

Abstract
Human rhinovirus type 2 (HRV2) is internalized by members of the low-density lipoprotein (LDL) receptor (LDLR) family. It then progresses into late endosomes, where it undergoes conversion from D- to C-antigenicity at pH < 5.6. Upon uncoating, the viral RNA is transferred into the cytoplasm across the endsosomal membrane. However, C-antigenic particles fail to attach to LDLR; this raised the question of whether the virus remains attached to the receptors and is carried to late compartments or rather falls off at the higher pH in early endosomes. We therefore determined the pH dependence of virus-receptor dissociation and virus conversion to C-antigen under conditions preventing endocytosis. 35S-HRV2 was attached to HeLa cells at 4°C and incubated in buffers of pH 7.4 to 5.0; levels of native virus and C-antigenic particles remaining cell associated or having been released into the medium were determined by immunoprecipitation. At pH 6.0, HRV2 was readily released from plasma membrane receptors in its native form, whereas at pH ≤ 5.4, it was entirely converted to C-antigen, which, however, only dissociated from the surface upon prolonged incubation. The antigenic conversion occurred at the same pH regardless of whether HRV2 was free in solution or bound to its receptors. These data suggest that, in vivo, the virus is no longer bound to its receptors when the antigenic conversion and uncoating occur in more acidic late endosomes. When virus was bound to HeLa cells at 4°C, converted into C-antigen by exposure to pH 5.3, and subsequently warmed to 34°C in the presence of bafilomycin (to prevent endosomal uncoating), viral de novo synthesis was detected. This study demonstrates for the first time that a nonenveloped virus such as HRV2 can infect from the plasma membrane when artificially exposed to low pH. This implies that the viral RNA can gain access to the cytoplasm from the plasma membrane.
Picornaviruses (small RNA viruses) comprise a number of important human and animal pathogens, such as poliovirus, hepatitis A virus, foot-and-mouth disease virus, and human rhinovirus (HRV), the main causative agent of the common cold. A total of 101 HRV serotypes are currently known. These can be divided into a major group and a minor group, based on the receptor used for cell entry and infection. Ninety-one serotypes attach to intercellular adhesion molecule 1 (ICAM-1), and 10 serotypes attach to members of the low-density lipoprotein (LDL) receptor (LDLR) superfamily (15, 37). Based on recent sequence comparisons, HRV87, originally considered a rhinovirus, has been reclassified as an enterovirus (34).
Picornaviruses are nonenveloped. An icosahedral protein coat built from 60 copies each of four capsid proteins, VP1 to VP4, encases the RNA genome, which is a single-stranded molecule of positive polarity about 7,100 nucleotides in length (for review, see reference 32). As demonstrated for HRV2, a minor group virus, upon infection, the native capsid is changed from D-antigenicity to C-antigenicity (22). Whereas native virions sediment at 150S and are D-antigenic, loss of VP4 results in subviral A particles sedimenting at 135S, and subsequent uncoating of the RNA genome results in B particles sedimenting at 80S. These are both C-antigenic. A C-antigen-specific epitope of HRV2 exclusively present on A and B particles is recognized by monoclonal antibody (MAb) 2G2 (27). Both kinds of subviral particles can also be generated in vitro upon incubation at >50°C; A particles are preferentially obtained by incubation in low pH buffer (19, 21, 22). C-antigenic particles fail to attach to cells and are therefore not infectious (28).
Infection is initiated upon interaction of host cell receptors with the viral binding site. Whereas ICAM-1 binds to major group viruses within the canyon, a cleft encircling the fivefold axes of icosahedral symmetry (18, 29), LDLRs bind to minor group viruses at the BC and HI loops of VP1, which are part of the star-shaped dome at the fivefold axis (14). Therefore, residues originating from VP1, VP2, and VP3 are involved in receptor interaction in major group HRVs, whereas residues of VP1 only are involved in receptor binding in minor group HRVs (18, 29).
