

Abstract
In vivo virologic compartments are cell types or tissues between which there is a restriction of virus flow, while virologic reservoirs are cell types or tissues in which there is a relative restriction of replication. The distinction between reservoirs and compartments is important because therapies that would be effective against a reservoir may not be effective against viruses produced by a given compartment, and vice versa. For example, the use of cytokines to “flush out” long-lived infected cells in patients on highly active antiretroviral therapy (T. W. Chun, D. Engel, M. M. Berrey, T. Shea, L. Corey, and A. S. Fauci, Proc. Natl. Acad. Sci. USA 95:8869-8873, 1998) may be successful for a latent reservoir but may not impact a compartment in which virus continues to replicate because of poor drug penetration. Here, we suggest phylogenetic criteria to illustrate, define, and differentiate between reservoirs and compartments. We then apply these criteria to the analysis of simulated and actual human immunodeficiency virus type 1 sequence data sets. We report that existing statistical methods work quite well at detecting viral compartments, and we learn from simulations that viral divergence from a calculated most recent common ancestor is a strong predictor of viral reservoirs.
Human immunodeficiency virus type 1 (HIV-1) persists in cells and tissues long after highly active antiretroviral therapy (HAART) has suppressed plasma HIV-1 RNA levels below the limit of detection. Even in patients who have been on suppressive therapy for several years, virus has been observed to rebound to pretherapy levels during therapy interruptions. Rebounding virus may be derived from latent reservoirs (organs or tissues that harbor potentially infectious virus in a near-dormant state for long periods) and/or organs or tissues in which there is ongoing viral replication (i.e., anatomical sites with low drug penetration). Sites of low drug penetration are generally thought to be restricted to specific cell types, organs, and/or tissues. Such tissues may also act as virologic compartments in which there is limited exchange of virus with other organs and tissues. Identifying viral compartments of low drug penetration and determining whether a given tissue or cell type is acting as a virus reservoir are important steps in developing targeted treatment strategies to enhance antiretroviral efficacy.
A number of examples of HIV-1 compartmentalization have been presented (e.g., references 9, 10, 19-21, and 28). In some reports, compartmentalization was detected upon visual inspection of a phylogenetic tree because compartment-specific sequences were grouped into separate clades on the tree (10, 21). However, when sequences from different compartments do not obviously group into separate clades, statistical support for viral compartmentalization can be obtained from quantitative cladistic (24, 28, 36, 37, 39) and maximum-likelihood (1) methods.
It is possible for a tissue compartment to maintain a certain level of latency and thus be classified as both a reservoir and a compartment. Peripheral blood mononuclear cells (PBMC) have been noted to harbor a small latent reservoir of infectious virus under HAART (3, 40). This reservoir has been quantified by induction of replication-competent virus from blood cells removed for study during effective therapy (13). However, PBMC may not contribute the majority of the rebounding HIV-1 upon discontinuation of HAART (5); hence, other viral reservoirs are thought to exist. It is important to note that induction of competent virus would be quite difficult or impossible to directly monitor in a wide range of tissue types outside the blood. Thus, a phylogenetics-based method for detecting a reservoir would be a valuable tool for targeting therapy, evaluating therapeutic efficacy, and defining and eliminating residual sources of virus replication in treated individuals.
We begin with a schematic depiction of a set of sequences sampled longitudinally (i.e., samples obtained serially throughout the course of infection) from the blood, and in the last time frame, we sample from both the blood and the putative reservoir. It is known that blood sequences sampled at one time point tend to cluster together, to the exclusion of other time points, and that later time points tend to fall further from the most recent common ancestor (MRCA) than do earlier time points (34). We propose that, given enough evolutionary time, viral sequences from a reservoir should have genetic characteristics that would distinguish them from contemporaneous sequences (Fig. 1A and B).
FIG. 1.

