

Abstract
Protein synthesis requires a large commitment of cellular resources and is highly regulated. Previous studies have shown that a number of factors that mediate the initiation and elongation steps of translation are regulated by phosphorylation. In this report, we show that a factor involved in the termination step of protein synthesis is also subject to phosphorylation. Our results indicate that eukaryotic release factor 1 (eRF1) is phosphorylated in vivo at serine 421 and serine 432 by the CK2 protein kinase (previously casein kinase II) in the budding yeast Saccharomyces cerevisiae. Phosphorylation of eRF1 has little effect on the efficiency of stop codon recognition or nonsense-mediated mRNA decay. Also, phosphorylation is not required for eRF1 binding to the other translation termination factor, eRF3. In addition, we provide evidence that the putative phosphatase Sal6p does not dephosphorylate eRF1 and that the state of eRF1 phosphorylation does not influence the allosuppressor phenotype associated with a sal6Δ mutation. Finally, we show that phosphorylation of eRF1 is a dynamic process that is dependent upon carbon source availability. Since many other proteins involved in protein synthesis have a CK2 protein kinase motif near their extreme C termini, we propose that this represents a common regulatory mechanism that is shared by factors involved in all three stages of protein synthesis.
Protein synthesis is carried out in three distinct steps: initiation, elongation, and termination. While the first two steps have been extensively studied, our understanding of the termination process has lagged behind. Two classes of release factors mediate translation termination in the budding yeast Saccharomyces cerevisiae and other eukaryotes. Eukaryotic release factor 1 (eRF1) (encoded by the SUP45 gene) is a class I release factor that recognizes any of the three stop codons when they are located in the ribosomal A site (5, 10). Following stop codon recognition, eRF1 also induces polypeptide chain release by activating the peptidyl transferase center of the ribosome. eRF3 (encoded by the SUP35 gene) is a class II release factor that facilitates stop codon recognition and stimulates the termination reaction in a GTP-dependent manner (17, 37).
Protein synthesis requires a major commitment of cellular energy and resources. It is not surprising that translation is regulated by multiple mechanisms in response to external stimuli such as nutrient abundance or environmental stress. One mechanism of modulating translational efficiency is through the phosphorylation of various translation factors. Probably the most well-characterized example of this regulation is the phosphorylation of serine 51 of the α subunit of the mammalian translation initiation factor eukaryotic initiation factor 2 (eIF2α). When eIF2α is phosphorylated, it competitively inhibits GTP exchange by eIF2B (39). As a result, the ternary complex of 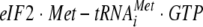 cannot be replenished, and translation initiation is inhibited. The importance of this regulatory mechanism is underscored by its strong conservation throughout essentially all eukaryotic species.
cannot be replenished, and translation initiation is inhibited. The importance of this regulatory mechanism is underscored by its strong conservation throughout essentially all eukaryotic species.
The elongation step of translation is also subject to regulation by phosphorylation in eukaryotes. For example, eukaryotic elongation factor 1A (eEF1A) can be phosphorylated on threonine 431 by protein kinase Cδ in mammalian cells (26). Insulin can also increase the phosphorylation of eEF1A, as well as the phosphorylation of the α and γ subunits of its exchange factor, eEF1B, through the action of the multipotential S6 kinase (9). These modifications are thought to enhance translation rates. In contrast, phosphorylation of threonine 56 in eEF2 by the novel eEF2 kinase strongly inhibits translation in mammalian cells (8). These examples illustrate the complexity of phosphorylation of specific translation factors on the overall efficiency of the translation process.
In this report, we describe the novel observation that eRF1 is phosphorylated in the budding yeast S. cerevisiae. Since human eRF1 was previously found to reside in a stable complex with the catalytic subunit of protein phosphatase 2A (PP2A) (2), these results raise the possibility that the termination step of translation, like initiation and elongation, may be subject to regulation by phosphorylation. Here, we characterize the site of yeast eRF1 phosphorylation, show that it is phosphorylated by the CK2 protein kinase, and demonstrate that this posttranslational modification is dependent upon carbon source availability.
MATERIALS AND METHODS
Strains and growth conditions.
The Saccharomyces cerevisiae strains used in this study are described in Table 1. Strains YDB447 and YDB640 were derived from YDB340 by using standard genetic techniques. Strain YDB640 was constructed using a one-step gene replacement strategy. The entire SAL6 open reading frame was removed by transforming yeast strains YDB340 and YDB447 with a PCR fragment containing the TRP1 gene from Candida glabrata with flanking homology upstream and downstream of SAL6. The C. glabrata TRP1 gene was amplified from pCGTRP1 (27). The UPF1 gene was deleted in strain YDB641 by using a similar strategy. Candidate deletion strains were screened by PCR to verify the insertion. Construction of strains YDB340 andYDB447 have been described elsewhere previously (37). Strains YDH6 and YDH8 were kind gifts from Claiborne V. C. Glover III, and their construction has been described previously (21).
TABLE 1.
Strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| YDB340 | MATα ura3-52 leu2-3,112 his3-Δ200 trp1-Δ901 lys2-80 suc2-Δ901[psi−] | 37 |
| YDB447 | MATα ura3-52 leu2-3,112 his3-Δ200 trp1-Δ901 lys2-80 suc2-Δ901 sup45::HIS3[psi−] | 37 |
| YDB640 | MATα ura3-52 leu2-3,112 his3-Δ200 trp1Δ-901 lys2-80 suc2-Δ901 sup45Δ::HIS3 sal6Δ::TRP1[psi−] | This study |
| YDB641 | MATα ura3-52 leu2-3,112 his3-Δ200 trp1Δ-901 lys2-80 suc2-Δ901 sup45Δ::HIS3 upf1Δ::TRP1[psi−] | This study |
| YDH6 | MATacka1-Δ1::HIS3 cka2-Δ1::TRP1 CKA2::LEU2 | 21 |
| YDH8 | MATacka1-Δ1::HIS3 cka2-Δ1::TRP1 cka2-8ts::LEU2 | 21 |
Yeast extract-peptone-dextrose is a rich medium supplemented with 2% glucose; synthetic medium supplemented with 2% dextrose (glucose) and amino acids was also used. Wickerham's minimal medium is a defined minimal medium (43). For [32P]orthophosphate labeling, the phosphate concentration in Wickerham's minimal medium was reduced to 100 μM. Other specific nutritional supplements were added as needed.
Plasmids.
The centromeric plasmid pDB800 (with a LEU2 selectable marker) carries the wild-type SUP45 gene with 1,568 bp upstream of the AUG start codon and 1,036 bp downstream of the UAA stop codon. pDB800 was also used as a template for site-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene). The S421A mutation was introduced into the SUP45 gene and verified by sequencing, and a SpeI-HindIII fragment was then subcloned back into pDB800 to generate pDB841. Using a similar approach, plasmids encoding the following SUP45 mutant alleles were constructed: S432A (pDB859), S421A/S432A (pDB902), S421D (pDB857), S432D (pDB860), and S421D/S432D (pDB858). Similarly, the I222S mutation was introduced into the SUP45 gene and subcloned back into pDB800 using a BlpI-SpeI fragment to generate pDB970.
