

Abstract
The host range of African trypanosomes is influenced by innate protective molecules in the blood of primates. A subfraction of human high-density lipoprotein (HDL) containing apolipoprotein A-I, apolipoprotein L-I, and haptoglobin-related protein is toxic to Trypanosoma brucei brucei but not the human sleeping sickness parasite Trypanosoma brucei rhodesiense. It is thought that T. b. rhodesiense evolved from a T. b. brucei-like ancestor and expresses a defense protein that ablates the antitrypanosomal activity of human HDL. To directly investigate this possibility, we developed an in vitro selection to generate human HDL-resistant T. b. brucei. Here we show that conversion of T. b. brucei from human HDL sensitive to resistant correlates with changes in the expression of the variant surface glycoprotein (VSG) and abolished uptake of the cytotoxic human HDLs. Complete transcriptome analysis of the HDL-susceptible and -resistant trypanosomes confirmed that VSG switching had occurred but failed to reveal the expression of other genes specifically associated with human HDL resistance, including the serum resistance-associated gene (SRA) of T. b. rhodesiense. In addition, we found that while the original active expression site was still utilized, expression of three expression site-associated genes (ESAG) was altered in the HDL-resistant trypanosomes. These findings demonstrate that resistance to human HDLs can be acquired by T. b. brucei.
Three subspecies of Trypanosoma brucei have been described previously based on host range and course of infection (17, 18, 19, 26). Trypanosoma brucei brucei causes nagana in cattle and livestock in sub-Saharan Africa, Trypanosoma brucei rhodesiense causes acute human African sleeping sickness in East Africa, and Trypanosoma brucei gambiense causes chronic human African sleeping sickness in West Africa. The phylogenetic relationship of these three organisms has been difficult to resolve, particularly for T. b. brucei and T. b. rhodesiense, which are morphologically indistinguishable and infect the same wild-animal hosts (17).
A distinguishing feature of T. b. brucei is its inability to infect humans. This innate protection is provided by trypanolytic activity found in normal human blood and also in the blood of most apes and Old World monkeys (24, 31, 46, 56). The trypanosome lytic factor (TLF) is a minor subfraction of heterogeneous human high-density lipoprotein (HDL) particles containing apoliprotein A-I (apoA-I), apoliprotein A-II (apoA-II), apolipoprotein L-I (apoL-I), and haptoglobin-related protein (Hpr) (23, 29, 38, 45, 48, 49, 50, 51, 52, 57, 59, 60, 64, 65, 69). The mechanism of TLF killing of T. b. brucei has been controversial. Initially, Hpr was suggested to be the active component in TLF, based on the evolution of the Hpr gene during primate evolution, the correlation of the trypanolytic activity in primate sera, and the presence of the Hpr gene in T. b. brucei-resistant animals (33, 56, 59). More recently, highly purified, recombinant apoL-I has been shown to have trypanosome-killing activity (45, 69). It now appears that Hpr and apoL-I both have trypanosome-killing activity and that the assembly of these proteins into the same HDL particle is synergistic, resulting in a naturally occurring supercomplex with enhanced activity compared with that of HDLs lacking either Hpr or apoL-I (57). The biochemical mechanism of TLF killing of T. b. brucei also continues to be debated (4, 36, 37, 45). However, there is good agreement that trypanosome killing by TLF follows a pathway beginning with high-affinity binding to a receptor in the flagellar pocket of the trypanosome, followed by endocytosis and localization to the lysosome. Within the acidic lysosome, TLF is activated and causes cell death. Inhibition of TLF trafficking to the lysosome or neutralization of the lysosomal pH abolishes TLF-mediated killing of T. b. brucei (13, 20, 21, 28, 30, 41, 49, 58, 70).
The discovery of a human serum resistance-associated gene (SRA) in T. b. rhodesiense, but not in T. b. brucei, provided a molecular explanation for human infectivity by these parasites (9, 10, 11). Several experiments have shown a correlation between the expression of the SRA gene and resistance to human serum (11, 35), and transfection of this gene into T. b. brucei conferred resistance to human serum and TLF (41, 74). In addition, the SRA gene has proven to be a reliable marker for T. b. rhodesiense in epidemiological studies throughout East Africa (16, 18, 39, 47, 72, 73).
The expression of the SRA gene is unstable when T. b. rhodesiense is maintained outside the human host. Repeated passage of human serum-resistant T. b. rhodesiense in mice results in a loss of SRA expression and a corresponding loss of serum resistance. When these human serum-susceptible lines are placed under TLF selection, some cells revert to the TLF resistance phenotype by reactivation of SRA gene expression (22, 35, 74).
Although differing in susceptibility to TLF, all three subspecies of T. brucei evade the host adaptive immune system by periodically changing the expressed variant surface glycoprotein (VSG) (7, 12, 32, 68). The expressed VSG gene lies at the end of a telomere in one of 20 polycistronically transcribed expression sites (ES). These expression sites also contain genes known as expression site-associated genes (ESAGs) that code for bloodstream-specific proteins, including the transferrin receptor (Tf-R). It has been postulated that genes in ES may be grouped and arranged to help the trypanosome survive in different hosts and environments (1, 44). In T. b. rhodesiense, SRA is an expression site-associated gene and shares some features with VSGs (74). Because of the similarities between T. b. rhodesiense and T. b. brucei, the origins of human serum resistance have been debated. However, it is generally thought that the SRA gene originated from a VSG gene in an ancestral, human serum-sensitive trypanosome (8, 11).
We wanted to determine whether repeated exposure of T. b. brucei to human HDLs would generate resistance to human HDL killing without the presence of the SRA gene. Several HDL-resistant populations emerged during the selection period, and the molecular and cellular characteristics of one of these trypanosome populations are reported here. Endocytosis of TLF did not occur in these trypanosomes, and they could be cultivated in media containing high concentrations of human HDL or normal human serum. Surprisingly, the selection for human HDL resistance correlated with a change in the expressed VSG and recombination events in the 221 expression site (221ES), leading to changes in the expression of ESAG1, -2, and -3. These in vitro-generated lines may provide important insight into the mechanism and evolution of human infectivity by African trypanosomes.
MATERIALS AND METHODS
Trypanosome strains and in vitro selection.
Bloodstream stages of the T. b. brucei Lister strain 427 (MiTat 1.2), expressing VSG221, were used in these studies. Cells were cultured in HMI-9 medium with the addition of heat-inactivated fetal bovine serum (FBS) (10%) and Serum Plus (10%) (JRH Biosciences, Kansas). T. b. brucei 427-221 is an antigenically stable line and contains a single copy of the VSG221 gene within the 221ES (3).
At a cell density of approximately 1 × 106 cells/ml, T. b. brucei 427-221 cells were exposed to various amounts of human HDLs for 24 h in a six-well plate (see Table 1 for amounts). Surviving trypanosomes were counted using a hemacytometer and then diluted into fresh HMI-9 medium and allowed to recover for 5 to 14 days. Once the cells had grown to a density of approximately 1 × 106 cells/ml, they were again incubated with human HDLs. Each round of selection was performed with increasing concentrations of human HDLs, and freezer stocks were prepared for each surviving population. Over 9 months we conducted eight rounds of human HDL selection, resulting in a population of T. b. brucei that survived incubation with 800 μl of human HDLs (160 lytic U).
TABLE 1.
In vitro selection for human HDL-resistant T. b. brucei
| Amt(s) human HDL (μl)/wella | Population designation |
|---|---|
| 0 | 427-221 |
| 0, 0.1, 0.5, 1, 1.5, 2 | 427-0.1 |
| 0, 0.1, 0.5, 1, 1.5, 2 | 427-0.5 |
| 0, 0.5, 1, 1.5, 2, 2.5 | 427-1 |
| 0, 1, 3, 5, 7, 10 | 427-3 |
| 0, 5, 10, 12, 15, 20 | 427-12 |
| 0, 12, 15, 20, 25, 30 | 427-20 |
| 0, 15, 30, 60, 190, 120 | 427-60 |
| 0, 60, 100, 200, 300, 400 | 427-300 |
| 0, 300, 500, 700, 800, 1,000 | 427-800 |
Shown are the amounts of human HDL added to each well in a 3-ml culture of T. b. brucei 427-221 for 24 h. The highest concentration of HDL that yielded viable cells during a round of selection is indicated in bold. These cells were allowed to recover for 5 to 14 days and then were used in the next round of selection.
Purification of human HDLs and trypanosome lysis assays.
Normal human serum was obtained from a healthy donor and total serum HDLs prepared by sequential flotation on sodium bromide gradients (1.063 and 1.26 g/ml) as described previously (23). The HDL samples were stored in small aliquots at −70°C to minimize the number of freeze-thaw cycles. At the beginning of the study, the specific activity of the purified HDL was determined to be 1 trypanolytic unit (1 U) per 0.1 μl. The concentration of HDL was approximately 2.35 μg per μl. For the in vitro lysis assays, a unit is defined as the amount of HDL needed to lyse 50% of the trypanosomes during a 2-h incubation at 37°C. The lysis reactions contained 1 × 107 bloodstream-form trypanosomes in 300 μl of F12 media containing 15% heat-inactivated FBS and 1% glucose, buffered with 25 mM HEPES (pH 7.5). After 9 months, the activity of this human HDL preparation decreased to 1 U per 5 μl. The naming of the various HDL-resistant populations indicates the maximum amount of HDL the cells survived during selection. For example, T. b. brucei 427-800 was able to survive overnight in a total culture volume of 3 ml containing 800 μl of HDLs. Since the specific activity of the HDL decreased over the course of the selection experiment, cell populations were retested for TLF susceptibility at the end of the study.