Binding to the specific receptor is followed by internalization. Cell entry leads to exposure of the virus to a low-pH environment in endosomes. For HRV2, this has been shown to trigger the conformational changes indicated above. For major group HRVs, the low pH appears to be dispensable with the receptor catalyzing the uncoating process. As seen from cryoelectron microscopy image reconstruction, the amino terminus of VP1 is externalized in B particles generated in vitro (12). Assuming that the mechanism of membrane interaction is similar to that observed for poliovirus (3), this hydrophobic region might become inserted into the membrane; furthermore, this process is believed to take part in the opening of a pore (in the case of HRV2) or in disruption of the membrane (in the case of HRV14) (31, 36). Finally, the viral RNA is released into the cytoplasm: in the case of HRV2, from late endosomes, with the empty B particles becoming degraded in lysosomes (2, 30), in the case of HRV14, from the particles themselves, which have gained access to the cytosol upon disruption of the endosomal membrane (36).
For some major group viruses, interaction in vitro with a soluble recombinant fragment of ICAM-1 at physiologic temperatures has been shown to result in uncoating without the necessity for the virus to become exposed to low pH (9, 16). In contrast, interaction of the minor group virus HRV2 with recombinant soluble LDLR fragments is reversible, and infectivity can be restored from the noninfectious viral aggregates upon dissociation of the receptors (15). This strongly suggests that the receptors are not directly involved in minor group HRV uncoating.
The LDLR (molecular mass, ∼115 kDa), the very-low-density lipoprotein receptor (VLDLR; molecular mass, ∼95 kDa), and, particularly, LDLR-related protein (LRP; ∼600 kDa) are molecules of considerable length (more than 12 nm) (17), making it highly unlikely that the virus remains attached to the receptor during the supposed insertion of the hydrophobic N terminus of VP1 into the endosomal membrane. We therefore wanted to investigate whether the minor group receptors facilitate the conformational modification leading to the uncoating of HRV2, or whether the virus rather dissociates from its receptors under conditions prevailing in early endosomes prior to initiating RNA transfer into the cytosol from late compartments. Given that the conformational modification of the virus can also be induced by low-pH buffer at 4°C, we further determined whether productive uncoating can take place at the plasma membrane.
MATERIALS AND METHODS
Chemicals.
All chemicals were obtained from Sigma (Sigma, St. Louis, Mo.) unless specified otherwise. Bafilomycin A1 (Alexis Corp., Lausen, Switzerland) was dissolved in dimethyl sulfoxide (DMSO) at 20 mM and stored at −20°C.
Buffer solutions and media.
Cells were grown in minimal essential medium (MEM) tissue culture medium supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine, 100 U of penicillin G sodium per ml, and 100 μg of streptomycin sulfate per ml (all from Gibco Invitrogen Corp., Paisley, United Kingdom). Infection was carried out in infection medium (MEM culture medium containing 30 mM MgCl2 and 2% FCS). Isotonic buffers were prepared by adjusting a mixture of 30 mM sodium acetate buffer (pH 5.0), 10 mM sodium phosphate buffer (pH 5.0), 110 mM NaCl, and 5 mM KCl with 0.1 M NaOH to pH values of between 7.4 and 5.4. PBS++ is phosphate-buffered saline (PBS) containing 1 mM CaCl2 and 1 mM MgCl2. Radioimmunoprecipitation assay (RIPA) buffer is 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl, 1 mM EDTA, 1% Na-deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1% Triton X-100.
Cell culture and viruses.
HeLa H1 cells (American Type Culture Collection, Manassas, Va.) were used throughout. HRV2, originally obtained from the American Type Culture Collection, was propagated and labeled with [35S]cysteine/methionine (American Radiolabeled Chemicals, Inc., St. Louis, Mo.) as described previously (27).
HRV2 conformational modification in solution.
For each sample, approximately 1.5 × 104 cpm of 35S-labeled HRV2 was added to 500 μl of isotonic buffers adjusted to pH values of 7.4 to 5.0 (described above) and incubated for 20 min at 4 or 34°C. Solutions were then neutralized with 10 mM Tris-base and processed for sequential immunoprecipitation to detect C-antigenic particles and native virus, respectively, as described below.
Virus binding, conformational modification, and immunoprecipitation.