To make a construct to express the eRF1 mutant lacking the C-terminal 19 amino acids (eRF1-CΔ19), an SpeI site was generated immediately 5′ of the stop codon of SUP45. A BlpI-HindIII fragment was then subcloned back into pDB800, digested with SpeI, and ligated together to precisely delete the last 19 codons. The resulting plasmid was named pDB843. The plasmid used for protein purification of N-terminal His6-tagged eRF1-CΔ19 was made by digesting plasmid pDB843 with SpeI and HindIII and subcloned into SUP45-pET-3A vector pDB698 to create pDB897. Similarly, plasmid pDB858 was digested with SpeI-HindIII and ligated into pDB698 to introduce the eRF1 S421D/S432D double mutant into the pET-3A vector (pDB941).
Metabolic labeling and Western blots.
Yeast strains were grown in Wickerham's low-phosphate minimal medium at 30°C. Cells were grown to mid-log phase (0.5 to 1.0 A600 units/ml), and 2.2 optical density (OD) units of each strain were harvested and resuspended at a cell density of 4 OD units/ml. Cells were then labeled with 200 μCi/ml of EXPRESS 35S protein labeling mix (Perkin-Elmer) for 1 h or with 400 μCi/ml of [32P]orthophosphate (Amersham) for 2 h unless otherwise specified. After labeling, 2.5 OD units of cells were harvested for each sample, and 100% trichloroacetic acid was added to achieve a final trichloroacetic acid concentration of 7%. Cells were incubated on ice for 20 min, harvested by centrifugation, washed twice with 100% acetone, and dried under a vacuum. Cells were then resuspended in 50 μl of sodium dodecyl sulfate (SDS) boiling buffer (1% SDS, 50 mM Tris, pH 7.5, 1 mM EDTA), lysed by agitation with glass beads, and boiled for 5 min. Next, 800 μl of Tween-IP buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Tween 20, 0.1 mM EDTA) was added to the samples. After a brief centrifugation, 600 μl of the supernatant was removed and combined with either 4 μl (eRF3) or 6 μl (eRF1) polyclonal antiserum and incubated overnight at 4°C. Next, 50 μl of a protein A-agarose bead suspension was added, incubated for 1 h at 4°C, and washed twice with Tween-IP buffer and twice with a 1% solution of 2-mercaptoethanol. The samples were then boiled in SDS gel loading buffer for 5 min. The samples were removed with a Hamilton syringe and loaded onto an 8% SDS-polyacrylamide gel. Gels were developed using a PhosphorImager (GE Healthcare).
Cells for Western blotting were grown and lysed using conditions identical to those described above, but the radioisotopes were omitted from the growth medium. Protein concentrations were determined by the Lowry method (29). Twenty-five micrograms of protein was loaded into each lane of an 8% SDS-polyacrylamide gel. Protein was then transferred to a polyvinylidene difluoride membrane using a Transblot apparatus (Bio-Rad). The membranes were blocked in 0.3% Tween 20-phosphate-buffered saline buffer containing 5% nonfat milk. Membranes were incubated in the same buffer with a 1:400 dilution of eRF1 polyclonal antiserum for 2 h at room temperature or overnight at 4°C. Bands were detected using 125I-protein A, and results were visualized using a PhosphorImager (GE Healthcare).
Glucose depletion metabolic labeling.
The glucose depletion experiment was carried out as described above, with the following modifications. Cells were grown in Wickerham's low-phosphate medium with 2% glucose to mid-log phase (0.5 to 1.0 A600 units/ml). Equal aliquots of cells were harvested and resuspended at 4 A600 units/ml in Wickerham's low-phosphate medium with or without 2% glucose. After further incubation at 30°C for 2 h, 400 μCi/ml [32P]orthophosphate was added, and incubation was continued for 2 h. Cells were then harvested and processed as described above. For the chase experiment, cells were grown in Wickerham's low-phosphate medium in the presence of 2% glucose to mid-log phase (0.5 to 1.0 A600 units/ml). Cells were then harvested and labeled for 2 h at 4 A600 units/ml in fresh Wickerham's low-phosphate medium containing 2% glucose. After 2 h, equal aliquots of cells were harvested and resuspended to 4 A600 units/ml in Wickerham's medium with phosphate in the presence or absence of 2% glucose to initiate the chase period. Samples were taken at 0, 0.5, 1, 2, and 4 h after initiating the chase period and processed as described above.
To assess the role of CK2 protein kinase in eRF1 phosphorylation, strains YDH6 and YDH8 were grown to early log phase at 25°C in Wickerham's low-phosphate medium. Equal aliquots of cells were then shifted to 37°C or maintained at 25°C with shaking for an additional 12 h (approximately three generations). The cells were then harvested and resuspended at 4 A600 units/ml in fresh medium, and aliquots were metabolically labeled as described above at either 25°C or 37°C for two more hours. The cells were then lysed, and samples were processed as described above.
Analytical ultracentrifugation.
Purification of N-terminal His6-tagged eRF1 and His6-tagged eRF3 (residues 254 to 685) from the soluble fraction of Escherichia coli lysates has been described previously (37). Immediately following purification, proteins were dialyzed in phosphate-buffered saline (140 mM NaCl, 20 mM NaPO4, pH 7.4). Protein concentrations were determined using Beer's law: A = elc, where A is the absorbance, e is the molar absorptivity, l is the light path through the sample, and c is the concentration. Protein concentrations for analysis were 29 μM for individual proteins or 58 μM total for the mixtures of eRF1 and eRF3. For complex formation, eRF1 and eRF3 were mixed in 1:1 molar ratios and incubated on ice for 20 min prior to centrifugation. Sedimentation values were obtained using a Beckman XL-A ultracentrifuge with a four-channel An-60 Ti rotor. Samples were centrifuged at 50,000 rpm at 4°C for a total of 150 scans. Distributions of the sedimentation coefficients were obtained using the software program SEDFIT (available at http://www.analyticalultracentrifugation.com).
Readthrough assays.
The stop codon readthrough assays were performed using a bicistronic reporter construct consisting of a Renilla luciferase gene followed by an in-frame firefly luciferase gene. Separating the two genes is either a tetranucleotide termination signal (e.g., UAA A) or a similar sequence containing a sense codon (e.g., CAA A). The construct is driven by the PGK1 promoter and has a CYC1 poly(A) addition signal. Cells were grown in synthetic medium supplemented with 2% dextrose and the appropriate amino acids. Percent readthrough was determined by taking the ratio of the firefly/Renilla activities obtained from the termination signal construct relative to the firefly/Renilla activities for the control (sense codon) construct. Samples were processed in quintuplicate, and each experiment was repeated at least twice (for further details, see reference 25).
