T, Hiatt R A, Wang G, Wickramasinghe S N, Everson R B, Ames B N. Proc Natl Acad Sci USA. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harrison L, Hatahet Z, Wallace S S. J Mol Biol. 1999;290:667–684. doi: 10.1006/jmbi.1999.2892. [DOI] [PubMed] [Google Scholar]
- 43.Beranek D T. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 44.Gadhia P K. Mutagenesis. 1998;13:151–152. doi: 10.1093/mutage/13.2.151. [DOI] [PubMed] [Google Scholar]
- 45.Moriwaki S, Ray S, Tarone R E, Kraemer K H, Grossman L. Mutat Res. 1996;364:117–123. doi: 10.1016/0921-8777(96)00029-8. [DOI] [PubMed] [Google Scholar]
- 46.Wei Q, Matanoski G M, Farmer E R, Hedayati M A, Grossman L. Proc Natl Acad Sci USA. 1993;90:1614–1618. doi: 10.1073/pnas.90.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kennah H E, 2nd, Coetzee M L, Ove P. Mech Ageing Dev. 1985;29:283–298. doi: 10.1016/0047-6374(85)90068-5. [DOI] [PubMed] [Google Scholar]
- 48.Niedermuller H, Hofecker G, Skalicky M. Mech Ageing Dev. 1985;29:221–238. doi: 10.1016/0047-6374(85)90064-8. [DOI] [PubMed] [Google Scholar]