HeLa cells were grown to 80% confluence in 75-cm2 tissue culture flasks, detached by treatment with PBS containing 10 mM EDTA at 4°C, and washed. Cells (106 cells per sample) were resuspended in 500 μl of MEM containing 30 mM MgCl2, approximately 2 × 104 cpm of 35S-labeled HRV2 was added, and the mixture was incubated for 1 h at 4°C. The cells were washed twice with ice-cold PBS++ and incubated in 500-μl isotonic buffers adjusted to the pH values indicated for 20 to 60 min at 4°C. Cells were pelleted, and the supernatants were removed and neutralized with 10 mM Tris base. The cell pellets were lysed in 500 μl of RIPA buffer for 10 min on ice, and cell debris was removed by centrifugation. The lysed cell pellets and supernatant media (described above) were processed separately for sequential immunoprecipitation: C-antigenic particles were recovered by incubation with MAb 2G2 immunocomplexes. D-antigenic particles remaining in the supernatant were then precipitated with rabbit antiserum against HRV2 bound to Staphylococcus aureus cells. Pellets were washed twice with RIPA buffer and twice with PBS and suspended in 10 volumes of emulsifier-safe scintillation cocktail (Packard-Perkin-Elmer Life Sciences Inc., Boston, Mass.) for counting. Immunocomplexes between S. aureus and MAb 2G2 or rabbit anti-HRV2 antiserum were made as described elsewhere (27).
Dissociation of native HRV2 from the plasma membrane.
HeLa cells were grown and detached from culture flasks as described above. 35S-labeled HRV2 (2 × 104 cpm) was bound to 106 cells in 500 μl of MEM containing 30 mM MgCl2 for 1 h at 4°C. Unbound virus was removed by washing with ice-cold PBS++, and cells were incubated for 20 min at 4°C in 500 μl of isotonic buffers adjusted to pH 7.4, 6.6, 6.0, and 5.9, respectively. Cells were pelleted and lysed, and C-antigenic (as a control so that no conformational change occurred) and native viruses were recovered by sequential immunoprecipitation as described above.
Fluorescence microscopy.
HeLa cells were plated at low density on eight-well Lab-Tek glass chamber slides (Nalge Nunc International, Rochester, N.Y.), grown overnight until half-confluent, and preincubated in serum-free MEM for 30 min at 37°C in the absence or presence of 20 nM bafilomycin. They were then challenged with HRV2 (multiplicity of infection [MOI] of 10) in the absence or presence of the drug for the times indicated, cooled to 4°C, washed with ice-cold PBS++, fixed with 4% paraformaldehyde (PFA) in PBS at room temperature for 30 min, and quenched with 50 mM NH4Cl in PBS. HRV2 was detected with MAb 8F5 (13) followed by Alexa 488-conjugated goat anti-mouse immunoglobulin G (IgG) (Molecular Probes, Inc., Eugene, Oreg.). Cells were mounted in Immuno Floure mounting medium (ICN Pharmaceuticals, Inc., Costa Mesa, Calif.) and viewed with a Zeiss Axioplan 2 (Carl Zeiss, Jena, Germany).
To monitor virus-specific protein synthesis, cells were grown in chamber slides as described above. Virus at an MOI of 1 was preincubated with buffer solutions of different pHs for 20 min at 4°C and incubated with the cells in the same buffers on ice for 1 h. The cells were washed extensively with PBS++ and incubated in infection medium for 1 h. Fresh medium was added, and the incubation was continued at 34°C for another 16 h. Cells were washed, fixed with 4%PFA in PBS, and quenched with 50 mM NH4Cl in PBS. Virus-specific proteins were detected by indirect immunofluorescence microscopy.
Dissociation of infectious HRV2 from the plasma membrane.
HeLa cells grown to 80% confluency in 24-well plates were incubated with HRV2 at an MOI of 10 in 400 μl of infection medium for 1 h at 4°C. Unbound virus was removed, and cells were treated for 1 h at 4°C with isotonic buffers of pHs 6.0 and 7.4. The cells were then washed twice for 5 min at 4°C with MEM (pH 7.4), and cell-associated infectious virus was determined as 50% tissue culture infective dose (TCID50) as described below.
Infection from the plasma membrane.