Northern blots.
RNA was extracted from cells using a hot-phenol extraction method (38). Total RNA (25 μg/lane) from the indicated strains was resolved on a 1% agarose-formaldehyde gel and transferred onto a nitrocellulose membrane using a Posi-Blot pressure blotter (Stratagene). Samples were baked in a vacuum oven at 80°C and then probed with a 32P-labeled DNA probe for the CYH2 gene. After results were visualized using a PhosphorImager (GE Healthcare), the membrane was stripped by incubation in stripping solution (10 mM Tris-HCl, pH 7.5, 0.2% SDS) for 1.5 h at 75°C. The membrane was then probed with an ACT1 probe for the loading control, and results were again visualized using a PhosphorImager (GE Healthcare).
RESULTS
eRF1 is phosphorylated by CK2 protein kinase in vivo.
Due to the frequent phosphorylation of translation initiation and elongation factors, we began this study by asking whether either one of the translation termination factors eRF1 and eRF3 is phosphorylated in the budding yeast Saccharomyces cerevisiae. Yeast cultures were metabolically labeled with [32P]orthophosphate or [35S]methionine for 2 h, and immunoprecipitation experiments were carried out using polyclonal antibodies to either eRF1 or eRF3. Figure 1 shows that while both eRF1 and eRF3 readily incorporated [35S]methionine, only eRF1 could be labeled with [32P]orthophosphate. These results demonstrate that eRF1 is a phosphoprotein in yeast cells.
FIG. 1.

eRF1 is phosphorylated in vivo. Yeast strain YDB447 expressing wild-type eRF1 (from plasmid pDB800) was labeled with [35S]methionine (35S) or [32P]orthophosphate (32P). eRF1 or eRF3 was immunoprecipitated from radiolabeled cells using rabbit polyclonal antiserum specific for each factor.
Using the NetPhos 2.0 phosphorylation site predictor (http://www.cbs.dtu.dk/services/NetPhos), we found that 10 serine and 3 threonine residues reside within predicted phosphorylation sites in eRF1. We compared this information with a search of the literature for other proteins involved in translation that are also phosphorylated. Table 2 shows a list of seven proteins that are phosphorylated near their C termini in consensus CK2 protein kinase motifs. Strikingly, eRF1 also has two consensus CK2 protein kinase motifs within its last 17 amino acids (see Fig. 3B). Our analysis suggested that a number of other proteins involved in translation also have CK2 protein kinase motifs near their extreme C termini (see Discussion). The conserved nature of these potential CK2 protein kinase motifs led us to next test whether the CK2 protein kinase is responsible for eRF1 phosphorylation in vivo.
TABLE 2.
Known CK2 phosphorylation sites near the C termini of translation factors
| Gene | Protein | Phosphorylation sitea | Function | Reference |
|---|---|---|---|---|
| RPP2B | P2β | . . .KEESDDDM. . . | Component of ribosomal stalk | 3 |
| RPP2A | P2α | . . .AEESDDDM. . . | Component of ribosomal stalk | 3 |
| RPP1B | P1β | . . .AEESDDDM. . . | Component of ribosomal stalk | 3 |
| RPP1A | P1α | . . .KEESDDDM. . . | Component of ribosomal stalk | 3 |
| RPP0 | P0 | . . .EEESDDDM. . . | Component of ribosomal stalk | 3 |
| TIF5 | eIF5 | . . .IISEEE. . . | Promotes GTP hydrolysis by eIF2 | 30 |
| . . .AESDDDEEDDE* | ||||
| SUI2 | eIF2α | . . .DSEDDEDESDDE* | α subunit of eIF2 | 16 |
An asterisk (*) indicates the position of the stop codon.
FIG. 3.

eRF1 is phosphorylated at serine residues 421 and 432. (A) [32P]orthophosphate labeling in strains expressing wild-type eRF1 (WT), eRF1-S421A, eRF1-S432A, or eRF1-S421A/S432A. (B) Amino acid sequence of the C-terminal 28 residues of eRF1. The locations of serine residues that reside in consensus CK2 protein kinase phosphorylation sites (S-X-X-D/E) that were mutated in A are underlined (31). The asterisk (*) indicates the position of the stop codon.
CK2 protein kinase activity is essential for cell viability in S. cerevisiae and is encoded by two genes, CKA1 and CKA2 (35). Consequently, we used a strain, YDH8, that retains only temperature-sensitive CK2 protein kinase activity due to a disruption of the CKA1 gene and the presence of a temperature-sensitive mutation in the CKA2 gene (cka2ts) to test the hypothesis that eRF1 is phosphorylated by the CK2 protein kinase. This cka2ts strain was previously used to determine whether other yeast phosphoproteins are substrates for the CK2 protein kinase (4, 7, 44). Yeast cultures were grown at the permissive temperature (25°C) to mid-log phase, shifted to the restrictive temperature of 37°C, and grown for an additional 12 h (approximately three generations). Cells were then labeled with [32P]orthophosphate, and eRF1 was immunoprecipitated to determine its level of phosphorylation (Fig. 2). Cells from parallel cultures were harvested for Western blot analysis to correct for any differences in the steady-state level of eRF1 under these conditions. The results show a 70% decrease in the phosphorylation state of eRF1 in the temperature-sensitive mutant (relative to the wild-type strain) at the restrictive temperature. These results suggest that yeast eRF1 is an in vivo target of the CK2 protein kinase.
FIG. 2.

eRF1 is phosphorylated by CK2 protein kinase in vivo. (A) Metabolic labeling of eRF1 in wild-type strain YDH6 (WT) or the CK2 temperature-sensitive mutant YDH8 (TS). Western blot data (WB) to determine steady-state protein levels and [32P]orthophosphate metabolic labeling (32P) were used to generate the ratios of 32P incorporated per unit of eRF1 protein shown in B. Twenty-five micrograms of total protein was loaded into each lane for each Western blot, while 2.5 A600 units of cells were immunoprecipitated with an eRF1-specific antiserum in each 32P-labeling experiment. The data represent the mean values (± the standard deviation) of multiple experiments normalized to the wild-type strain grown at 25°C.
The experiments presented above, as well as the results from the NetPhos 2.0 analysis, suggested that eRF1 phosphorylation occurred at one or both CK2 protein kinase sites located within the C-terminal 17 amino acids of eRF1. To test this hypothesis, site-directed mutagenesis was used to change each of the serine residues at positions 421 and 432 to alanine, both individually and together, and plasmids encoding the mutant forms of eRF1 were introduced into a sup45Δ strain expressing wild-type eRF1 from a plasmid carrying the URA3 gene as a selectable marker. Plasmid shuffling (6) was then carried out by plating the transformants onto medium supplemented with 5-fluoroorotic acid, a uracil analogue that allows the formation of colonies only from cells that have lost the wild-type SUP45 plasmid with the URA3 marker. Each strain was then tested for the ability to phosphorylate eRF1 (Fig. 3). The results show that the introduction of either the S421A or S432A mutation reduced the level of eRF1 phosphorylation, while both mutations together eliminated most (if not all) eRF1 phosphorylation. These results indicate that the serine residues at positions 421 and 432 are the major sites of eRF1 phosphorylation in vivo.