Genetic characteristics of virus from persistent reservoirs and nonreservoir compartments. Panel A illustrates the characteristics predicted for a persistent virus reservoir. The phylogenetic tree depicts viral sequences from blood over four time points (thin lines for times 1 to 3, thick lines for time 4) plus a putative virus reservoir sampled at time 4 (thick gray lines). The relationships between the putative reservoir sequences are summarized in panel B, and the relationships between blood sequences (i.e., nonreservoir) sampled at time point 4 are summarized in panel C. The reservoir sequences in panel A have less temporal structure (indicated by the length of the brackets in panels B and C), higher levels of sequence diversity (the length of lines without arrowheads in panels B and C), and a lower average divergence from the MRCA than contemporaneous virus from the blood (compare arrow lengths in panels B and C). Of these three criteria, the reduction in sequence divergence is the most important for detection of a reservoir. Panels D to F illustrate some examples of samples from a putative reservoir (thick gray lines) that do not fit all of our criteria. The sequences in panel D are temporally structured, since they are phylogenetically intermingled with the contemporaneous blood-derived virus. More importantly, they have high mean divergence from the MRCA. The sequences in panel E have higher mean diversity but also have high levels of divergence equivalent to blood-derived sequences. These relationships would be expected from a compartment having high asymmetric migration from the blood to the compartment with turnover as fast as that of virus in the blood. The sequences in panel F have high divergence and high diversity, as well as high temporal structure. These relationships would be expected for a compartment seeded early in infection and evolving separately from the virus in the blood.
(i) A sampling of sequences obtained from a reservoir should have reduced temporal structure compared to a sample of sequences obtained from a nonreservoir because they would be more likely to share close ancestry outside the time they were sampled. Therefore, sequences from a reservoir should be dispersed throughout a phylogenetic tree of viruses sampled over time from an individual. It is important to recognize that the criterion of reduced temporal structure can only be evaluated with samples taken serially over the course of infection.
(ii) A sampling of sequences obtained from a reservoir in patients who have been infected for long periods of time will typically have higher levels of diversity than those from sites not serving as a reservoir, reflecting their dispersed phylogenetic lineages and the presence of both contemporary and historical viral sequences.
(iii) Most importantly, reservoir viral sequences should have diverged, on average, less from the MRCA than contemporaneous virus from a nonreservoir. This is due to the reservoir being fed at both early and late times of infection and persisting in approximately the same state because of a lack of ongoing replication.
The strongest signal of a reservoir comes from characteristic iii, largely because it is less dependent on the length of time of infection. For characteristics i and ii, but i in particular, to be readily detected requires either directional selection through time and/or the passage of more generations than needed for a sample to coalesce to its MRCA. Temporal structure can be quantified by using clustering algorithms (2, 36, 37), while diversity and divergence can be estimated by using phylogenetic algorithms available in the PHYLIP (12) and PAUP* (38) software packages.
In contrast to virus in a reservoir, blood-borne virus sampled contemporaneously (t = 4 in Fig. 1A) should have relatively strong temporal structure, low diversity, and high mean divergence from the MRCA (Fig. 1C). The sequences in Fig. 1D fail to meet our criteria for a viral reservoir because of strong temporal structure, low viral diversity, and a large mean distance from the MRCA. This phylogenetic structure indicates substantial comingling of virus in the candidate reservoir with virus in the blood (i.e., a high migration rate). The sequences in Fig. 1E also fail to meet the criteria for a reservoir because of their high mean divergence from the MRCA. Thus, although the candidate reservoir was seeded with virus over time (i.e., because of asymmetric migration from the blood to the site), virus in the candidate reservoir is evolving at the same rate as virus in the blood. The sequences in Fig. 1F fail to meet our criteria for a reservoir because of a high mean divergence from the MRCA and because of their strong temporal structure. Such a pattern would be expected in a compartment seeded early in infection but containing virus replicating separately from virus in the blood.
We next performed a computer simulation to establish a baseline for the differences in genetic diversity and divergence that could be expected between reservoirs and compartments. We used the serially sampled coalescent (11) to model the evolutionary process in the genealogy of a viral population sample involving both contemporary virus and reservoir virus. The coalescent describes the accumulation of neutral mutations occurring as Poisson events during the evolution of a sample of individuals from a large ideal (Wright-Fisher) population (15). Modifications of a basic simulator (16) make it possible to sample the ideal population at various times during evolution. To model the evolution of reservoir virus, we suppose that virus infects reservoir cells and becomes latent at different times throughout infection.