Cells were grown to 80% confluency in 24-well plates overnight and preincubated without or with 40 nM bafilomycin in 200 μl of infection medium. Two hundred microliters of infection medium containing HRV2 to obtain an MOI of 1 or 10 was added, and virus was allowed to bind to the cells for 1 h at 4°C. The cells were washed, treated for 1 h at 4°C with isotonic buffers of pH 7.4 (with or without 20 nM bafilomycin) and 5.3 (with 20 nM bafilomycin) as indicated, washed, and incubated in fresh medium without or with 20 nM bafilomycin for 0 or 17 h at 34°C. The plates where then subjected to three freeze/thaw cycles and sonicated, cell debris was removed by low-speed centrifugation, and the TCID50 of the virus in the supernatants was determined (4).
RESULTS
HeLa cells express several members of the LDLR family, of which at least LDLR, LRP, and VLDLR are being used as attachment proteins for HRV2 (15, 24). The viral entry pathway and infection mechanism have been extensively characterized in these cells (2, 30). In order to study whether the antigenic conversion of HRV2 at low pH is facilitated when the virus is attached to its cellular receptors, internalization has to be avoided. This can be achieved when the cells are kept at 4°C.
pH-dependent conformational changes of HRV2 are similar at 4 and 34°C.
We first determined whether lowering the temperature from 34°C to 4°C would influence the pH-dependent antigenic conversion of the virus when free in solution. 35S-HRV2 was incubated in isotonic acetate-phosphate buffers of pHs 7.4 to 5.0 for 20 min at 4 and 34°C. Then the fraction of C-antigenic particles generated was determined by immunoprecipitation with MAb 2G2 followed by HRV2 antiserum to recover residual native virus. As shown in Fig. 1A, the levels of pH dependence of the antigenic conversion of native HRV2 are very similar at 4 and 34°C, and the percentages of C-antigenic particles generated at both temperatures are not significantly different. In agreement with earlier data (10), <100% of input virus was recovered with MAb 2G2 at the lowest pH; this is not due to incomplete conversion, since determination of infectious virus revealed the absence of any infectious particles (see also below). We also measured the inactivation of viral infectivity as TCID50 at pH 5.3 at 4 and 34°C and at 20 and 60 min with essentially the same results (data not shown). From this, it is clear that decreasing the temperature from 34°C to 4°C has no gross effect on the low-pH-dependent conformational modification of HRV2 in solution. Therefore, it is legitimate to carry out the experiments described below at 4°C.
FIG. 1.

Influence of temperature and receptor binding on the pH-dependent conformational change of HRV2 in solution. (A) 35S-labeled HRV2 (1.5 × 104 cpm) was incubated in 500 μl of isotonic buffers at the pHs indicated for 20 min at 4 or 34°C. The percentage of C-antigenic particles formed was then determined by sequential immunoprecipitation with MAb 2G2 and rabbit antiserum against HRV2 and scintillation counting. The data are expressed as the percentage of total 35S-labeled HRV2. The data represent the mean ± standard deviation for three independent experiments. (B) 35S-labeled HRV2 (2 × 104 cpm) was bound to 106 HeLa cells at 4°C for 60 min, and nonbound virus was washed away with cold PBS. Cell-bound virus or free virus (1.5 × 104 cpm) was then incubated at 4°C for 20 min in isotonic buffers at the pH indicated. The percentage of C-antigen (cell associated plus supernatant in the case of cell-bound virus [for details, see Materials and Methods]) was determined as described for panel A. The data represent the mean ± standard deviation for 11 independent experiments carried out in triplicate.
Receptor binding does not promote C-antigen formation.
To study whether low pH and receptor cooperate in the conformational change, 35S-labeled virus was bound to the cells for 1 h at 4°C, nonattached virus was washed away, and the cells were incubated in buffers of various pHs at 4°C for 20 min. Cells were then lysed with detergent, and the percentage of C-antigenic particles was determined as described above (Fig. 1B). For comparison, 35S-labeled HRV2 (in the absence of cells) was incubated under the same conditions, and the amount of C-antigen formed was analyzed by immunoprecipitation. The pH-dependent formation of C-antigen was independent of whether the virus was cell bound or free in solution (i.e., in the absence of receptors). At a pH of >6.0, the virus remained native, with some conversion starting at pH 5.8. The percentage of conformationally altered particles increased to 50% at pH 5.6 with maximal conversion (80%) at a pH of ≤5.4 (Fig. 1B). The conformational change is thus not enhanced when the virus is bound to the cell surface. These results suggest that the receptors do not take part in the conformational change of HRV2.