The phosphorylation state of eRF1 has little effect on the efficiency of translation termination or nonsense-mediated mRNA decay (NMD).
Having identified the two major sites of eRF1 phosphorylation, we used the phosphorylation mutants described above to characterize the role that eRF1 phosphorylation plays in the cell. The double mutant eRF1-S421A/S432A allowed us to examine how the loss of eRF1 phosphorylation affects the function of this protein. We also constructed another mutant, eRF1-S421D/S432D, in which the phosphorylated serine residues were changed to aspartic acid to mimic constitutive phosphorylation. We first used these mutants to determine whether phosphorylation plays a role in translation termination. To do this, we used a dual luciferase reporter system (19, 25, 37). Briefly, this system utilizes tandem Renilla and firefly luciferase genes that are separated by a single in-frame stop codon (Fig. 4). The activity of firefly luciferase, encoded by the distal open reading frame, provides a quantitative measure of the readthrough of the stop codon that separates the two open reading frames. The activity of Renilla luciferase, encoded by the proximal open reading frame, serves as an internal control for mRNA abundance and translation initiation rates. Since the efficiency of translation termination is influenced not only by the stop codon but also by the first nucleotide after a stop codon (together referred to as the tetranucleotide termination signal), we examined the effect of the eRF1 phosphorylation mutants on all possible tetranucleotide termination signals (Table 3). We found that the mutations that eliminated phosphorylation (eRF1-S421A/S4342A) caused a small (1.2- to 1.5-fold) increase in readthrough at the UAA A, UAA U, and UAG G termination signals. Similarly, the mutations that mimicked constitutive phosphorylation (eRF1-S421D/S432D) exhibited a small (1.3- to 1.6-fold) increase in readthrough at the UAA A, UAA U, UAG C, and UGA G termination signals but had little effect on stop codon recognition at other signals. These increases are significant (P ≤ 0.01) but relatively small compared to an eRF1 mutant lacking the extreme C-terminal 19 amino acids (3.3- to 17.6-fold) (data not shown). Taken together, these results indicate that the phosphorylation state of eRF1 has little effect on the efficiency of stop codon recognition in vivo.
FIG. 4.

The dual luciferase reporter system used to monitor the readthrough of stop codons in vivo.
TABLE 3.
Effect of eRF1 phosphorylation status on readthrough of stop codonsa
| Termination signal | % Readthrough ± SDb
|
Fold change (relative to WT)
|
|||
|---|---|---|---|---|---|
| WT | S421A/S432A | S421D/S432D | S421A/S432A | S421D/S432D | |
| UAA A | 0.30 ± 0.03 | 0.41 ± 0.02 | 0.38 ± 0.03 | 1.37c | 1.27c |
| UAA C | 0.51 ± 0.08 | 0.51 ± 0.03 | 0.63 ± 0.06 | 1.00 | 1.24 |
| UAA G | 0.37 ± 0.05 | 0.34 ± 0.05 | 0.37 ± 0.05 | 0.92 | 1.02 |
| UAA U | 0.23 ± 0.01 | 0.28 ± 0.02 | 0.36 ± 0.06 | 1.21c | 1.56c |
| UAG A | 0.21 ± 0.01 | 0.23 ± 0.02 | 0.25 ± 0.03 | 1.10 | 1.19 |
| UAG C | 0.49 ± 0.04 | 0.46 ± 0.02 | 0.69 ± 0.06 | 0.94 | 1.41c |
| UAG G | 0.23 ± 0.02 | 0.34 ± 0.02 | 0.27 ± 0.02 | 1.49c | 1.17 |
| UAG U | 0.16 ± 0.01 | 0.16 ± 0.01 | 0.16 ± 0.02 | 1.00 | 0.98 |
| UGA A | 0.67 ± 0.09 | 0.58 ± 0.08 | 0.67 ± 0.09 | 0.87 | 1.00 |
| UGA C | 1.33 ± 0.14 | 1.32 ± 0.25 | 1.23 ± 0.08 | 0.99 | 0.92 |
| UGA G | 0.58 ± 0.06 | 0.62 ± 0.05 | 0.80 ± 0.09 | 1.07 | 1.38c |
| UGA U | 0.22 ± 0.03 | 0.21 ± 0.03 | 0.20 ± 0.02 | 0.95 | 0.91 |
All measurements were carried out in sup45Δ strain YDB447 carrying pDB800 (eRF1 wild type [WT]), pDB902 (eRF1-S421A/S432A), or pDB858 (eRF1-S421D/S432D).
Percent readthrough is expressed as mean ± standard deviation.
Significantly different from the wild-type strain (P ≤ 0.01), as determined by the Mann-Whitney statistical test.
NMD rapidly degrades mRNAs that contain premature stop mutations in a process that is intimately linked with translation termination (1, 25). The induction of NMD in yeast requires a termination complex (eRF1 and eRF3 bound to the 80S ribosome at a stop codon), three NMD factors (Upf1p, Upf2p/Nmd2p, and Upf3p), and an aberrant 3′ untranslated region in the mRNA (1, 18). Furthermore, eRF1 interacts with Upf1p (12). Deletion of any of the three Upf factors will abrogate NMD and prevent the rapid turnover of mRNAs that contain premature stop codons. We used the CYH2 mRNA to test the hypothesis that phosphorylation of eRF1 plays a role in NMD. This mRNA has an intron that is spliced inefficiently. The intron results in an in-frame premature stop codon that normally makes this pre-mRNA a substrate for the NMD pathway. Furthermore, it has been shown that defects in the NMD pathway prevent the rapid turnover of the CYH2 pre-mRNA (13). To determine whether the phosphorylation of eRF1 plays a role in NMD, we carried out Northern blot analysis using a probe that recognized both the precursor and mature forms of the CYH2 mRNA (Fig. 5). We observed a significant accumulation of the unspliced CYH2 transcript in a upf1Δ strain but not in the eRF1-S421A/S432A or the eRF1-S421D/S432D double mutant. These results indicate that the phosphorylation status of eRF1 does not adversely affect the NMD pathway.
FIG. 5.

The phosphorylation state of eRF1 does not affect NMD. Northern blot analysis of the CYH2 transcript was carried out on total RNA from the following strains: wild type (WT), upf1Δ, eRF1 S421A/S432A, and eRF1 S421D/S432D. For Northern blots, 25 μg of total RNA from the indicated strains was loaded into each lane.