We thus consider reservoir viruses to have arrived in the reservoir at times earlier than the actual patient sampling time, undergoing little or no further mutation accumulation up to the time of patient sampling. Under this scheme, the genealogy of a reservoir sample should follow a serially sampled coalescent. Each individual in a reservoir sample has a sampling time given by a random variate drawn from a plausible age distribution of latently infected cells. In the Appendix, we describe the derivation of this latent-cell age distribution based on a simplified variant of an often-cited model for the dynamics of virus and latently infected cells (27). The entire contemporary sample has coalescent time zero, equivalent to the present or actual patient sampling time. The simulated genealogy includes both contemporary virus and reservoir virus. Lineages that had not coalesced at the time of infection were forced to coalesce instantaneously at that point (18); this approximately models the rapid outgrowth of virus upon infection.
We sought to model the impact of reduced replication in the reservoir on the levels of diversity and divergence in the reservoir, compared to those in a contemporary nonreservoir sample. Each iteration of the simulation, therefore, consisted of the following steps: for n reservoir individuals and n contemporary individuals, (i) choose n random sampling times in the past for the reservoir sample, according to the latent-cell age distribution; (ii) use the serial coalescent to create a genealogy of all 2n sampled individuals, using the random sampling times for the reservoir and time zero for the contemporary individuals; (iii) place mutations on the genealogy at a specified constant rate; and (iv) calculate the average pairwise diversity within the samples and the average divergence from the root of the genealogy for both the reservoir and the contemporary samples and record the differences. Two hundred fifty iterations were performed for each set of parameters, giving rise to a total of 6,000 simulations.
To perform the aforementioned simulations, we required estimates of several evolutionary and virological parameters. Because our intent was to demonstrate the qualitative behavior of diversity and divergence in viral reservoirs, we did not exhaustively explore the parameter space but attempted to use biologically reasonable point estimates for most parameters. Fixed parameters were the scaled mutation rate θ = 2Neu (where Ne is the effective population size and u is the mutation rate per site per generation), 0.190 (based on estimates from the data of Shankarappa et al. [34]); a sample size (n) for both reservoir and contemporary samples of 10; and a viral generation time of 1.2 days/generation (31). Viral-load parameters were selected such that the load rose from approximately 100 virions/ml of plasma upon infection to 106 virions/ml 14 days postinfection. Acute high viremia resolved by 30 days postinfection, and the viral set point was approximately 103 virions/ml (see Appendix). We varied three parameters: Ne (1,000 or 2,500 [23]), latent-cell half-life (6, 24, or 44 months [30, 35, 41]), and length of infection (12, 30, 60, or 120 months).
Figure 2 displays the simulation results. The hypothesis that reservoir divergence is less than contemporary divergence was borne out in all cases; the central 95% of the empirical distributions of the difference was always less than zero. Diversity in the reservoir can become greater than contemporary diversity, on average, provided that the infection has continued long enough and the latent-cell half-life is long. Earlier in infection, before there has been enough time for mutations to become fixed in the population, diversity was generally reduced in the reservoir.
FIG. 2.

Diversity and divergence comparisons between reservoir and contemporary virus samples. The median difference between the reservoir sample divergence (black triangles) and diversity (gray squares) and contemporary divergence and diversity is graphed for three latent-cell half-lives (6, 24, and 44 months) and two effective population sizes (1,000 and 2,500). Each simulation represents 250 iterations. The 95th percentiles of the diversity and divergence distributions are indicated by gray and black bars, respectively.
Finally, we analyzed two HIV sequence data sets from different patients to demonstrate how our assessment method can be used to evaluate viral reservoirs. The first patient comes from a study of sequences derived from a known and well-defined reservoir, and the second comes from a study in which the sequences were not derived from a known reservoir but nonetheless exhibit reservoir-like behavior.
A latent-virus reservoir composed of HIV-1-infected resting CD4+ T cells that can only be detected in patients who have been on suppressive combination therapy (i.e., HAART) has been previously described (5-7, 13, 14, 30). We used our reservoir criteria to determine if the phylogenetic signal we predict would arise from such a sampling of data (Fig. 3). Imamichi et al. (17) published sequences from a 40-year-old male who had been infected with HIV-1 for at least 6 years before initiation of HAART. His levels of HIV RNA declined to <50 copies/ml during therapy. Figure 3 shows that the DNA sample taken from PBMC after 5 months of effective HAART had higher levels of genetic diversity but lower levels of divergence, as we would predict for a reservoir. Stronger inferences concerning temporal structure could have been made if samples from more time points prior to the onset of therapy had been available. Nevertheless, this result is consistent with what we expect from a reservoir population. We recognize that this sample could represent a pool of defective archival sequences that would not contribute to the rebounding virus pool when HAART is discontinued (4, 22). However, the process that leads to latent infection is likely to be one of the same processes that drive the development of the defective archival pool. Thus, the evolutionary patterns in the archival pool should be similar to that found in the latent-reservoir pool.