To verify that the observed resistance to human HDL was specific to TLF, several cell lines (427-221, 427-12, 427-20, 427-60, and 427-800) were retested for susceptibility to both highly purified TLF and intact human serum. TLF was purified from total human HDLs by immunoaffinity chromatography using an Hpr-specific monoclonal antibody (13). The anti-Hpr affinity column was prepared using Affi-gel 10 (Bio-Rad) according to the manufacturer's recommendations. Total human HDL (density, 1.063 to 1.26 g/ml) was purified by flotation on sodium bromide, dialyzed against phosphate-buffered saline (PBS) containing 3 mM EDTA, pH 7.5, and concentrated to 0.4 mg/ml with an Amicon filtration concentrator (Amicon no. 4306). Hpr-containing HDLs were bound to the anti-Hpr column, washed with PBS containing 3 mM EDTA, pH 7.5, and eluted using 100 mM glycine at pH 2.0. Following elution, this highly purified preparation of TLF was neutralized by the addition of 1 M Tris, pH 7.5. The Hpr-specific monoclonal antibody reacts with human Hpr, whether free or in Hpr-containing HDLs, but shows no cross-reactivity with other human serum proteins (57). This TLF was used for all standard lysis assays presented.
SDS-polyacrylamide gel electrophoresis and Western blotting.
For the analysis of total cell protein, trypanosomes were incubated in lysis buffer (100 mM Tris, pH 8.0, 10 mM EDTA, 0.5% sodium dodecyl sulfate [SDS]), and an equivalent of 3 × 106 cells per lane was run on 8% or 10% SDS-polyacrylamide gels. Following electrophoresis, proteins were transferred to a nitrocellulose filter membrane (0.45 μm; Protran, Schleicher and Schuell, Dassel, Germany) for 1.5 h at 60 V and probed for VSG221 by standard Western blotting methods.
Alexa labeling of TLF and immunofluorescence microscopy.
Cultured trypanosomes were collected by centrifugation at 2,500 × g for 10 min at room temperature. Cells were then resuspended in HMI-9 media at a density of 3 × 107 cells/ml. TLF (∼100 μg) was fluorescently labeled using an Alexa Fluor 488 protein labeling kit (Molecular Probes, California). Alexa-labeled TLF (6 μg) was added to 3 × 107 trypanosomes and then incubated at 37°C for 1.5 h. Cells were then collected and washed three times with ice-cold PBS containing 1% glucose. After the last wash, the cell pellet was resuspended in ice-cold PBS containing 1% glucose at one-fourth the original volume of media. The cells were smeared onto a glass slide and fixed with methanol. Once the slide was dry, it was blocked with PBS containing 10% FBS for 1 h at room temperature. The slides were incubated with primary antibodies, either a polyclonal antiserum against VSG221 or a monoclonal antibody against lysosomal protein p67 (1:4,000 and 1:1,000 dilution, respectively), for 1 h at room temperature and washed with PBS containing 1% FBS. Secondary antibodies were then added at a dilution of 1:1,000 and incubation continued at room temperature for 1 h. In the last 10 min of the incubation, DAPI (4′,6′-diamidino-2-phenylindole) was added at a concentration of 2 μg/ml to stain the nucleus and kinetoplast. Slides were washed in PBS containing 1% FBS, Fluoromount G was added, and a coverslip was mounted. The slides were viewed and images were captured using a Zeiss Axiocam camera. Pictures were taken using automated gain as well as manual gain times. To negate any difference due to image capture, the gain time for TLF exposures was kept constant. To block acidification of the trypanosome lysosome, samples were pretreated with 50 μM chloroquine for 30 min at 37°C prior to TLF incubation. This treatment blocked TLF-mediated lysis and allowed maximal accumulation of TLF in the lysosome (58).
Identification of the expressed VSGs and transferrin receptor.
The expressed VSGs and transferrin receptor (ESAG6 and ESAG7) were identified by cDNA cloning of reverse transcriptase PCR (RT-PCR) products. A universal 3′ VSG primer (5′-GGGTAACTTACGTGTTAAAATATATCAG-3′) and a 5′ spliced leader RNA primer (5′-CGCTATTATTAGAACAGTTTCTGTAC-3′) were used to amplify and clone the expressed VSG cDNA. ESAG6/7 primers (5′-GCGAATTCCCTTTACAAAATTGAGGATTC-3′ and 5′-GCTCTAGACATCACTGCATTTTTGCTTC-3′) were used to amplify and clone the expressed ESAG6/7 cDNA. Following reverse transcription and PCR, amplified bands were excised from the gel, purified, and cloned into the TA vector for DNA sequencing.
RNA isolation, RT-PCR, probe labeling, and Northern blot analysis.
Total trypanosome RNA was isolated using Tripure isolation reagent (Roche, Mannheim, Germany), and RT-PCR was performed on 1 to 3 μg by use of a Superscript III first-strand synthesis system for RT-PCR (Invitrogen, Carlsbad, CA). Primers for ESAG1 (5′-AAGGACAGTGTGGGAGGATG-3′ and 5′-GAAACGGCTTTGGAGTGAAG-3′), ESAG2 (5′-CCCGATTGGGACACAATTAC-3′ and 5′-CATGTGAATCCCGCAGTATG-3′), ESAG3 (5′-TCATGCAACACAAGGATGGT-3′ and 5′-AATAGGCTGTCCGGGAAAAT-3′), and ESAG8 (5′-CCTTGAGGCTTTGTCTCTGG-3′ and 5′-CAAATTACGCAAATTCCCGAC-3′) were used to amplify the expressed ESAG from 427-221 and 427-800 trypanosomes. After RT-PCR, products were gel purified and cloned into the TA vector for sequencing. For Northern blot analysis, 2.5 μg total RNA was separated on a 1% agarose formaldehyde gel and electroblotted at 15 V for 1 h and then for an additional 2 h at 50 V onto a charged nylon membrane. The blot was hybridized overnight with a digoxigenin-labeled probe at 40°C in Easy Hyb buffer (Roche, Mannheim, Germany). Immunological detection of digoxigenin-labeled probe by chemiluminescence was performed according to the manufacturer's instructions.
Radiolabeled probes were generated using a random primer labeling kit (Stratagene, La Jolla, CA). The SRA probe was generated from the cloned T. b. rhodesiense SRA gene (KETRI 2482) by using SRA-specific primers (5′-CACACCTCTAAGAATCACAATAG-3′ and 5′-AATTCATGAAAATGTGTTAAAG-3′). VSG probes were generated from cloned VSG221 and VSG800 by using primers specific for VSG221 (5′-CGAGGAGCTAGACGACCAAC-3′ and 5′-TCGCTGTTGCAGTAGCTGTT-3′) and VSG800 (5′-ATGCGGCCTAACTGATGAAC-3′ and 5′-TATCTTTGTAGGCCGCTGCT-3′).
Pulsed-field gel electrophoresis and Southern blot analysis.
Chromosome separation was performed by contour-clamped homogenous electric field (CHEF) electrophoresis with a Bio-Rad CHEF-DR II system (Bio-Rad, Hercules, CA). Genomic plugs were prepared by immobilizing trypanosomes at a concentration of 2 × 107 cells per 1 ml in 0.7% low-melting-temperature agarose. The plugs were poured, allowed to solidify at 4°C, and then treated with proteinase K (1 mg/ml) for 48 h before use. Agarose gels were prepared in 1× TAFE (0.01 M Tris, 0.5 mM Titriplex II EDTA, 0.025% acetic acid) and were electrophoresed at 12°C with buffer recirculation. Hansenula wingei (Bio-Rad) chromosomes were run concurrently as a standard. The switch time was fixed at 900 s. The overall running time was 72 h at 2.0 V/cm, and the included angle was 120° in 1× TAFE. Radiolabeled probes for VSG221 and VSG800 for Southern blotting were prepared as described above. The GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene was used as a control probe for trypanosome chromosome VIa (34).
SAGE.