Phosphorylation of eRF1 does not affect its ability to bind eRF3.
The release factor eRF1 interacts with its functional partner eRF3 via a binding site near its C terminus (14, 34). Since our data indicate that phosphorylation sites are also located near the C terminus of eRF1, it is possible that the phosphorylation state of eRF1 may influence its ability to interact with eRF3. To test this hypothesis, we examined the association between eRF1 and eRF3 by performing sedimentation velocity experiments using analytical ultracentrifugation (Fig. 6). His-tagged forms of full-length eRF1 or a fragment of eRF3 consisting of amino acid residues 254 to 685 were purified from E. coli extracts using Ni2+-nitrilotriacetic acid affinity chromatography. Since E. coli does not contain protein kinases that recognize CK2 protein kinase sites, wild-type eRF1 purified from E. coli was assumed to be in the dephosphorylated form. As a negative control for eRF1-eRF3 complex formation, we used an eRF1 mutant lacking the last 19 amino acids (eRF1-CΔ19) that does not efficiently bind eRF3 (15). Preliminary experiments found that eRF1, eRF1-CΔ19, and eRF3 each have sedimentation coefficients ranging from 2.07S to 2.09S (Fig. 6). When wild-type (unphosphorylated) eRF1 and eRF3 were incubated together for 20 min at 4°C before centrifugation, we observed a new 3.06S complex that represented the eRF1-eRF3 complex. This indicated that a lack of phosphorylation did not prevent the formation of the eRF1-eRF3 complex. As expected, this 3.06S peak was not observed when eRF1-CΔ19 was incubated with eRF3, indicating that the deletion of the C-terminal 19 amino acids of eRF1 disrupted eRF1-eRF3 complex formation. Incubation of eRF3 with the eRF1 mutant that mimics constitutive phosphorylation (eRF1-S421D/S432D) resulted in the appearance of the 3.06S peak, indicating that complex formation still occurred when acidic residues were present at the sites of eRF1 phosphorylation. Taken together, these results indicate that the phosphorylation state of eRF1 does not play a significant role in eRF1-eRF3 heterodimer formation.
FIG. 6.

Phosphorylation status of eRF1 does not affect its association with eRF3. Sedimentation coefficients of the indicated proteins (indicated in boxes) were generated by analytical ultracentrifugation. The values indicated above the peaks represent the S values derived using the SEDFIT program. S, Svedberg units; c(S) Distribution, differential distribution (fringes/Svedberg).
The state of eRF1 phosphorylation is not affected by the putative phosphatase encoded by SAL6 and does not affect the allosuppressor phenotype associated with a sal6Δ mutant.
Mutations in the SUP45 and SUP35 genes, encoding eRF1 and eRF3, respectively, were originally identified based on their ability to cause readthrough of stop mutations in various biosynthetic genes of S. cerevisiae (22). These mutations were termed omnipotent suppressors because they cause readthrough at all three stop codons. Later, the SAL6 gene was identified in a screen for mutants that enhanced the readthrough phenotypes associated with omnipotent suppressor mutations (40). Mutations in the SAL6 gene alone did not result in a suppressor phenotype, but they exacerbated the readthrough associated with omnipotent suppressor mutations in the SUP35 or SUP45 gene. This enhanced suppression phenotype is referred to as an allosuppressor effect. The SAL6 gene was later sequenced and was found to encode a protein with significant homology to PP1-like protein phosphatases (11, 42). However, it is presently still not clear how the protein encoded by the SAL6 gene mediates the allosuppressor effect.
Given our finding that eRF1 is a phosphoprotein, we next tested the hypothesis that the allosuppressor phenotype associated with the sal6Δ mutation is mediated through its effect on the phosphorylation state of eRF1. To do this, we used the dual luciferase reporter system to compare termination efficiencies in strains expressing the phosphorylation mutants in SAL6 and sal6Δ backgrounds (Fig. 7). Since the allosuppressor effect requires the presence of an omnipotent suppressor mutation, the phosphorylation site mutations were combined with the eRF1-I222S mutation, which was originally identified as the sup45-2 omnipotent suppressor allele (41). When these eRF1 mutant proteins were expressed from a low-copy-number plasmid in a sup45Δ strain, we found that expression of the eRF1-I222S allele as the sole source of eRF1 resulted in a 3.3-fold increase in readthrough (Fig. 7A). When eRF1-I222S was expressed in the sal6Δ strain, we observed a 6.6-fold increase in readthrough, indicating that the presence of the sal6Δ mutation caused an additional twofold increase in readthrough. This observation recapitulated the previously observed allosuppressor effect associated with SAL6 mutations (40). However, we did not detect any significant change in readthrough when either the S421A/S432A or the S421D/S432D phosphorylation site mutation was combined with the eRF1-I222S mutation in the sal6Δ strain. If Sal6p was the phosphatase for eRF1, the sal6Δ mutation might increase the phosphorylation state of eRF1. This hypothesis predicts that the mutant protein that mimics constitutive phosphorylation, eRF1-S421D/S432D, will phenocopy the allosuppressor effect in a SAL6 strain, since the presence of the aspartic acid residues mimics the maximal phosphorylation state of eRF1. However, such an allosuppressor effect was not observed in the SAL6 strain expressing eRF1-I222S/S421D/S432D (Fig. 7A). These results suggest that the allosuppressor effect associated with the sal6Δ mutation is not mediated by its effect on the phosphorylation state of eRF1.
FIG. 7.

The SAL6 allosuppressor phenotype is unaffected by the phosphorylation state of eRF1. (A) Stop codon readthrough in SAL6 or sal6Δ strains was measured using the dual luciferase reporter with the UAA U termination signal. These strains expressed either wild-type eRF1 (WT), eRF1-I222S (the sup45-2 omnipotent suppressor allele), eRF1-I222S/S421A/S432A, or eRF1-I222S/S421D/S432D. (B) [32P]orthophosphate labeling of eRF1 in wild-type (WT) and sal6Δ strains and Western blots (WB) of total protein indicate that 32P incorporated into eRF1 per unit of steady-state eRF1 protein is unaffected by the sal6Δ mutation.
The absolute level of eRF1 phosphorylation might also be expected to increase in a sal6Δ strain if Sal6p is the phosphatase responsible for the dephosphorylation of eRF1 (assuming that it is not already fully phosphorylated under normal conditions). To test this hypothesis, we used our metabolic labeling assay to determine whether any change in the phosphorylation state of eRF1 could be detected in the sal6Δ mutation. As shown in Fig. 7B, we did not observe any significant difference in the phosphorylation state of eRF1 between the wild-type and sal6Δ strains. We cannot rule out the possibility that eRF1 is completely phosphorylated under the growth conditions under which this experiment was carried out. If that were the case, the deletion of its phosphatase would not be expected to show a detectable increase in phosphorylation. However, when considered together with the allosuppressor readthrough data, these results argue that Sal6p is not the phosphatase responsible for the dephosphorylation of eRF1, and the state of eRF1 phosphorylation does not appear to be important for the allosuppressor effect associated with sal6 mutations.
eRF1 phosphorylation and dephosphorylation are dynamic processes that require cell growth.