FIG. 3.

Analysis of sequence data obtained prior to and after ∼5 months of effective therapy. (A) A maximum-likelihood tree with a model of evolution determined to be HKY + G. The tree was constructed by using a neighbor-joining start with an SPR algorithm for branch swapping in PAUP*. (B) Diversity and divergence of sequences within this patient. We note that these quantities are neither normally distributed nor independent measures of distances, and thus, it is not appropriate to use parametric statistics. Instead, we chose to use resampling methods to determine significance levels. The confidence intervals and the mean test are based on 1,000 bootstrap replicates. We found that the samples taken after ∼5 months of HAART had the same evolutionary patterns found in the simulation (Ne of 1,000 and latency half-life of 44 months), whereas diversity was higher and divergence was lower for the latent-reservoir pool. OG, outgroup; BS, bootstrap.
The second HIV-1-infected patient also had pleural tuberculosis (TB) due to Mycobacterium tuberculosis (8). Sequences from the C2-C3 coding region of HIV-1 env were obtained from PBMC and pleural fluid mononuclear cells, as well as from virions from plasma and pleural fluid. The main conclusion was that pleural TB raises HIV-1 genetic variation largely in the pleural cavity and somewhat systemically. We critically reanalyzed the sequences from patient 57 with the methods outlined in Fig. 3 and found that HIV-1 sequences from the pleural space were less divergent (P = 0.051) and more diverse (P = 0.005) than sequences from the blood, suggesting that pleural TB stimulates the production of HIV-1 from a latent-reservoir pool. Overall, HIV-1 sequences from the pleural space from five of the eight patients described by Collins et al. (8) appear to have this pattern. Those authors suggested that the increase in diversity they observed came from new rounds of viral replication and selection. However, a more parsimonious explanation of their observation is that pleural TB infection releases archived virus from the latent-reservoir pool, probably because of immune stimulation. This implies to us that, at least with respect to the pleural cavity, a latent-reservoir pool resides at this anatomical site that may be composed of macrophages (G. S. Laco, D. C. Nickle, S. J. Brodie, A. K. Ghosh, A. J. Melvin, K. Mohan, L. M. Frenkel, and J. I. Mullins, unpublished data). Additional experiments (i.e., cell sorting) are required to further examine this potential newly identified latent reservoir.
Reservoirs have been studied previously by observing viral DNA decay from specific cell types after the onset of effective therapy. We have shown through simulations that a reduction in divergence in the reservoir sample is a useful genetic assessment of a reservoir. Ruff et al. (32) used some of these concepts (i.e., lack of temporal structure) to detect the viral reservoir in pediatric AIDS patients that were on effective therapy. More importantly, their result, like the result we obtained with the data of Imamichi et al. (17), was largely driven by the lack of divergence in the reservoir sample. Our plans are to exploit this notion of depressed divergence in the reservoir sample and develop a statistical test for the identification of a reservoir. The evolutionary definitions proposed herein allow the detection of potential reservoirs in tissues or cell types in which viral dynamics cannot be accurately quantified (e.g., the pleural cavity).
FIG. A1.

Representative latent-cell age distribution.
FIG. A2.
V(t) and component logistic curves for representative parameters.
Acknowledgments
We thank Mark Fishbein, Geoffrey Gottlieb, Brett Riddle, Gary S. Laco, and Katherine Davis for helpful comments.
Support for this study was provided by a New Investigator Award from the National Institutes of Health-funded University of Washington Center for AIDS Research (CFAR). M.A.J. is supported by a University of Washington CFAR STD/AIDS training grant. Further support was provided by grants from the U.S. Public Health Service and the University of Washington CFAR to J.I.M.
APPENDIX
Age distribution of latently infected cells.
In order to perform the simulations described below, we required a biologically reasonable approximation of the age structure of the latently infected cell (or latent-cell) population at any given time. A probability distribution can be derived from the age structure, and random variates can be sampled from that distribution by using the rejection method (29).
Derivation of age distribution.