Serial analysis of gene expression (SAGE) libraries were constructed using ∼30 μg total RNA, purified as described above and DNase treated, from T. b. brucei 427-221 and 427-800 populations following the I-SAGE Long kit protocol (Invitrogen, Carlsbad, CA). Recombinant pZEro1 clones produced by SAGE were purified using GeneMachines RevPrep Orbit (Genomic Solutions, Ann Arbor, MI) and were sequenced on an ABI 3730xl DNA sequencer (Applied Biosystems, Foster City, CA). Sequences collected were analyzed with software created at the MBL specifically for T. b. brucei SAGE analysis. The SAGE software extracts ditag sequences from the ABI 3730xl results according to the SAGE sequence grammar, parses out individual SAGE tags, excludes tags with sequence ambiguities, and reduces all SAGE tags to a look-up table of unique SAGE tag sequences and their observed frequencies among all of the T. b. brucei SAGE libraries. SAGE tags not found more than once in at least one SAGE library were excluded from analyses as putative sequencing error, unless the tag sequence had a perfect match to available T. b. brucei genomic data. The unique tag sequences were mapped to all available T. b. brucei DNA sequences to determine the identities of expressed genes. For this, draft assembly contigs and open reading frame (ORF) calls from the TIGR and Sanger sequencing centers were downloaded from their respective FTP sites and supplemented with nonredundant T. b. brucei genome sequences available at NCBI (113 total contigs summing to 25,496,703 bp with 9,737 total predicted ORFs). The 21-bp SAGE tags were mapped to assembly contigs based on exact sequence matches. Tags were then visualized in a generic model organism database using the Gbrowse interface (61) in the context of genome sequences and predicted open reading frames. As SAGE tags are generated from the most 3′ NlaIII restriction site of transcripts, tags with significant differential expression among the SAGE libraries were manually assigned to predicted genes based on their position relative to predicted ORFs. These genes were annotated by BLAST and other homology detection methods. SAGE tags were scored for differential expression among the 427-221 and 427-800 libraries by use of the Stekel et al. (62) R statistic, a log likelihood ratio statistic which scores tags by their deviation from the null hypothesis of equal frequencies given the tag sampling depth for each SAGE library. Higher scores represent a greater deviation from the null hypothesis, while scores close to zero represent near-constitutive expression. To reduce the effects of sampling error in highlighting differential expression, only tags with an R value of 4 or greater were considered for RT-PCR and other post-SAGE analysis.
RESULTS
In vitro generation of HDL-resistant T. b. brucei.
In the initial round of selection for HDL-resistant T. b. brucei, cells were treated with low concentrations of human HDL (Table 1). Cultures exposed to concentrations of HDL above 0.03% (0.5, 1.0, 1.5, and 2.0 μl) failed to yield viable cells. Trypanosomes that survived the initial round of selection (427-0.1) were expanded and a second round of human HDL selection started. During each round of selection, the population of cells able to grow at the highest HDL concentration was expanded and treated with increasing amounts of human HDL. Following eight rounds of selection over a 9-month period of time, a population of T. b. brucei resistant to 800 μl of human HDL added to growth media was obtained. The 427-800 cell line is highly resistant not only to human HDL but also to purified TLF and is able to grow in the presence of 25% normal human serum (data not shown).
To determine whether the observed HDL resistance was a stable trait, trypanosome populations and clonal lines of each were expanded and tested for susceptibility to lysis by human HDLs (Fig. 1). Trypanosome populations selected with increasing concentrations of human HDLs were resistant to human HDLs at concentrations corresponding to those used in the selection protocols. Both the 427-12 and 427-20 populations were intermediate in susceptibility to human HDL killing compared to the T. b. brucei 427-221 population and the highly resistant T. b. brucei 427-800 and T. b. rhodesiense populations (Fig. 1).
FIG. 1.
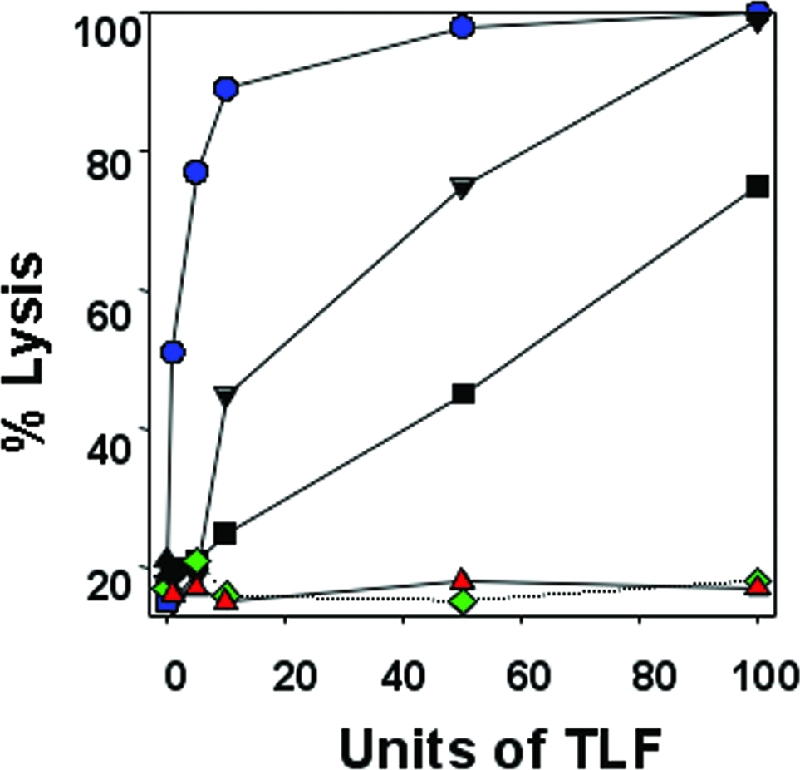
T. b. brucei 427-800 cells are resistant to human HDLs. The percentages of trypanosomes lysed following incubation for 4 h at 37°C with designated trypanolytic units of human HDLs are shown. A unit of HDL is defined as the amount of HDL needed to lyse 50% of 1 × 107 trypanosomes during a 2-h incubation at 37°C. T. b. brucei 427-221 (blue circles), 427-12 (black inverted triangles), 427-20 (black squares) and 427-800 (red triangles) and T. b. rhodesiense (KETRI 2482) (green diamonds) were analyzed.
Uptake and localization of TLF.
The uptake and localization of TLF may contribute to the resistance of T. b. rhodesiense to normal human serum killing (22). To investigate TLF uptake by T. b. brucei 427-800, TLF was conjugated with Alexa-488 and incubated with both 427-221 and 427-800 cells (Fig. 2). T. b. brucei 427-221 rapidly endocytosed the labeled TLF. Intracellular, fluorescent granules were initially visible within 30 min and accumulated during the 1.5-h incubation at 37°C (Fig. 2A, C, D, and E). In contrast, T. b. brucei 427-800 cells failed to take up TLF (Fig. 2B, F, G, and H). We had shown previously that treatment of T. b. brucei with low concentrations of chloroquine (50 μM) inhibited acidification of the lysosome, ablated TLF killing, and resulted in the accumulation of TLF in the lysosome (21, 58). Therefore, even low levels of TLF uptake would be detectable under these conditions, since it would accumulate in the lysosome of the chloroquine-treated cells. When T. b. brucei 427-221 cells were preincubated with chloroquine, Alexa-conjugated TLF levels were elevated relative to cells not treated with chloroquine. Consistent with previous results, the Alexa-conjugated TLF colocalized with the lysosomal protein p67 in the T. b. brucei 427-221 cells (Fig. 3) (21, 58). No TLF uptake or accumulation was detected in the T. b. brucei 427-800 cells. These results contrast our findings for T. b. rhodesiense, where TLF was taken up by human HDL-resistant trypanosomes but failed to localize to the lysosome (22).
FIG. 2.
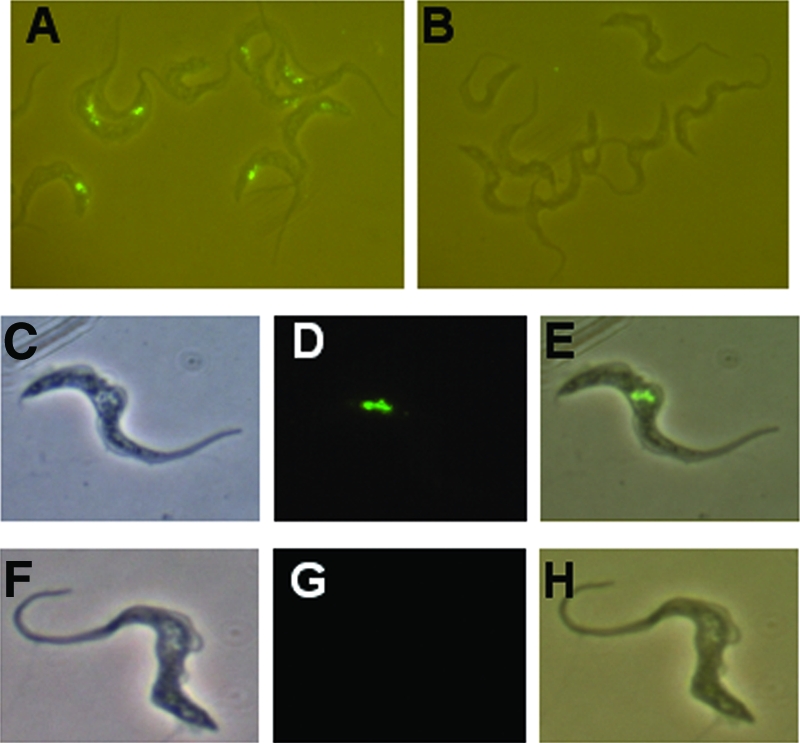
T. b. brucei 427-800 does not take up TLF. Cells were preincubated with chloroquine for 30 min and then exposed to Alexa-labeled TLF (green) for 1.5 h at 37°C. Panels A, C, D, and E show T. b. brucei 427-221 cells. Panels B, F, G, and H show T. b. brucei 427-800 cells. Panels A, B, E, and H are merged phase-contrast and Alexa-488 images. Panel C and F are phase-contrast images only, and panels D and G are Alexa-488 images only.
FIG. 3.

Lysosomal localization of TLF in T. b. brucei 427-221. T. b. brucei 427-221 and 427-800 were preincubated with chloroquine for 30 min and then exposed to Alexa-488-labeled TLF (green) for 1.5 h at 37°C. Cells were then fixed with methanol and incubated with antibodies to the lysosomal protein p67 (red). DAPI (blue) staining was used to localize the nucleus (n) and kinetoplast (k).