The results described above indicate that the phosphorylation state of eRF1 does not significantly influence its termination activity. Consistent with this finding, mutations that eliminated the CK2 phosphorylation sites in the five proteins that form the ribosomal stalk shown in Table 1 also did not affect either ribosome activity or the formation of the stalk structure (3, 36). Similarly, studies that altered the CK2 phosphorylation sites in eIF2α and eIF5 did not have any detectible effects on translation initiation (16, 30). However, the existence of CK2 phosphorylation sites in these and other proteins involved in translation suggests that this modification may carry out some common function. To gain further insight into the nature of eRF1 phosphorylation, we next asked whether this modification required active cell growth. Wild-type cells were grown to mid-log phase and harvested, and identical aliquots were resuspended in the same growth medium in the presence or absence of 2% glucose. The cultures were incubated for 2 h at 30°C to deplete any residual glucose in the glucose-depleted culture, and [32P]orthophosphate was then added to each culture. Incubation at 30°C was continued for an additional 2 h, and the cells were harvested. As expected, glucose depletion under these conditions effectively blocked cell growth (Fig. 8A). When we examined the phosphorylation state of eRF1, we found that growth inhibition by glucose depletion essentially eliminated the phosphorylation of eRF1 (Fig. 8B). These results indicate that ongoing cell growth is required for eRF1 phosphorylation to occur.
FIG. 8.

Phosphorylation of eRF1 is reduced upon carbon source starvation. (A) Growth curve of the sup45Δ strain expressing wild-type eRF1 (YDB447 carrying pDB800) during experiments in the presence (+) or absence (−) of glucose. (B) 32P metabolic labeling and Western blot (WB) analysis of eRF1 from cells harvested after incubation in medium with or without glucose. Cells were harvested at 4 h as shown in A.
There are two possible explanations for this result. First, eRF1 may only be phosphorylated once during (or immediately after) its synthesis. By subjecting the cells to glucose depletion, we were inhibiting de novo protein synthesis and consequently the phosphorylation of newly synthesized eRF1. Alternatively, eRF1 may cycle between phosphorylated and dephosphorylated forms during normal growth. When cell growth is inhibited by glucose depletion, this phosphorylation cycle may arrest with cells that contain predominantly the dephosphorylated form of eRF1. To distinguish between these possibilities, we performed a metabolic chase experiment (Fig. 9). Cells were labeled for 2 h with [32P]orthophosphate and then shifted to growth medium with excess unlabeled orthophosphate (with or without glucose) to monitor the persistence of the 32P incorporated into eRF1. We found that eRF1 phosphorylation decreased more than 10-fold during the first 4 h in cells incubated with glucose. Since the doubling time for this strain under these conditions is ∼3.5 h, the large decrease in preexisting phosphorylated eRF1 that we observed was much greater than the twofold decrease expected following one round of cell division. In contrast, eRF1 phosphorylation decreased only by about twofold in cells incubated in the absence of glucose (most of which occurred during the first 30 min, while residual glucose was being metabolized). From these results, we conclude that eRF1 phosphorylation is a dynamic process that is dependent upon active cell growth.
FIG. 9.

eRF1 undergoes dynamic phosphorylation during cell growth. (A) Cells grown in Wickerham's low-phosphate medium were metabolically labeled with [32P]orthophosphate for 2 h. To initiate the chase period, cells were harvested and resuspended in Wickerham's medium with excess unlabeled orthophosphate. The chase was carried out for 4 h in the presence or absence of glucose, and samples were collected at the indicated times for immunoprecipitation with an eRF1-specific antiserum (32P). Samples were harvested at the same times from unlabeled cultures (but otherwise grown under identical conditions) for Western blot analysis (WB). (B) Logarithmic plot of 32P incorporation per unit of eRF1 from A. The data shown are representative of two independent experiments.
DISCUSSION
Previous studies have shown that several factors involved in the initiation and elongation steps of translation undergo posttranslational modification by phosphorylation. In this study, we show that the translation termination factor eRF1 is also a phosphoprotein in yeast, which shows for the first time that all three steps in translation are modified in this manner. We found that eRF1 is phosphorylated in two sites near its C terminus by the CK2 protein kinase as previously shown for several other yeast proteins involved in translation, including all five proteins of the ribosomal stalk (3), and initiation factors eIF5 (30) and eIF2α (16) (Table 2). Surprisingly, we found that the introduction of mutations that either eliminated eRF1 phosphorylation or mimicked constitutive eRF1 phosphorylation had little effect on the efficiency of stop codon recognition in vivo (as measured by a translational readthrough assay). The level of eRF1 phosphorylation was also unaffected by the disruption of the SAL6 gene, which encodes a putative PP1-like phosphatase. Furthermore, the allosuppressor phenotype associated with the sal6Δ mutation was insensitive to the state of eRF1 phosphorylation. These results indicate that the phosphorylation of eRF1 does not play a significant role in its ability to mediate efficient translation termination under the growth conditions used in the current study.
We also found that the phosphorylation state of eRF1 did not influence its ability to bind eRF3, even though phosphorylation occurred within the portion of eRF1 that is responsible for eRF3 binding. In addition, our results indicate that phosphorylation of eRF1 does not influence nonsense-mediated mRNA decay. From these data, we can infer that the phosphorylation state of eRF1 probably does not affect Upf1p binding, since the association of Upf1p with the termination complex is required for optimal translation termination efficiency (12, 25).
Based on the results summarized above, the physiological role for CK2-mediated phosphorylation of eRF1 remains obscure. Previous studies of other proteins involved in translation that undergo CK2 phosphorylation near their C termini have also had difficulty establishing a function for this modification. A study of the P1β protein of the ribosomal stalk suggested that phosphorylation, in conjunction with an N-terminal signal, may stimulate protein degradation. This hypothesis is consistent with the previous observation that phosphorylation can function as a degradation signal for the vacuolar and proteasome turnover pathways (23). However, it was subsequently shown that P1β was not degraded by either of these pathways. Instead, it was shown that a failure of the P1 dimer to associate with P2 causes a rapid turnover of P1 (33). Therefore, phosphorylation of P1 may simply play a role in its association with P2, with enhanced degradation occurring due to a lack of assembly. In another example, yeast eIF2α is phosphorylated by the CK2 protein kinase at three serine residues near its C terminus (amino acids 292, 294, and 301). Although mutation of these serine residues to alanine did not cause a detectible phenotype, it did exacerbate the growth defects observed when these mutations were combined with other mutations that reduced the efficiency of nucleotide exchange, suggesting that phosphorylation of the CK2 protein kinase sites in eIF2α may be required for optimum eIF2 activity (16). Similarly, we found that changes in the phosphorylation state of eRF1 resulted in only subtle effects on stop codon recognition that may help to fine-tune the termination process at certain termination signals (Table 3). Finally, it was shown that eIF5 is phosphorylated at multiple sites near its C terminus by the CK2 protein kinase (30), but a role for this modification was not found. Taken together, these results suggest that phosphorylation of various translation factors by the CK2 protein kinase does not play a strong role in their specific functions related to translation under steady-state growth conditions. Thus, the purpose of this common posttranslational modification remains somewhat obscure.