Kinetics of the dissociation of C-antigenic particles from the plasma membrane.
C-antigen does not bind to the viral receptors (28). Therefore, we wondered whether virus particles bound to plasma membrane receptors and exposed to a pH sufficiently low to change their conformation would dissociate or rather remain bound to the membrane via hydrophobic domains becoming exposed as a consequence of the structural modification. 35S-labeled HRV2 was bound to HeLa cells at pH 7.4 for 60 min at 4°C and subsequently incubated in pH 5.0 or 7.4 buffer. At the times indicated, C-antigenic particles remaining cell associated or having been released into the supernatant were determined (Fig. 2). In agreement with the data shown in Fig. 1B, at 20 min, 80% of the bound virus was recovered as C-antigen, corresponding to 100% conversion (complete loss of infectivity [see Fig. 1A and Fig. 6]). The subviral particles were gradually released from the plasma membrane until all could be precipitated as C-antigen from the supernatant at 60 min. (Note that the procedure results in recovery of only 80%.) For control purposes, we determined the time-dependent dissociation of native virus at pH 7.4; at this pH, virus bound to the cells was not released to any significant extent within 60 min (data not shown). These data suggest that low-pH treatment of membrane-bound virus leads to formation of C-antigenic particles that subsequently fall off their receptors. However, newly exposed hydrophobic domains may transiently interact with the plasma membrane.
FIG. 2.

Kinetics of dissociation of C-antigenic HRV2 from HeLa cells. 35S-labeled HRV2 (2 × 104 cpm) was bound to 106 HeLa cells at 4°C for 60 min, nonbound virus was washed away with cold PBS, and the cells were incubated in 500 μl of isotonic buffer at pH 5.0 and 7.4 (control [data not shown]) at 4°C. At the times indicated, the supernatant was removed, and the cells were lysed. C-antigen present in the supernatant and in the cell lysate was determined as described for Fig. 1B. Data represent the mean ± standard deviation for three independent experiments, presented as a percentage of total virus bound to the cells at time 0. Note that only 80% of total C-antigen can be immunoprecipitated, corresponding to 100% conversion (Fig. 1A).
FIG. 6.
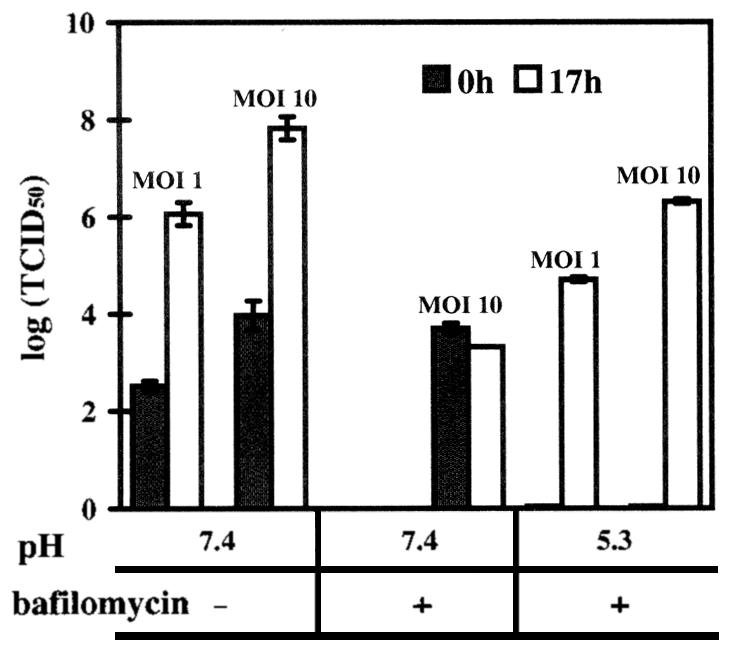
HRV2 infection can occur from the plasma membrane at low pH. Cells grown in 24-well plates were preincubated without and with bafilomycin (40 nM) for 30 min, challenged with HRV2 at an MOI of 1 or 10 at 4°C, and exposed to buffers at pH 7.4 (control) or 5.3. After further incubation in infection medium for 17 h, the viral titer was compared to that at time 0. Where indicated, bafilomycin was maintained at a concentration of 20 nM. The data represent the mean ± standard deviation for three independent experiments.