Identification of expressed VSGs.
Since T. b. brucei 427 lacks the SRA gene, we took an unbiased approach to look for differences in the 427-221 and 427-800 trypanosomes that might explain the HDL resistance trait. When total cell protein from the 427-221, 427-1, 427-3, 427-12, 427-20, and 427-800 populations was fractionated on SDS-polyacrylamide gels, changes in the overall protein banding patterns were observed (Fig. 4A). A major protein migrating at approximately 50 kDa (Fig. 4A) in the 427-221, 427-1, and 427-3 cells was absent in the 427-12, 427-20, and 427-800 cells. A new protein migrating at approximately 62 kDa was present in the 427-12 and 427-20 cells, while a protein migrating at approximately 55 kDa was present in the 427-800 cells (Fig. 4A). As we will show later, these differences correspond to changes in VSG expression. Western blotting and immunofluorescence microscopy confirmed that the 427-221, 427-1, and 427-3 trypanosomes express VSG221 (Fig. 4B and D). Low levels of VSG switching were expected to occur during the course of the in vitro selection experiment, but it was unexpected that the trypanosomes that had switched VSGs would become the predominant cells in the resistant populations in the absence of immune selection. This suggested that expression of a different VSG provided a selective survival advantage in the presence of human HDL.
FIG. 4.
Changes in proteins expressed in HDL-selected T. b. brucei 427. (A) Coomassie-stained SDS-polyacrylamide gels of total cell protein from T. b. brucei 427-221, 427-1, 427-3, 427-12, 427-20, and 427-800. A prominent protein corresponding to the expressed VSG is indicated with an arrowhead: red, VSG221; green, VSGV02; and blue, VSG800 (GenBank accession no. DQ667685). Sizes in kilodaltons are given at right. (B) Western blot analysis of protein sample with anti-VSG221. (C and D) Immunofluorescence analysis of 427-221, 427-3, 427-12, and 427-800 cells stained with DAPI (C) and anti-VSG221 (D).
In order to determine the VSG expressed by the T. b. brucei cell line highly resistant to human HDL, we cloned and sequenced the expressed VSG cDNA from 427-800 cells. The cDNAs for the VSG expressed by the 427-221 and 427-800 cells were amplified from total cell RNA by reverse transcriptase PCR with primers specific to the conserved sequence within the 3′ untranslated region of all VSG genes and the spliced leader RNA that is trans-spliced to the 5′ end of all trypanosome mRNAs. These cDNAs were cloned and sequenced. The 427-221 cDNA corresponded to the expected VSG221, while the 427-800 cDNA was unique. To verify that the 427-800 VSG corresponded to the expressed protein, the VSG was purified using conditions specific for the release of glycosylphosphatidylinositol (GPI)-anchored proteins (43). The 55-kDa protein was trypsin treated and analyzed by mass spectroscopy. We were able to unambiguously identify the VSG800 based on the determined mass of 20 peptides covering roughly half of the predicted VSG800 sequence (data not shown).
The in vitro selection of human HDL-resistant 427-800 cells raised the possibility that VSG800 might be functionally or structurally similar to the SRA protein expressed by T. b. rhodesiense (9). To examine this, the predicted protein sequences for VSG221, VSG800, and SRA were aligned using the computer program MUSCLE (14) (Fig. 5). As reported by others, SRA contains a large internal deletion relative to other known VSG genes (8). Overall, VSG and SRA sequences show limited sequence similarity outside of the conserved C-terminal portion of the proteins. However, SRA and VSGs do share several structural and functional features, including an N-terminal signal sequence, a C-terminal GPI anchor site, and extended regions of predicted α-helices (Fig. 5). Comparison of SRA with VSG221 and VSG800 did not suggest that VSG800 had greater homology to SRA than other VSGs.
FIG. 5.

Alignment of the predicted amino acid sequences of VSG221, VSG800, and SRA, produced using the computer program MUSCLE (14), with manual correction by eye. Predicted signal sequences are indicated in blue, predicted alpha-helical regions are underlined, and putative GPI anchor sites are shown in red. Numbers on the right indicate amino acids.
Based on the cDNA sequences of SRA, VSG221, and VSG800, specific primers were designed to determine whether these genes were present in the 427-221 and 427-800 cell lines as well as the KETRI 2482 line of T. b. rhodesiense (Fig. 6A). The VSG221 and VSG800 genes were detected in T. b. brucei 427-221 genomic DNA, but the VSG221 gene was absent from T. b. brucei 427-800. This is consistent with studies from other labs that have reported that the VSG221 gene is single copy and that gene conversion within the 221ES could lead to loss of the VSG221 gene (53). PCR amplification of genomic DNA failed to detect a copy of the SRA gene in either T. b. brucei 427-221 or T. b. brucei 427-800. However, the SRA gene was readily amplified from total genomic DNA from T. b. rhodesiense (KETRI 2482) (Fig. 6A).
FIG. 6.

Analysis of the VSG221, VSG800, and SRA genes and transcripts in T. b. brucei and T. b. rhodesiense. (A) PCR analysis of genomic DNA from T. b. brucei 427-221 (lane 1) and 427-800 (lane 2) and T. b. rhodesiense (lane 3) for VSG221, VSG800, and SRA genes. The VSG221 gene is deleted in the T. b. brucei 427-800 line, and the SRA gene is present only in T. b. rhodesiense. (B) Northern blot analysis of VSG221, VSG800, and SRA expression in T. b. brucei 427-221 (lane 1), 427-800 (lane 2), and T. b. rhodesiense (lane 3).
The expression of VSG221 and VSG800 was confirmed by Northern blot hybridization with cDNA probes and total RNA from T. b. brucei 427-221 and 427-800, respectively (Fig. 6B). The SRA mRNA was detected in the T. b. rhodesiense total mRNA but not in RNA samples from either of the T. b. brucei cell lines (Fig. 6B). These findings show that our in vitro-generated, human HDL-resistant T. b. brucei line does not express SRA and that acquisition of HDL resistance correlates with VSG switching.
Transcriptome analysis by SAGE.
SAGE offers a powerful, unbiased approach for the analysis of genome-wide mRNA abundance differences in different cell populations. After the removal of putative sequencing error, a total of 38,545 and 38,334 SAGE tags (21 bp each) were sampled from 427-221 and 427-800, respectively. The sampled tags represented 26,385 unique 21-bp sequences, of which roughly 85% mapped to a single location in the genome. The SAGE analysis revealed that a major difference between the 427-221 and 427-800 transcriptomes was the presence of VSG221 and VSG800 transcripts in the respective cell lines (Table 2). We also found that SAGE tags specific for ESAG1, ESAG2, the terminal ESAG3, and ESAG8b encoded in the 221ES were absent in 427-800. Other, differentially abundant tags corresponded to transcripts for housekeeping proteins including tubulin, translation elongation factors, ribosomal proteins, and enolase. These were present in both the 427-221 and 427-800 libraries and may reflect differences in cell density at the time when RNA was extracted for SAGE library construction. These results showed that selection for HDL resistance accompanied VSG switching and possible loss of ESAG1, ESAG2, the terminal copy of ESAG3, and ESAG8b expression from the 221ES.
TABLE 2.
Differences in mRNA abundance between T. b. brucei 427-221 and 427-800, as detected by SAGEa
| Tag ID no.b | Tag sequence | Annotationc | Frequency in:
|
R valued | Fold change | |
|---|---|---|---|---|---|---|
| 427-221 library | 427-800 library | |||||
| 43548 | CATGGCAGATGGCGGCTGACA | vsg800 primary tag | 0 | 513 | 155.44 | P/Ae |
| 21744 | CATGGACTGCTAAAGAAGGTC | vsg800 secondary tag | 0 | 68 | 20.60 | P/A |
| 22017 | CATGGGCGGTTACCTATCTTT | vsg800 secondary tag | 0 | 15 | 4.54 | P/A |
| 43207 | CATGCAAATGGCATAACGATG | vsg221 primary tag | 447 | 0 | 133.68 | P/A |
| 26 | CATGATGGACACCAGCGGAAC | vsg221 secondary tag | 67 | 0 | 20.04 | P/A |
| 56 | CATGTGAGGGAGAACTAAAAA | esag1 | 44 | 0 | 13.16 | P/A |
| 177 | CATGAAAATCCTGAGGCAGAA | esag3f | 20 | 0 | 5.98 | P/A |
| 263 | CATGTGCACTGGACTATGAGC | esag8b | 15 | 0 | 4.49 | P/A |
| 615 | CATGTCTTTTATTCTACTTTC | Putative 40S ribosomal protein S23 | 7 | 33 | 4.04 | 4.74 |
| 43734 | CATGGTCAGATGCATCACTGC | Elongation factor 2 | 36 | 110 | 8.68 | 3.07 |
| 43193 | CATGGATACGAAGGTAAGCCC | Enolase | 30 | 84 | 5.89 | 2.82 |
| 42626 | CATGCTCAGGGACTAAAAAAA | Unresolved SAGE tag | 25 | 67 | 4.40 | 2.69 |
| 41477 | CATGGTTGCATAACTGTATGC | Putative 40S ribosomal protein S13 | 27 | 69 | 4.21 | 2.57 |
| 42586 | CATGATAAAATATTTGTGGGT | Elongation factor 1 alpha | 78 | 167 | 7.36 | 2.15 |
| 43126 | CATGATGTGGGATGTATTGGG | Beta tubulin | 142 | 266 | 8.56 | 1.88 |
Tags are sorted by change (n-fold), which has been calculated with correction for total sampling effort. Data presented in bold represent tags from the 221ES.