The database searches carried out during this study also revealed that at least two other yeast proteins involved in translation, eIF2Bɛ and eIF4γ, also have CK2 phosphorylation motifs near their extreme C termini. In addition, a number of other translation factors, including eEF1β, eIF1A, Tif35p, eIF2Bγ, and eIF4B, have consensus CK2 phosphorylation motifs within 50 amino acids of their C termini. We hypothesize that these proteins are also phosphorylated by the CK2 protein kinase. Based on the results of the phosphorylation studies described above, the phosphorylation of these factors (if it occurs) may not play a dramatic role in regulating their activities. Instead, they may be phosphorylated by the CK2 protein kinase in response to favorable growth conditions in a manner similar to that of the five ribosomal stalk proteins, eIF2α, eIF5, and eRF1. Here, we have provided evidence that (i) the phosphorylation of eRF1 is dynamic and goes through a phosphorylation/dephosphorylation cycle rather then being constitutively phosphorylated following its synthesis, (ii) active cell growth is required for the phosphorylation/dephosphorylation cycle, and (iii) inhibition of this phosphorylation cycle by eliminating the sites of phosphorylation does not have adverse effects on translation or steady-state growth when cells are grown with glucose as a carbon source. It is possible that this phosphorylation pathway is important when the cells are grown under adverse conditions where subtle changes in the activity of the translational apparatus or the function of other related factors may be important. It is also possible that this conserved modification may act to down-regulate translation under conditions that are not favorable for cell growth.
A significant body of evidence also indicates that many factors have alternate cellular functions. For example, eEF1A not only plays a role in translation elongation but has also been shown to bind actin and influence the distribution of the actin cytoskeleton (20, 24, 32). Ccr4p, a subunit of the major cytoplasmic deadenylase complex, also acts as a transcription factor that becomes active in response to DNA damage (28). It is currently not known whether a posttranslational modification such as phosphorylation regulates any (or all) of the alternate functions of these factors. Further studies will be required to examine this possibility.
Acknowledgments
We thank Claiborne V. C. Glover III for providing strains and John Rodgers, Bill Marion, and Peter Prevelige for assistance with the analytical ultracentrifugation experiments.
This work was supported by NIH grant RO1 GM 68854 (D.M.B.).
REFERENCES
- 1.Amrani, N., R. Ganesan, S. Kervestin, D. A. Mangus, S. Ghosh, and A. Jacobson. 2004. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432:112-118. [DOI] [PubMed] [Google Scholar]
- 2.Andjelkovic, N., S. Zolnierowicz, C. Van Hoof, J. Goris, and B. A. Hemmings. 1996. The catalytic subunit of protein phosphatase 2A associates with the translation termination factor eRF1. EMBO J. 15:7156-7167. [PMC free article] [PubMed] [Google Scholar]
- 3.Ballesta, J. P., M. A. Rodriguez-Gabriel, G. Bou, E. Briones, R. Zambrano, and M. Remacha. 1999. Phosphorylation of the yeast ribosomal stalk. Functional effects and enzymes involved in the process. tFEMS Microbiol. Rev. 23:537-550. [DOI] [PubMed] [Google Scholar]
- 4.Bandhakavi, S., R. O. McCann, D. E. Hanna, and C. V. Glover. 2003. Genetic interactions among ZDS1,2, CDC37, and protein kinase CK2 in Saccharomyces cerevisiae. FEBS Lett. 554:295-300. [DOI] [PubMed] [Google Scholar]
- 5.Bertram, G., H. A. Bell, D. W. Ritchie, G. Fullerton, and I. Stansfield. 2000. Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA 6:1236-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boeke, J. D., F. LaCroute, and G. R. Fink. 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197:345-346. [DOI] [PubMed] [Google Scholar]
- 7.Cardenas, M. E., Q. Dang, C. V. Glover, and S. M. Gasser. 1992. Casein kinase II phosphorylates the eukaryote-specific C-terminal domain of topoisomerase II in vivo. EMBO J. 11:1785-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlberg, U., A. Nilsson, and O. Nygard. 1990. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 191:639-645. [DOI] [PubMed] [Google Scholar]
- 9.Chang, Y. W., and J. A. Traugh. 1998. Insulin stimulation of phosphorylation of elongation factor 1 (eEF-1) enhances elongation activity. Eur. J. Biochem. 251:201-207. [DOI] [PubMed] [Google Scholar]
- 10.Chavatte, L., L. Frolova, L. Kisselev, and A. Favre. 2001. The polypeptide chain release factor eRF1 specifically contacts the s(4)UGA stop codon located in the A site of eukaryotic ribosomes. Eur. J. Biochem. 268:2896-2904. [DOI] [PubMed] [Google Scholar]
- 11.Chen, M. X., Y. H. Chen, and P. T. Cohen. 1993. PPQ, a novel protein phosphatase containing a Ser + Asn-rich amino-terminal domain, is involved in the regulation of protein synthesis. Eur. J. Biochem. 218:689-699. [DOI] [PubMed] [Google Scholar]
- 12.Czaplinski, K., M. J. Ruiz-Echevarria, S. V. Paushkin, X. Han, Y. Weng, H. A. Perlick, H. C. Dietz, M. D. Ter-Avanesyan, and S. W. Peltz. 1998. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 12:1665-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahlseid, J. N., J. Puziss, R. L. Shirley, A. L. Atkin, P. Hieter, and M. R. Culbertson. 1998. Accumulation of mRNA coding for the ctf13p kinetochore subunit of Saccharomyces cerevisiae depends on the same factors that promote rapid decay of nonsense mRNAs. Genetics 150:1019-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebihara, K., and Y. Nakamura. 1999. C-terminal interaction of translational release factors eRF1 and eRF3 of fission yeast: G-domain uncoupled binding and the role of conserved amino acids. RNA 5:739-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eurwilaichitr, L., F. M. Graves, I. Stansfield, and M. F. Tuite. 1999. The C-terminus of eRF1 defines a functionally important domain for translation termination in Saccharomyces cerevisiae. Mol. Microbiol. 32:485-496. [DOI] [PubMed] [Google Scholar]