HRV2 is released from its receptor at mildly acidic pH.
In vivo the virus gradually encounters a lower pH when transported from early to late compartments. It is well established that LDL dissociates from its receptor at the mildly low pH of early endosomes, allowing for recycling of the LDLRs to the plasma membrane (5). We therefore wondered whether HRV2 can be released prior to undergoing the conformational modification.
35S-HRV2 was bound to HeLa cells at 4°C for 60 min and then incubated with isotonic buffers with pHs between 7.4 and 5.9 for 20 min at 4°C. Under these conditions, virus free in solution is not converted to C-antigen to any significant extent (10). The amounts of native and C-antigenic particles present in the cell pellet and in the supernatant were then determined by immunoprecipitation. A significant amount of virus was released in its native conformation by decreasing the pH of the incubation buffer from 6.6 to 5.9; this resulted in about 50% dissociation at the lowest pH of 5.9 (Fig. 3A). This pH value is in good accordance with the pH prevailing in early endocytic compartments in HeLa cells D. Schober, N. Bayer, R. F. Murphy, D. Blaas, and R. Fuchs, (abstract from the 38th American Society for Cell Biology Annual Meeting, 1998, Mol. Biol. Cell 9:466a, 1998). This finding suggests that the virus dissociates from its receptors in early endosomes and is further transported to later compartments in the absence of the receptors.
FIG. 3.

Dissociation of native HRV2 from the cell surface. (A) 35S-HRV2 (2 × 104 cpm) was bound to 106 HeLa cells for 1 h at 4°C, unbound virus was removed, and cells were incubated in isotonic buffers at different pHs for 20 min at 4°C. Native virus in the cell pellet or in the supernatant was determined by immunoprecipitation. The average of three independent experiments carried out in triplicate ± standard deviation is shown. (B) HeLa cells grown in 24-well plates were preincubated in infection medium for 30 min at 34°C. HRV2 at an MOI of 10 was added and allowed to attach for 1 h on ice. Cells were washed and exposed to isotonic buffers at pHs 6.0 and 7.4 (control, 100%) for 1 h on ice; cell-associated infectious virus was then determined as TCID50. The data represent the mean ± standard deviation for three independent experiments.
The influence of the pH on the dissociation of cell-bound HRV2 was also studied by using a biological assay. HeLa cells grown in 24-well plates were cooled to 4°C and challenged with HRV2 at an MOI of 10. After 1 h of incubation, unbound particles were removed, and cells were exposed to isotonic buffer of pH 6.0 or 7.4 at 4°C. Virus released into the supernatant was removed, the cells were lysed, and the titer of cell-associated, infectious virus was determined. As shown in Fig. 3B, native virus present in the cell pellet was reduced to 57% after incubation in pH 6.0 buffer compared to that of the control (incubation at pH 7.4). These data suggest that the affinity of the receptor for the virus is decreased below a pH of 6.0. This is corroborated by data from indirect immunofluorescence when virus binding was carried out at pH 6.0 (Fig. 4). Virus was preincubated in buffer of pH 6.0 or 7.4 for 20 min at 4°C and then added to HeLa cells at an MOI of 1 and at the same temperature for 60 min. Unbound virus was removed, cells were incubated in infection medium for 17 h, and de novo-synthesized virus was determined by indirect immunofluorescence microscopy. The cells were counted after staining the cell nuclei with Hoechst dye. The number of cells engaged in viral synthesis was found to be reduced to 20% of the control (incubation at pH 7.4). Taken together, a pH of 1.4 pH units below the pH of the medium leads to a reduction in the affinity between virus and receptor. For these reasons, the virus most probably detaches from the receptors during passage through early endosomes, where it becomes part of the fluid phase and is further transported to late compartments, where it finally undergoes conformational modifications in the low-pH environment that promote membrane interaction.
FIG. 4.