ID, identification.
Primary tags are those from the most 3′-proximal NlaIII site of transcripts, whereas secondary tags are produced for highly abundant transcripts by incomplete NlaIII digestion during SAGE library construction.
SAGE is performed without replicates upon a single RNA extract, making traditional statistics unworkable. As such, SAGE is best viewed as a measure of relative change. To reduce the effects of sampling and sequencing error in highlighting differential expression, only tags with log likelihood (R) values of 4 or greater are shown. Higher log likelihood scores represent a greater deviation from the null hypothesis of equal abundance among the two SAGE libraries.
Tags completely absent in either the 427-221 or 427-800 library are presented as P/A (present/absent).
Two copies of esag3 exist in the 221ES. SAGE tag no. 177 maps to the terminal copy only.
Expression site characterization.
Since VSG switching may occur by either gene conversion or activation of a new, previously silent expression site, we examined the chromosomal location of VSG800 in T. b. brucei 427-221 and 427-800 (Fig. 7). CHEF blotting confirmed loss of VSG221 and duplication of VSG800 in the 427-800 cell line. The new copy of VSG800 localized to chromosome VIa, which contains the 221ES. Each chromosome, however, contains two ES. It was therefore necessary to directly determine whether the 221ES was transcribed in the 427-800 cell line. The ESAG6/7 genes of T. b. brucei have been extensively characterized, and their sequences are diagnostic for the different bloodstream expression sites. cDNA clones were prepared for a hypervariable region of ESAG6/7 from 427-800 (data not shown). The sequence of these clones was identical to that of the 221ES copy of ESAG6/7.
FIG. 7.

Analysis of chromosomal locations of VSG221 and VSG800 by CHEF and Southern blotting. (A) Ethidium bromide-stained CHEF gel of total genomic DNA from T. b. brucei 427-221 and 427-800. H. wingei chromosomes were used as markers (M). (B) Southern blot of genomic DNA from T. b. brucei 427-221 and 427-800 probed for VSG221. The VSG221 gene is absent in the T. b. brucei 427-800 genome. (C) Southern blot of genomic DNA from T. b. brucei 427-221 and 427-800 probed for VSG800. One copy of the VSG800 gene is present in both populations, while a second copy is present only in T. b. brucei 427-800 and localizes to chromosome VIa, which contains the 221ES.
To better evaluate the transcribed ES in T. b. brucei 427-800, we reexamined the SAGE tags of the 427-221 and 427-800 cell lines (Table 3). Both cell lines contained similar numbers of SAGE tags throughout the ES, from ESAG6/7 to ESAG8b (Fig. 8A). Starting at ESAG8b, three 221ES-specific SAGE tags (no. 263, no. 177, and no. 56) were observed only in the 427-221 cells, suggesting the duplicated VSG800 recombined at or near ESAG8b. RT-PCR analysis of RNA from 427-221 and 427-800 cells revealed the presence of ESAG1, -2, -3, and -8 transcripts in both cell lines but suggested that the levels of ESAG1 and -2 in the 427-800 cells were reduced while ESAG3 and ESAG8 transcript levels remained largely unchanged (Fig. 8B). To determine whether the transcripts amplified in these reactions came from the 221ES, we prepared and sequenced cDNAs from 427-800 and 427-221 cells by using ESAG1-, ESAG2-, ESAG3-, and ESAG8-specific primers. Comparison of the cDNAs indicated that the 427-800-derived transcripts were distinct from the 221ES-encoded ESAG1 and -2 transcripts (Fig. 8C). We sequenced three ESAG1, seven ESAG2, and 113 ESAG3 cDNA clones from the 427-800 cells. No sequences identical to that of the 221ES ESAG1 were identified in the 427-800 cDNA clones. However, we did identify one ESAG1-like sequence with 97% nucleotide similarity to the 221ES ESAG1 sequence. A second cDNA had extensive similarity to small regions of the 221ES ESAG1 but only 7% overall similarity (Fig. 8C). We also did not find cDNAs with sequences identical to that of the 221ES ESAG2 in the 427-800 cell line. However, we did identify two ESAG2-like sequences with 97% and 93% nucleotide similarity to the 221ES ESAG2 sequence (Fig. 8C). The analysis of ESAG3 expression is complicated by the presence of three copies of ESAG3 in the 221ES. Within the 221ES is an upstream ESAG3 pseudogene (ESAG3Ψ) and two nearly identical copies of ESAG3. We designated these copies ESAG3 (for the upstream copy of the gene) and ESAG3b (for the downstream copy) (Fig. 8A). As expected from the SAGE analysis, we identified cDNAs for ESAG3, -3b and -3Ψ in 421-221 cells (Fig. 8C). While SAGE detected the possible presence of ESAG3 transcripts in 427-800 cells, we could detect transcripts only from ESAG3Ψ and a non-221ES copy of an ESAG3-like transcript by RT-PCR. While RT-PCR analysis of ESAG8 revealed the two products expected for ESAG8 and -8b, sequencing failed to resolve whether one or both copies of ESAG8 were expressed (data not shown).
TABLE 3.
Mapping of SAGE tags to the 221ESa
| Tag ID no.b | Tag sequence | Annotation | Frequency in:
|
R value | |
|---|---|---|---|---|---|
| 427-221 library | 427-800 library | ||||
| 41524 | CATGTGGTATGTACAAGCTAC | esag6/7c | 44 | 25 | 1.13 |
| 41878 | CATGGGGAACACTGCTGTGCT | esag6 | 32 | 32 | 0.00 |
| 43692 | CATGTAAGGAAACGGCAACAG | Downstream of esag6 | 58 | 45 | 0.34 |
| 63 | CATGATATAAAACGTGGATGC | esag5Ψ | 40 | 45 | 0.07 |
| 87 | CATGAAAATCCTGGGGCAGCA | esag3/3Ψd | 34 | 28 | 0.12 |
| 42811 | CATGTGGGGAGGGGTGACTTT | esag4 | 46 | 38 | 0.16 |
| 42492 | CATGATCTTTTTTTTCTGATT | Hypothetical protein H25N7.21 | 38 | 39 | 0.00 |
| 501 | CATGGAATTGATGTCTCTTCC | esag8/8be | 10 | 2 | 1.25 |
| 41933 | CATGTGCCTTGTGCTTCTTTT | Hypothetical protein H25N7.25 | 15 | 2 | 2.43 |
| 263 | CATGTGCACTGGACTATGAGC | esag8b | 15 | 0 | 4.50 |
| 177 | CATGAAAATCCTGAGGCAGAA | esag3 | 20 | 0 | 6.00 |
| 56 | CATGTGAGGGAGAACTAAAAA | esag1 | 44 | 0 | 13.16 |
Tags are approximately sorted by position on the 221ES (GenBank accession no. AL671259) (Fig. 8). Data presented in bold represent tags with sequences entirely unique to the 221ES. All other tag sequences can be mapped to other known T. b. brucei expression sites but not non-ES locations.
ID, identification.
Tag no. 41524 cannot discriminate between esag6 and esag7 transcripts.
Tag no. 87 maps to both the esag3 pseudogene and the functional esag3 copy in the middle of the ES. It does not map to the terminal copy of esag3.
Tag no. 501 maps to both copies of esag8 as well as to esag8b. As tag no. 263 independently suggests an absence of esag8b transcripts in 427-800, we assume esag8b does not contribute to the frequency of tag no. 501 in 427-800.
FIG. 8.

Mapping of SAGE tags from T. b. brucei 427-221 and 427-800 to the 221ES (GenBank accession no. AL671259) and analysis of ESAG1, -2, and -3 expression. (A) Mapping of SAGE tags to the 221ES. Included on the map are ESAG, VSGs, and their pseudogenes present before (427-221) and after (427-800) recombination. SAGE tags mapping to more than one location on the 221ES are underlined. SAGE tag identification numbers and frequencies can be found in Tables 2 and 3. Tags unique to 427-221 (red) and 427-800 (blue) are indicated. (B) Analysis of transcripts from 221ES genes from 427-221 and 427-800. RT-PCR products were prepared using the specific primer pairs described in Material and Methods and were fractionated by agarose gel electrophoresis and stained with ethidium bromide. (C) Sequence analysis of ESAG1, -2, and -3 cDNAs from 427-221 and 427-800 cells. Only a portion of the derived cDNA sequence is shown for each of the cDNA clones. A single ESAG1 transcript was present in the 427-221 cells, and its sequence was identical to that of the ESAG1 in the 221ES. In contrast, two ESAG1-like sequences not present in the 221ES were obtained from the 427-800 cDNA clones. For ESAG2, a single transcript was expressed in the 427-221 cells and its sequence was identical to the ESAG2 sequence in the 221ES. In contrast, two ESAG2-like sequences not found in the 221ES were obtained from the 427-800 cDNA clones. Transcripts from the 221ES ESAG3ψ, -3, and -3b were present in the 427-221 cells. ESAG3ψ transcripts were also present in the 427-800 cells, in addition to an ESAG3-like transcript not encoded by the 221ES.