We assume that the dynamics of the latent-cell population are described by a simple first-order differential equation:
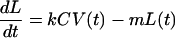 |
(1) |
where L(t) is the total number of latent cells per unit of blood volume at time t, V(t) is the number of virions per unit volume, C is the number of susceptible cells, k is the rate of latent infection upon encounter with a virion, and m is the exponential decay rate of the latent cell. As in reference 26, we assume that kC is constant over the period of infection we consider, i.e., that the number of susceptible cells has reached a steady state of production of cells on the one hand and destruction or infection of cells on the other. V(t) can be arbitrarily specified. Rather than establish further differential equations describing it, we chose the functional form of V to mimic the typical viral-load phenomenology. This is described below.
With an arbitrary V, the solution to equation 1 is simply
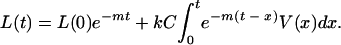 |
(2) |
Assume that L(0) = 0, i.e., that there are no latent cells at the onset of infection. Then the solution (equation 2) can be interpreted as follows: for any time t, the total number of latent cells is the sum of contributions kCV(t) over all times x from 0 to t, each discounted by exponential decay over the time since the contribution was made, e−(t − x ). That is, the number of latent cells between ages x and x + dx existing at time t is proportional to the integrand in equation 2. Since L(t) is that total, the distribution of latent-cell ages x at time t is
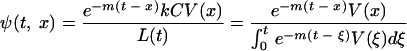 |
(3) |
and the proportion of latent cells with ages between x1 and x2 at time t is
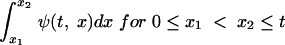 |
(4) |
Choice of viral-load function.
The plasma viral load in HIV-1 patient blood rapidly attains a peak between 105 and 107 virions/μl after infection and then quickly drops several logs, to a level (102 to 105 virions/μl) that remains relatively stable for up to several years (25, 33). This behavior can be approximated by subtracting two logistic curves with offset critical points. Let f(a, k, r, t) = (akert)/(1 + aert) and write V(t) = f(a1, k1, r1, t) − f(a2, k2, r2, t), where a1 > a2, k1 > k2, and r1 > r2. Figure A1 depicts V(t) and component logistic curves for representative parameters. Figure A2 depicts a representative latent-cell age distribution when V as described is employed in equation 3.
Implementation of rejection method.
To generate random deviates from the latent-cell distribution, we employed the rejection method as outlined in reference 29. Briefly, this is an algorithm that allows the transformation of uniform deviates into deviates drawn from an arbitrary distribution. It requires the construction of an envelope function f that is greater than or equal to the distribution function everywhere on its domain but also for which a closed-form antiderivative F and inverse antiderivative F−1 may be found. A random pair (x, y) can be drawn uniformly from under the envelope function by drawing first a value z uniformly from the interval (0, A), where A is the area under the envelope function, and letting x = F−1(z), then drawing y uniformly from the interval [0, f(x)]. If y ≤ ψ(t, x), we accept the deviate (x, y); otherwise, we reject it and repeat. The set of accepted x's are random deviates distributed as ψ and form a set of random times used in the simulations. We constructed f as follows. First, note that for any t, we can choose the smallest xɛ, large enough that
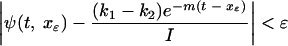 |
where I is the integral denominator in equation 3. Next, we can find xcrit numerically such that ψ(t, xcrit) = M, where M is the local maximum at the left of the distribution induced by the early viral-load peak (Fig. A2). Then the function
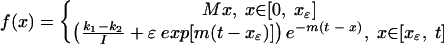 |
is everywhere greater than or equal to ψ(t, x) on [0, t] and is easily integrated and inverted in closed form. Routines to make the above-described calculations and implement the rejection method were written in Mathematica 4.0 (Wolfram Research, Champaign, Ill.).
REFERENCES
- 1.Beerli, P., and J. Felsenstein. 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 98:4563-4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beerli, P., N. C. Grassly, M. K. Kuhner, D. Nickle, O. Pybus, M. Rain, A. Rambaut, A. G. Rodrigo, and Y. Wang. 2000. Population genetics of HIV: parameter estimation using genealogy-based methods, p. 217-258. In A. G. Rodrigo and G. H. Learn (ed.), Computational and evolutionary analyses of HIV sequences. Kluwer Academic Publishers, Boston, Mass.