- 16.Feng, L., H. Yoon, and T. F. Donahue. 1994. Casein kinase II mediates multiple phosphorylation of Saccharomyces cerevisiae eIF-2α (encoded by SUI2), which is required for optimal eIF-2 function in S. cerevisiae. Mol. Cell. Biol. 14:5139-5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frolova, L., X. Le Goff, G. Zhouravleva, E. Davydova, M. Philippe, and L. Kisselev. 1996. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA 2:334-341. [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez, C. I., W. Wang, and S. W. Peltz. 2001. Nonsense-mediated mRNA decay in Saccharomyces cerevisiae: a quality control mechanism that degrades transcripts harboring premature termination codons. Cold Spring Harb. Symp. Quant. Biol. 66:321-328. [DOI] [PubMed] [Google Scholar]
- 19.Grentzmann, G., J. A. Ingram, P. J. Kelly, R. F. Gesteland, and J. F. Atkins. 1998. A dual-luciferase reporter system for studying recoding signals. RNA 4:479-486. [PMC free article] [PubMed] [Google Scholar]
- 20.Gross, S. R., and T. G. Kinzy. 2005. Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat. Struct. Mol. Biol. 12:772-778. [DOI] [PubMed] [Google Scholar]
- 21.Hanna, D. E., A. Rethinaswamy, and C. V. Glover. 1995. Casein kinase II is required for cell cycle progression during G1 and G2/M in Saccharomyces cerevisiae. J. Biol. Chem. 270:25905-25914. [DOI] [PubMed] [Google Scholar]
- 22.Hawthorne, D. C., and U. Leupold. 1974. Suppressors in yeast. Curr. Top. Microbiol. Immunol. 64:1-47. [DOI] [PubMed] [Google Scholar]
- 23.Hershko, A., and A. Ciechanover. 1998. The ubiquitin system. Annu. Rev. Biochem. 67:425-479. [DOI] [PubMed] [Google Scholar]
- 24.Kandl, K. A., R. Munshi, P. A. Ortiz, G. R. Andersen, T. G. Kinzy, and A. E. Adams. 2002. Identification of a role for actin in translational fidelity in yeast. Mol. Genet. Genomics 268:10-18. [DOI] [PubMed] [Google Scholar]
- 25.Keeling, K. M., J. Lanier, M. Du, J. Salas-Marco, L. Gao, A. Kaenjak-Angeletti, and D. M. Bedwell. 2004. Leaky termination at premature stop codons antagonizes nonsense-mediated mRNA decay in S. cerevisiae. RNA 10:691-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kielbassa, K., H. J. Muller, H. E. Meyer, F. Marks, and M. Gschwendt. 1995. Protein kinase C delta-specific phosphorylation of the elongation factor eEF-alpha and an eEF-1 alpha peptide at threonine 431. J. Biol. Chem. 270:6156-6162. [DOI] [PubMed] [Google Scholar]
- 27.Kitada, K., E. Yamaguchi, and M. Arisawa. 1995. Cloning of the Candida glabrata TRP1 and HIS3 genes, and construction of their disruptant strains by sequential integrative transformation. Gene 165:203-206. [DOI] [PubMed] [Google Scholar]
- 28.Lenssen, E., N. James, I. Pedruzzi, F. Dubouloz, E. Cameroni, R. Bisig, L. Maillet, M. Werner, J. Roosen, K. Petrovic, J. Winderickx, M. A. Collart, and C. De Virgilio. 2005. The Ccr4-Not complex independently controls both Msn2-dependent transcriptional activation—via a newly identified Glc7/Bud14 type I protein phosphatase module—and TFIID promoter distribution. Mol. Cell. Biol. 25:488-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 30.Maiti, T., A. Bandyopadhyay, and U. Maitra. 2003. Casein kinase II phosphorylates translation initiation factor 5 (eIF5) in Saccharomyces cerevisiae. Yeast 20:97-108. [DOI] [PubMed] [Google Scholar]
- 31.Meggio, F., and L. A. Pinna. 2003. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17:349-368. [DOI] [PubMed] [Google Scholar]
- 32.Munshi, R., K. A. Kandl, A. Carr-Schmid, J. L. Whitacre, A. E. Adams, and T. G. Kinzy. 2001. Overexpression of translation elongation factor 1A affects the organization and function of the actin cytoskeleton in yeast. Genetics 157:1425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nusspaumer, G., M. Remacha, and J. P. Ballesta. 2000. Phosphorylation and N-terminal region of yeast ribosomal protein P1 mediate its degradation, which is prevented by protein P2. EMBO J. 19:6075-6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paushkin, S. V., V. V. Kushnirov, V. N. Smirnov, and M. D. Ter-Avanesyan. 1997. Interaction between yeast Sup45p (eRF1) and Sup35p (eRF3) polypeptide chain release factors: implications for prion-dependent regulation. Mol. Cell. Biol. 17:2798-2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poole, A., T. Poore, S. Bandhakavi, R. O. McCann, D. E. Hanna, and C. V. C. Glover. 2005. A global view of CK2 function and regulation. Mol. Cell. Biochem. 274:163-170. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Gabriel, M. A., M. Remacha, and J. P. Ballesta. 1998. Phosphorylation of ribosomal protein P0 is not essential for ribosome function but can affect translation. Biochemistry 37:16620-16626. [DOI] [PubMed] [Google Scholar]
- 37.Salas-Marco, J., and D. M. Bedwell. 2004. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol. Cell. Biol. 24:7769-7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt, M. E., T. A. Brown, and B. L. Trumpower. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18:3091-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonenberg, N., and T. E. Dever. 2003. Eukaryotic translation initiation factors and regulators. Curr. Opin. Struct. Biol. 13:56-63. [DOI] [PubMed] [Google Scholar]
- 40.Song, J. M., and S. W. Liebman. 1987. Allosuppressors that enhance the efficiency of omnipotent suppressors in Saccharomyces cerevisiae. Genetics 115:451-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stansfield, I., V. V. Kushnirov, K. M. Jones, and M. F. Tuite. 1997. A conditional-lethal translation termination defect in a sup45 mutant of the yeast Saccharomyces cerevisiae. Eur. J. Biochem. 245:557-563. [DOI] [PubMed] [Google Scholar]
- 42.Vincent, A., G. Newnam, and S. W. Liebman. 1994. The yeast translational allosuppressor, SAL6: a new member of the PP1-like phosphatase family with a long serine-rich N-terminal extension. Genetics 138:597-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wickerham, L. J. 1946. A critical evaluation of the nitrogen assimilation tests commonly used in classification of yeasts. J. Bacteriol. 52:293-301. [DOI] [PubMed] [Google Scholar]
- 44.Wilson, L. K., N. Dhillon, J. Thorner, and G. S. Martin. 1997. Casein kinase II catalyzes tyrosine phosphorylation of the yeast nucleolar immunophilin Fpr3. J. Biol. Chem. 272:12961-12967. [DOI] [PubMed] [Google Scholar]