Based on the combination of SAGE analysis and cDNA sequence analysis, we conclude that the duplication and transposition of VSG800 into the 221ES resulted in deletion of coding sequences for ESAG1, -2, and -3b. The transcripts in the 427-800 cells are the result of transcription of related genes from other copies in the 221ES, other expression sites, or internal chromosomal copies. We are currently preparing telomere clones of the 221ES from 427-800 cells to precisely define the ESAGs deleted during the transposition of VSG800 into the 221ES.
DISCUSSION
The resistance of humans to infection by T. b. brucei is a consequence of an innate, protective class of human HDLs containing apoL-1, Hpr, apoA-1, and apoA-II (23, 50, 57, 59, 60, 65, 69). African trypanosomes that cause human sleeping sickness circumvent the cytotoxic activity of this HDL subclass. The infectivity of T. b. rhodesiense for humans requires the expression of the VSG-like protein, SRA, which localizes to intracellular organelles of the endocytic pathway and the lysosome (9, 35, 41, 69, 74). While the mechanism of TLF inhibition by SRA is not completely understood, recent studies suggest that binding of SRA to apoL-1 neutralizes the trypanosome-killing activity of TLF (69). Both the SRA gene and its telomeric ES are highly conserved in T. b. rhodesiense (16). This has led to the suggestion that the SRA gene arose only once during evolution and subsequently spread through genetic exchange (17, 18).
Although compelling evidence supports the role of SRA in human infectivity by T. b. rhodesiense, several investigators have reported the occurrence of human serum-resistant lines of African trypanosomes that lack the SRA gene (2, 47, 66, 67). To investigate the mechanism of SRA-independent resistance, we generated human serum-resistant lines of T. b. brucei 427 (MiTat 1.2) by repeated in vitro exposure to human HDLs. We extensively characterized one human HDL-resistant line, T. b. brucei 427-800, which was also highly resistant to purified TLF and could grow in media containing 25% normal human serum. The resistance phenotype correlated with the absence of TLF binding and accumulation in T. b. brucei 427-800 cells (Fig. 2 and 3). Analysis of both protein and RNA profiles indicated that T. b. brucei 427-800 continued to transcribe the 221ES, but ESAG1, -2, -3, and -8b mRNAs (from the 221ES) were not detected and VSG800 was transcribed (Fig. 8; Table 3). Duplication and transposition of VSG800 into the 221ES were confirmed by CHEF blotting (Fig. 7). These studies show that human HDL-resistant variants of T. b. brucei 427 can be generated in vitro and that the resistance phenotype is directly linked to antigenic variation. Based on these findings, we propose that resistance to human HDLs is due to either the expression of VSG800, the deletion of ESAG1, -2, -3, or -8b from the 221ES, or some other unknown mechanism.
The potential for antigenic variation to directly influence human serum susceptibility or resistance was previously proposed based on studies of T. b. rhodesiense and T. b. gambiense isolates that had lost human serum resistance following VSG switching (25, 42, 74). Based on the genomic organization of the SRA gene within bloodstream expression sites, it is possible that antigenic switching by in situ activation of a new, SRA-lacking ES is the mechanism of loss of human serum resistance in T. b. rhodesiense (74).
By analogy to the role that SRA acts as an inhibitor of human HDL killing of T. b. rhodesiense, one might expect human HDL-resistant T. b. brucei 427-800 to express a protein that specifically interacts with TLF and ablates its activity. Since SRA is a member of the VSG family of proteins, it is possible that in vitro exposure of T. b. brucei to human HDL may have selected for the expression of a VSG that conferred resistance. The T. b. brucei genome contains a large number of silent VSG genes, many of which are clustered at internal locations on large chromosomes, with a minority of these genes distributed at telomeres of minichromosomes, intermediate chromosomes, and megabase chromosomes. The expression of VSG genes is tightly regulated to ensure that a single telomeric VSG is transcribed at a time. This leaves hundreds of silent VSG genes free of functional constraints and susceptible to random sequence drift contributing to the variability of VSGs. The high multiplicity of silent VSG genes provides a potential pool for selection of a gene that could confer resistance to human HDLs. Based on limited sequence comparison, we have been unable to identify features of VSG800 that might contribute to inhibition of human HDL killing of trypanosomes. However, the role of VSG800 in the resistance trait can be evaluated directly by replacement of the VSG221 gene in the human HDL-susceptible T. b. brucei 427-221 cell line. This experiment is under way.
In addition to SRA, there is at least one other example of a VSG that has evolved to take on a different function. The trypanosome Tf-Rs are encoded by ESAG6 and ESAG7 and assemble as a heterodimeric receptor anchored to the flagellar pocket by a single GPI on the ESAG6-encoded subunit (6, 27, 54, 63). The ESAG6 and ESAG7 subunits contain VSG-like N-terminal domains that facilitate packing within the VSG surface coat in the flagellar pocket and variable C-terminal sequences with differing affinities for mammalian transferrins (55). Growth of trypanosomes in different sera can select for VSG ES switching and results in the expression of different Tf-Rs (5, 15). A correlation to the relative transferrin binding affinity of the expressed trypanosome Tf-R and the host transferrin has been made previously (15, 71).
T. b. brucei 427-800 and T. b. rhodesiense clearly share the human HDL resistance phenotype in a standard lysis assay. Despite this superficial similarity, analysis of TLF uptake and localization revealed differences. Two reports show that TLF (22) or purified apoL-I (69) is taken up by T. b. rhodesiense. In a study with native purified TLF we have found that, although TLF accumulation was reduced in T. b. rhodesiense, flagellar pocket binding and perhaps endocytosis occurred. However, TLF did not traffic to the lysosome (22). Vanhamme and coworkers also found that T. b. rhodesiense bound recombinant apoL-I but, in contrast to our initial studies, found that apoL-I was efficiently taken up and colocalized to the trypanosome lysosome with SRA (69). In the studies reported here, we were unable to detect TLF accumulation in T. b. brucei 427-800 cells, raising the possibility that human HDL resistance in T. b. brucei 427-800 may result from loss of the TLF receptor. Consistent with this possibility, we found that ESAG1, -2, and -3 were potentially deleted during the transposition of VSG800 into the 221ES and that transcripts from these genes were absent in the SAGE library and also in cDNAs prepared from T. b. brucei 427-800 (Table 3; Fig. 8). ESAG1 and -2 are interesting candidates for the TLF receptor in T. b. brucei. ESAG2 is a GPI-anchored protein found exclusively in the flagellar pocket of bloodstream trypanosomes (40, 44). Less is known about ESAG1, but it is predicted to be a transmembrane cell surface protein in bloodstream trypanosomes (44). While the function of each of these proteins is unknown, it is interesting to note that another human serum-resistant line of T. b. brucei (TREU 927; GuTat 10.1) also lacks these genes in the active expression site (66, 67).
We have shown here that T. b. brucei can gain serum resistance after repeated exposure to human HDL. The genetic cause of resistance has not been thoroughly determined; we have shown, however, that the lack of TLF accumulation in the lysosome is the most likely cellular mechanism. This phenotype may be due to the expression of VSG800, which, by some unknown mechanism, inhibits uptake of TLF. Alternatively, TLF resistance might be a consequence of loss of a gene encoding the TLF receptor during recombination of VSG800 into the 221ES. These possibilities are currently being tested.
Acknowledgments
We thank members of the Global Infectious Disease Program at the Marine Biological Laboratory for critical discussion and comments on the manuscript. In addition, we thank the students of the Biology of Parasitism (BoP 2005) course at the MBL for their enthusiastic interest in this project.
These studies were supported by grants AI39033 and AI054596 from the National Institutes of Health and the Ellison Medical Foundation. Mass spectrometry was supported by NIH P20 RR17695 from the Institutional Development Award (IDeA) Program of the National Center for Research Resources. Computational resources were provided by the Josephine Bay Paul Center for Comparative Molecular Biology and Evolution (MBL) through funds provided by the W. M. Keck Foundation and the G. Unger Vetlesen Foundation.