- 3.Blankson, J. N., D. Persaud, and R. F. Siliciano. 2002. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 53:557-593. [DOI] [PubMed] [Google Scholar]
- 4.Bourinbaiar, A. S. 1994. The ratio of defective HIV-1 particles to replication-competent infectious virions. Acta Virol. 38:59-61. [PubMed] [Google Scholar]
- 5.Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. S. Justement, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757-761. [DOI] [PubMed] [Google Scholar]
- 6.Chun, T. W., D. Engel, M. M. Berrey, T. Shea, L. Corey, and A. S. Fauci. 1998. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 95:8869-8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun, T. W., and A. S. Fauci. 1999. Latent reservoirs of HIV: obstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA 96:10958-10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins, K. R., M. E. Quinones-Mateu, Z. Toossi, and E. J. Arts. 2002. Impact of tuberculosis on HIV-1 replication, diversity, and disease progression. AIDS Rev. 4:165-176. [PubMed] [Google Scholar]
- 9.Coombs, R., C. Speck, J. Hughes, W. Lee, R. Sampoleo, S. Ross, J. Dragavon, G. Peterson, T. Hooton, A. Collier, L. Corey, L. Koutsky, and J. Krieger. 1998. Association between culturable human immunodeficiency virus type 1 (HIV-1) in semen and HIV RNA levels in semen and blood: evidence for compartmentalization of HIV-1 between semen and blood. J. Infect. Dis. 177:320-330. [DOI] [PubMed] [Google Scholar]
- 10.Delwart, E., J. Mullins, P. Gupta, G. Learn, M. Holodniy, D. Katzenstein, B. Walker, and M. Singh. 1998. Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 72:617-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drummond, A., and A. G. Rodrigo. 2000. Reconstructing genealogies of serial samples under the assumption of a molecular clock using serial-sample UPGMA. Mol. Biol. Evol. 17:1807-1815. [DOI] [PubMed] [Google Scholar]
- 12.Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package) version 3.5c. University of Washington, Seattle, Wash.
- 13.Finzi, D., J. Blankson, J. D. Siliciano, J. B. Margolick, K. Chadwick, T. Pierson, K. Smith, J. Lisziewicz, F. Lori, C. Flexner, T. C. Quinn, R. E. Chaisson, E. Rosenberg, B. Walker, S. Gange, J. Gallant, and R. F. Siliciano. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5:512-517. [DOI] [PubMed] [Google Scholar]
- 14.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gallant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295-1300. [DOI] [PubMed] [Google Scholar]
- 15.Hartl, D. L., and A. G. Clark. 1989. Principles of population genetics, 2nd ed. Sinauer Associates, Sunderland, Mass.
- 16.Hudson, R. R. 1990. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 7:1-14. [Google Scholar]
- 17.Imamichi, H., K. A. Crandall, V. Natarajan, M. K. Jiang, R. L. Dewar, S. Berg, A. Gaddam, M. Bosche, J. A. Metcalf, R. T. Davey, Jr., and H. C. Lane. 2001. Human immunodeficiency virus type 1 quasispecies that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasispecies present before initiation of therapy. J. Infect. Dis. 183:36-50. [DOI] [PubMed] [Google Scholar]
- 18.Jensen, M. A., B. Charlesworth, and M. Kreitman. 2002. Patterns of genetic variation at a chromosome 4 locus of Drosophila melanogaster and D. simulans. Genetics 160:493-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keys, B., J. Karis, B. Fadeel, A. Valentin, G. Norkrans, L. Hagberg, and F. Chiodi. 1993. V3 sequences of paired HIV-1 isolates from blood and cerebrospinal fluid cluster according to host and show variation related to the clinical stage of disease. Virology 196:475-483. [DOI] [PubMed] [Google Scholar]
- 20.Kiessling, A. A., L. M. Fitzgerald, D. Zhang, H. Chhay, D. Brettler, R. C. Eyre, J. Steinberg, K. McGowan, and R. A. Byrn. 1998. Human immunodeficiency virus in semen arises from a genetically distinct virus reservoir. AIDS Res. Hum. Retrovir. 14(Suppl. 1):S33-S41. [PubMed] [Google Scholar]
- 21.Korber, B. T., K. MacInnes, R. F. Smith, and G. Myers. 1994. Mutational trends in V3 loop protein sequences observed in different genetic lineages of human immunodeficiency virus type 1. J. Virol. 68:6730-6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kusumi, K., B. Conway, S. Cunningham, A. Berson, C. Evans, A. K. N. Iversen, D. Colvin, M. V. Gallo, S. Coutre, E. G. Shpaer, D. V. Faulkner, A. DeRonde, S. Volkman, C. Williams, M. S. Hirsch, and J. I. Mullins. 1992. HIV-1 envelope gene structure and diversity in vivo and following cocultivation in vitro. J. Virol. 66:875-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leigh Brown, A. J. 1997. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad. Sci. USA 94:1862-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maddison, W. P., and D. R. Maddison. 2001. MacClade—analysis of phylogeny and character evolution—version 4. Sinauer Associates, Inc., Sunderland, Mass.
- 25.Mellors, J., L. Kingsley, C. Rinaldo, J. Todd, B. Hoo, R. Kokka, and P. Gupta. 1995. Quantitation of HIV-1 RNA in plasma predicts outcome after seroconversion. Ann. Intern. Med. 122:573-579. [DOI] [PubMed] [Google Scholar]
- 26.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582-1586. [DOI] [PubMed] [Google Scholar]
- 27.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1 infected compartments during therapy. Nature 387:189-191. [DOI] [PubMed] [Google Scholar]
- 28.Poss, M., A. G. Rodrigo, J. J. Gosink, G. H. Learn, D. de Vange Panteleeff, H. L. Martin, Jr., J. Bwayo, J. K. Kreiss, and J. Overbaugh. 1998. Evolution of envelope sequences from the genital tract and peripheral blood of women infected with clade A human immunodeficiency virus type 1. J. Virol. 72:8240-8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Press, W. H., and Numerical Recipes Software. 1994. Numerical recipes in C, version 2.04, 2nd ed. Cambridge University Press, New York, N.Y.
- 30.Ramratnam, B., J. E. Mittler, L. Zhang, D. Boden, A. Hurley, F. Fang, C. A. Macken, A. S. Perelson, M. Markowitz, and D. D. Ho. 2000. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 6:82-85. [DOI] [PubMed] [Google Scholar]
- 31.Rodrigo, A. G., E. G. Shpaer, E. L. Delwart, A. K. N. Iversen, M. V. Gallo, J. Brojatsch, M. S. Hirsch, B. D. Walker, and J. I. Mullins. 1999. Coalescent estimates of HIV-1 generation time in vivo. Proc. Natl. Acad. Sci. USA 96:2187-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruff, C. T., S. C. Ray, P. Kwon, R. Zinn, A. Pendleton, N. Hutton, R. Ashworth, S. Gange, T. C. Quinn, R. F. Siliciano, and D. Persaud. 2002. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J. Virol. 76:9481-9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schacker, T. W., J. P. Hughes, T. Shea, R. W. Coombs, and L. Corey. 1998. Biological and virologic characteristics of primary HIV infection. Ann. Intern. Med. 128:613-620. [DOI] [PubMed] [Google Scholar]
- 34.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siliciano, R. 2000. Can antiretroviral therapy ever be stopped? AIDS Read. 10:224-229. [PubMed] [Google Scholar]
- 36.Slatkin, M., and W. P. Maddison. 1989. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123:603-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slatkin, M., and W. P. Maddison. 1990. Detecting isolation by distance using phylogenies of genes. Genetics 126:249-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swofford, D. L. 1999. PAUP 4.0: Phylogenetic analysis using parsimony (and other methods), version 4.0, 2nd ed. Sinauer Associates, Inc., Sunderland, Mass.
- 39.Wares, J. P., S. D. Gaines, and C. W. Cunningham. 2001. A comparative study of asymmetric migration events across a marine biogeographic boundary. Evol. Int. J. Org. Evol. 55:295-306. [DOI] [PubMed] [Google Scholar]
- 40.Zhang, L., C. Chung, B. S. Hu, T. He, Y. Guo, A. J. Kim, E. Skulsky, X. Jin, A. Hurley, B. Ramratnam, M. Markowitz, and D. D. Ho. 2000. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J. Clin. Investig. 106:839-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A. Talal, P. Racz, A. S. Perelson, B. T. Korber, M. Markowitz, and D. D. Ho. 1999. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 340:1605-1613. [DOI] [PubMed] [Google Scholar]