REFERENCES
- 1.Barry, J. D., M. L. Ginger, P. Burton, and R. McCulloch. 2003. Why are parasite contingency genes often associated with telomeres? Int. J. Parasitol. 33:29-45. [DOI] [PubMed] [Google Scholar]
- 2.Berberof, M., D. Perez-Morga, and E. Pays. 2001. A receptor-like glycoprotein specific to Trypanosoma brucei gambiense. Mol. Biochem. Parasitol. 113:127-138. [DOI] [PubMed] [Google Scholar]
- 3.Berriman, M., N. Hall, K. Sheader, F. Bringaud, B. Tiwari, T. Isobe, S. Bowman, C. Corton, L. Clark, G. A. M. Cross, M. Hoek, T. Zanders, M. Berberof, P. Borst, and G. Rudenko. 2002. The architecture of variant surface glycoprotein gene expression sites in Trypanosoma brucei. Mol. Biochem. Parasitol. 122:131-140. [DOI] [PubMed] [Google Scholar]
- 4.Bishop, J. R., M. Shimamura, and S. L. Hajduk. 2001. Insight into the mechanism of trypanosome lytic factor-1 killing of Trypanosoma brucei brucei. Mol. Biochem. Parasitol. 118:33-40. [DOI] [PubMed] [Google Scholar]
- 5.Bitter, W., H. Gerrits, R. Kieft, and P. Borst. 1998. The role of transferrin-receptor variation in the host range of Trypanosoma brucei. Nature 391:499-502. [DOI] [PubMed] [Google Scholar]
- 6.Borst, P., and A. H. Fairlamb. 1998. Surface receptors and transporters of Trypanosoma brucei. Annu. Rev. Microbiol. 52:745-778. [DOI] [PubMed] [Google Scholar]
- 7.Borst, P., and S. Ulbert. 2001. Control of VSG gene expression sites. Mol. Biochem. Parasitol. 114:17-27. [DOI] [PubMed] [Google Scholar]
- 8.Campillo, N., and M. Carrington. 2003. The origin of the serum resistance associated (SRA) gene and a model of the structure of the SRA polypeptide from Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 127:79-84. [DOI] [PubMed] [Google Scholar]
- 9.De Greef, C., H. Imberechts, G. Matthyssens, N. Van Meirvenne, and R. Hamers. 1989. A gene expressed only in serum-resistant variants of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 36:169-176. [DOI] [PubMed] [Google Scholar]
- 10.De Greef, C., E. Chimfwembe, J. Kihang'a Wabacha, E. Bajyana Songa, and R. Hamers. 1992. Only the serum-resistant bloodstream forms of Trypanosoma brucei rhodesiense express the serum resistance associated (SRA) protein. Ann. Soc. Belge Med. Trop. 72(Suppl. 1):13-21. [PubMed] [Google Scholar]
- 11.De Greef, C., and R. Hamers. 1994. The serum resistance-associated (SRA) gene of Trypanosoma brucei rhodesiense encodes a variant surface glycoprotein-like protein. Mol. Biochem. Parasitol. 68:277-284. [DOI] [PubMed] [Google Scholar]
- 12.Donelson, J. E. 2003. Antigenic variation and the African trypanosome genome. Acta Trop. 85:391-404. [DOI] [PubMed] [Google Scholar]
- 13.Drain, J., J. R. Bishop, and S. L. Hajduk. 2001. Haptoglobin-related protein mediates trypanosome lytic factor binding to trypanosomes. J. Biol. Chem. 276:30254-30260. [DOI] [PubMed] [Google Scholar]
- 14.Edgar, R. C. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerrits, H., R. Mussmann, W. Bitter, R. Kieft, and P. Borst. 2002. The physiological significance of transferrin receptor variations in Trypanosoma brucei. Mol. Biochem. Parasitol. 119:237-247. [DOI] [PubMed] [Google Scholar]
- 16.Gibson, W., and V. Ferris. 2003. Conservation of the genomic location of the human serum resistance associated gene in Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 130:159-162. [DOI] [PubMed] [Google Scholar]
- 17.Gibson, W. 2002. Will the real Trypanosoma brucei rhodesiense please step forward? Trends Parasitol. 18:486-490. [DOI] [PubMed] [Google Scholar]
- 18.Gibson, W., T. Backhouse, and M. Griffiths. 2002. The human serum resistance associated gene is ubiquitous and conserved in Trypanosoma brucei rhodesiense throughout East Africa. Infect. Genet. Evol. 1:207-214. [DOI] [PubMed] [Google Scholar]
- 19.Gibson, W. C. 1986. Will the real Trypanosoma brucei gambiense please stand up? Parasitol. Today 2:255-257. [DOI] [PubMed] [Google Scholar]
- 20.Green, H. P., M. del Pilar Molina Portela, E. N. St. Jean, E. B. Lugli, and J. Raper. 2003. Evidence for a Trypanosoma brucei lipoprotein scavenger receptor. J. Biol. Chem. 278:422-427. [DOI] [PubMed] [Google Scholar]
- 21.Hager, K. M., M. A. Pierce, D. R. Moore, E. M. Tytler, J. D. Esko, and S. L. Hajduk. 1994. Endocytosis of a cytotoxic human high density lipoprotein results in disruption of acidic intracellular vesicles and subsequent killing of African trypanosomes. J. Cell Biol. 126:155-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hager, K. M., and S. L. Hajduk. 1997. Mechanism of resistance of African trypanosomes to cytotoxic human HDL. Nature 385:823-826. [DOI] [PubMed] [Google Scholar]
- 23.Hajduk, S. L., D. R. Moore, J. Vasudevacharya, H. Siqueira, A. F. Torri, E. M. Tytler, and J. D. Esko. 1989. Lysis of Trypanosoma brucei by a toxic subspecies of human high-density lipoprotein. J. Biol. Chem. 264:5210-5217. [PubMed] [Google Scholar]
- 24.Hawking, F., D. B. Ramsden, and S. Whytock. 1973. The trypanocidal action of human serum and of baboon plasma. Trans. R. Soc. Trop. Med. Hyg. 67:501-516. [DOI] [PubMed] [Google Scholar]
- 25.Hawking, F. 1976. The resistance to human plasma of Trypanosoma brucei, T. rhodesiense and T. gambiense. Trans. R. Soc. Trop. Med. Hyg. 70:504-512. [DOI] [PubMed] [Google Scholar]
- 26.Hoare, C. A. 1972. The trypanosomes of mammals. A zoological monograph. Blackwell Scientific Publications, Oxford, United Kingdom.
- 27.Ligtenberg, M. J. L., W. Bitter, R. Kieft, D. Steverding, H. Janssen, J. Calafat, and P. Borst. 1994. Reconstitution of a surface transferrin binding complex in insect form Trypanosoma brucei. EMBO J. 13:2565-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lorenz, P., P. E. Barth, W. Rudin, and B. Betschart. 1994. Importance of acidic intracellular compartments in the lysis of Trypanosoma brucei brucei by normal human serum. Trans. R. Soc. Trop. Med. Hyg. 88:487-488. [DOI] [PubMed] [Google Scholar]
- 29.Lorenz, P., R. W. James, J. S. Owen, and B. Betschart. 1994. Heterogeneity in the properties of the trypanolytic factor in normal human serum. Mol. Biochem. Parasitol. 64:153-164. [DOI] [PubMed] [Google Scholar]
- 30.Lorenz, P., J. S. Owen, and D. G. Hassall. 1995. Human serum resistant Trypanosoma brucei rhodesiense accumulates similar amounts of fluorescently-labeled trypanolytic human HDL3 particles as human serum sensitive T. b. brucei. Mol. Biochem. Parasitol. 74:113-118. [DOI] [PubMed] [Google Scholar]
- 31.Lugli, E. B., M. Pouliot, M. del Pilar Molina Portela, M. R. Loomis, and J. Raper. 2004. Characterization of primate trypanosome lytic factors. Mol. Biochem. Parasitol. 138:9-20. [DOI] [PubMed] [Google Scholar]
- 32.McCulloch, R. 2004. Antigenic variation in African trypanosomes: monitoring progress. Trends Parasitol. 20:117-121. [DOI] [PubMed] [Google Scholar]
- 33.McEvoy, S. M., and N. Maeda. 1988. Complex events in the evolution of the haptoglobin gene cluster in primates. J. Biol. Chem. 263:15740-15747. [PubMed] [Google Scholar]
- 34.Melville, S. E., V. Leech, M. Navarro, and G. A. M. Cross. 2000. The molecular karyotype of the megabase chromosomes of Trypanosoma brucei stock 427. Mol. Biochem. Parasitol. 111:261-273. [DOI] [PubMed] [Google Scholar]
- 35.Milner, J. D., and S. L. Hajduk. 1999. Expression and localization of serum resistance associated protein in Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 104:271-283. [DOI] [PubMed] [Google Scholar]
- 36.Molina-Portela, M. P., J. Raper, and S. Tomlinson. 2000. An investigation into the mechanism of trypanosome lysis by human serum factors. Mol. Biochem. Parasitol. 110:273-282. [DOI] [PubMed] [Google Scholar]
- 37.Molina-Portela, M. P., E. B. Lugli, E. Recio-Pinto, and J. Raper. 2005. Trypanosome lytic factor, a subclass of high-density lipoprotein, forms cation-selective pores in membranes. Mol. Biochem. Parasitol. 144:218-226. [DOI] [PubMed] [Google Scholar]
- 38.Muranjan, M., V. Nussenzweig, and S. Tomlinson. 1998. Characterization of the human serum trypanosome toxin, haptoglobin-related protein. J. Biol. Chem. 273:3884-3887. [DOI] [PubMed] [Google Scholar]
- 39.Njiru, Z. K., K. Nbung'u, G. Mateti, J. M. Ndungu, and W. C. Gibson. 2004. Detection of Trypanosoma brucei rhodesiense in animals from sleeping sickness foci in East Africa using the serum resistance associated (SRA) gene. Acta Trop. 90:249-254. [DOI] [PubMed] [Google Scholar]
- 40.Nolan, D. P., M. Geuskens, and E. Pays. 1999. N-linked glycans containing linear poly-N-acetyllactosamine as sorting signals in endocytosis in Trypanosoma brucei. Curr. Biol. 9:1169-1172. [DOI] [PubMed] [Google Scholar]
- 41.Oli, M. W., L. F. Cotlin, A. M. Shiflett, and S. L. Hajduk. 2006. Serum resistance-associated protein blocks lysosomal targeting of trypanosome lytic factor in Trypanosoma brucei. Eukaryot. Cell 5:132-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ortiz-Ordonez, J. C., J. B. Sechelski, and J. R. Seed. 1994. Mechanism of lysis of Trypanosoma brucei gambiense by human serum. J. Parasitol. 80:924-930. [PubMed] [Google Scholar]
- 43.Paturiaux-Hanocq, F., N. Zitzmann, J. Hanocq-Quertier, L. Vanhamme, S. Rolin, M. Geuskens, M. A. Ferguson, and E. Pays. 1997. Expression of a variant surface glycoprotein of Trypanosoma gambiense in procyclic forms of Trypanosoma brucei shows that the cell type dictates the nature of the glycosylphosphatidylinositol membrane anchor attached to the glycoprotein. Biochem. J. 324:885-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pays, E., S. Lips, D. Nolan, L. Vanhamme, and D. Pérez-Morga. 2001. The VSG expression sites of Trypanosoma brucei: multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol. Biochem. Parasitol. 114:1-16. [DOI] [PubMed] [Google Scholar]
- 45.Pérez-Morga, D., B. Vanhollebeke, F. Paturiaux-Hanocq, D. P. Nolan, L. Lins, F. Homble, L. Vanhamme, P. Tebabi, A. Pays, P. Poelvoorde, A. Jacquet, R. Brasseur, and E. Pays. 2005. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 309:469-472. [DOI] [PubMed] [Google Scholar]
- 46.Poelvoorde, P., L. Vanhamme, J. Van Den Abbeele, W. M. Switzer, and E. Pays. 2004. Distribution of apolipoprotein L-I and trypanosome lytic activity among primate sera. Mol. Biochem. Parasitol. 134:155-157. [DOI] [PubMed] [Google Scholar]
- 47.Radwanska, M., M. Chamekh, L. Vanhamme, F. Claes, S. Magez, E. Magnus, P. De Baetselier, P. Büscher, and E. Pays. 2002. The serum resistance-associated gene as a diagnostic tool for the detection of Trypanosoma brucei rhodesiense. Am. J. Trop. Med. Hyg. 67:684-690. [DOI] [PubMed] [Google Scholar]
- 48.Raper, J., R. Fung, J. Ghiso, V. Nussenzweig, and S. Tomlinson. 1999. Characterization of a novel trypanosome lytic factor from human serum. Infect. Immun. 67:1910-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raper, J., M. P. Molina Portela, E. Lugli, U. Frevert, and S. Tomlinson. 2001. Trypanosome lytic factors: novel mediators of human innate immunity. Curr. Opin. Microbiol. 4:402-408. [DOI] [PubMed] [Google Scholar]
- 50.Rifkin, M. R. 1978. Identification of the trypanocidal factor in normal human serum: high density lipoprotein. Proc. Natl. Acad. Sci. USA 75:3450-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rifkin, M. R. 1984. Trypanosoma brucei: biochemical and morphological studies of cytotoxicity caused by normal human serum. Exp. Parasitol. 58:81-93. [DOI] [PubMed] [Google Scholar]
- 52.Rifkin, M. R. 1991. Trypanosoma brucei: cytotoxicity of host high-density lipoprotein is not mediated by apolipoprotein A-I. Exp. Parasitol. 72:216-218. [DOI] [PubMed] [Google Scholar]
- 53.Rudenko, G., I. Chaves, A. Dirks-Mulder, and P. Borst. 1998. Selection for activation of a new variant surface glycoprotein gene expression site in Trypanosoma brucei can result in deletion of the old one. Mol. Biochem. Parasitol. 95:97-109. [DOI] [PubMed] [Google Scholar]
- 54.Salmon, D., M. Geuskens, F. Hanocq, J. Hanocq-Quertier, D. Nolan, L. Ruben, and E. Pays. 1994. A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell 78:75-86. [DOI] [PubMed] [Google Scholar]
- 55.Salmon, D., J. Hanocq-Qertier, F. P. Hanocq, A. Pays, P. Tebabi, D. P. Nolan, A. Michel, and E. Pays. 1997. Characterization of the ligand-binding site of the transferring receptor in Trypanosoma brucei demonstrates a structural relationship with the N-terminal domain of the variant surface glycoprotein. EMBO J. 16:7272-7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seed, J. R., J. B. Sechelski, and M. R. Loomis. 1990. A survey for a trypanocidal factor in primate sera. J. Protozool. 37:393-400. [DOI] [PubMed] [Google Scholar]
- 57.Shiflett, A. M., J. R. Bishop, A. Pahwa, and S. L. Hajduk. 2005. Human high density lipoproteins are platforms for the assembly of multi-component innate immune complexes. J. Biol. Chem. 280:32578-32585. [DOI] [PubMed] [Google Scholar]
- 58.Shimamura, M., K. M. Hager, and S. L. Hajduk. 2001. The lysosomal targeting and intracellular metabolism of trypanosome lytic factor by Trypanosoma brucei brucei. Mol. Biochem. Parasitol. 115:227-237. [DOI] [PubMed] [Google Scholar]
- 59.Smith, A. B., J. D. Esko, and S. L. Hajduk. 1995. Killing of trypanosomes by the human haptoglobin-related protein. Science 268:284-286. [DOI] [PubMed] [Google Scholar]
- 60.Smith, A. B., and S. L. Hajduk. 1995. Identification of haptoglobin as a natural inhibitor of trypanocidal activity in human serum. Proc. Natl. Acad. Sci. USA 92:10262-10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stein, L. D., C. Mungall, S. Shu, M. Caudy, M. Mangone, A. Day, E. Nickerson, J. E. Stajich, T. W. Harris, A. Arva, and S. Lewis. 2002. The generic genome browser: a building block for a model organism system database. Genome Res. 12:1599-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stekel, D. J., Y. Git, and F. Falciani. 2000. The comparison of gene expression from multiple cDNA libraries. Genome Res. 10:2055-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steverding, D., Y. D. Stierhof, H. Fuchs, R. Tauber, and P. Overath. 1995. Transferrin-binding protein complex is the receptor for transferrin uptake in Trypanosoma brucei. J. Cell Biol. 131:1173-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomlinson, S., A. M. Jansen, A. Koudinov, J. A. Ghiso, N. H. Choi-Miura, M. R. Rifkin, S. Ohtakiand, and V. Nussenzweig. 1995. High-density-lipoprotein-independent killing of Trypanosoma brucei by human serum. Mol. Biochem. Parasitol. 70:131-138. [DOI] [PubMed] [Google Scholar]
- 65.Tomlinson, S., M. Muranjan, V. Nussenzweig, and J. Raper. 1997. Haptoglobin-related protein and apolipoprotein AI are components of the two trypanolytic factors in human serum. Mol. Biochem. Parasitol. 86:117-120. [PubMed] [Google Scholar]
- 66.Turner, C. M. R., S. McLellan, L. A. G. Lindergard, L. Bisoni, A. Tait, and A. MacLeod. 2004. Human infectivity trait in Trypanosoma brucei: stability, heritability and relationship to sra expression. Parasitology 129:445-454. [DOI] [PubMed] [Google Scholar]
- 67.Vanhamme, L., H. Renauld, L. Lecordier, P. Poelvoorde, J. Abbeele, and E. Pays. 2004. The Trypanosoma brucei reference strain TREU927/4 contains T. b. rhodesiense-specific SRA sequences, but displays a distinct phenotype of relative resistance to human serum. Mol. Biochem. Parasitol. 135:39-47. [DOI] [PubMed] [Google Scholar]
- 68.Vanhamme, L., E. Pays, R. McCulloch, and J. D. Barry. 2001. An update on antigenic variation in African trypanosomes. Trends Parasitol. 17:338-343. [DOI] [PubMed] [Google Scholar]
- 69.Vanhamme, L., F. Paturiaux-Hanocq, P. Poelvoorde, D. P. Nolan, L. Lins, J. Van Den Abbeele, A. Pays, P. Tebabi, H. Van Xong, A. Jacquet, N. Moguilevsky, M. Dieu, J. P. Kane, P. De Baetselier, R. Brasseur, and E. Pays. 2003. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 422:83-87. [DOI] [PubMed] [Google Scholar]
- 70.Vanhamme, L., and E. Pays. 2004. The trypanosome lytic factor of human serum and the molecular basis of sleeping sickness. Int. J. Parasitol. 34:887-898. [DOI] [PubMed] [Google Scholar]
- 71.van Luenen, H. G. A. M., R. Kieft, R. Mussmann, M. Engstler, B. ter Riet, and P. Borst. 2005. Trypanosomes change their transferrin receptor expression to allow effective uptake of host transferrin. Mol. Microbiol. 58:151-165. [DOI] [PubMed] [Google Scholar]
- 72.Welburn, S. C., K. Picozzi, E. M. Fevre, P. G. Coleman, M. Odiit, M. Carrington, and I. Maudlin. 2001. Identification of human-infective trypanosomes in animal reservoir of sleeping sickness in Uganda by means of serum-resistance-associated (SRA) gene. Lancet 358:2017-2019. [DOI] [PubMed] [Google Scholar]
- 73.Welburn, S. C., and M. Odiit. 2002. Recent developments in human African trypanosomiasis. Curr. Opin. Infect. Dis. 15:477-484. [DOI] [PubMed] [Google Scholar]
- 74.Xong, H. V., L. Vanhamme, M. Chamekh, C. E. Chimfwembe, J. Van Den Abbeele, A. Pays, N. Van Meirvenne, R. Hamers, P. De Baetselier, and E. Pays. 1998. A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell 95:839-846. [DOI] [PubMed] [Google Scholar]