Infection at pH 6 is less efficient. HRV2 was preincubated for 20 min and subsequently bound to HeLa cells (MOI of 1) at pH 7.4 (control) and at 6.0 for 1 h at 4°C. Cells were washed, unbound virus was removed, and cells were further incubated in infection medium at 34°C for 17 h. (A) Cells were fixed, and HRV2 was stained with MAb 8F5 followed by antimouse Alexa 488-conjugated IgG. Nuclei were revealed with Hoechst stain. Bars, 100 μm. (B) The percentage of cells synthesizing viral antigen was determined in two different experiments with a total of 1,500 cells counted.
HRV2 can infect from the plasma membrane when exposed to low pH.
Low-pH-induced fusion of the enveloped Semliki Forest virus (SFV) with the plasma membrane can lead to infection, although the usual in vivo entry pathway involves endocytosis and fusion in acidic endosomes (11, 38). Similarly, under physiologic conditions, HRV2 internalization is required for infection; the subsequent structural modification leads to insertion of hydrophobic sequences into the membrane. Therefore, we wondered whether the latter process, when induced artificially at the plasma membrane, could result in infection. For this experiment, we chose to use bafilomycin to prevent endosomal uncoating by increasing the vesicular pH above the threshold for HRV2 uncoating (2). The influence of this low concentration of bafilomycin on HRV2 infection was first examined by indirect immunofluorescence microscopy. HeLa cells were preincubated with or without 20 nM bafilomycin, cells were cooled to 4°C, and HRV2 at an MOI of 10 was bound for 1 h. Unbound virus was removed, and cells were warmed in infection medium (± bafilomycin) to 34°C and incubated for 17 h. Viral antigen was then visualized by indirect immunofluorescence microscopy, and nuclei were stained with Hoechst dye (Fig. 5). As demonstrated for other picornaviruses (33, 35), the control incubation displayed viral protein staining with an endoplasmic reticulum-like pattern (Fig. 5, lower panels). In accordance with earlier results, no viral de novo synthesis was seen in the presence of bafilomycin; however, endosomal staining of the input virus was clearly visible even after 17 h (Fig. 5, lower panel).
FIG. 5.

HRV2 synthesis is blocked by 20 nM bafilomycin: Cells were preincubated without (control) and with 20 nM bafilomycin for 30 min, followed by incubation with an MOI of HRV2 of 10 at 4°C for 1 h (± bafilomycin). Cells were washed and further incubated for 17 h ± bafilomycin at 34°C. Cells were fixed, viral antigen was stained with MAb 8F5 followed by antimouse IgG conjugated to Alexa 488, and nuclei were stained with Hoechst dye. The upper panels show an overlay of nuclear staining and staining for viral antigen (bars, 200 μm). The lower panels show higher magnification of representative single cells with Alexa 488 staining only (bars, 10 μm).
These conditions were then used to study whether exposure of plasma membrane-bound virus would lead to infection. HeLa cells were preincubated with or without bafilomycin for 30 min at 34°C. Cells were cooled to 4°C, and HRV2 at an MOI of 10 or 1 was added and allowed to bind to the plasma membrane for 1 h on ice. Cells were washed and treated with buffers of pH 5.3 or 7.4 ± 20 nM bafilomycin at 4°C. After washing, infection medium ± bafilomycin was added, and cells were either analyzed immediately or further incubated at 34°C for 17 h. As shown in Fig. 6, at pH 7.4, in the absence of the drug, infection led to an increase of the TCID50 at 17 h postinfection of 4 logs as compared to that of input virus (time 0). As expected, bafilomycin completely blocked the infection; interestingly, the titer of the input virus remained largely constant over the period of 17 h, indicating that the virus was neither uncoated nor degraded. Conversely, when cell-bound HRV2 was exposed to pH 5.3, infectivity was no longer detectable. On the other hand, when cells were subsequently warmed to 34°C in the presence of bafilomycin, viral replication occurred, as indicated by a titer only 1.5 log less than that in the control incubation (pH 7.4 in the absence of bafilomycin). Considering that at time 0 no infectious virus was detected, this corresponds to viral de novo synthesis levels of 4 and 5 logs at MOIs of 1 and 10, respectively. When low-pH-treated virus (1 h at 4°C, pH 5.3 buffer) was incubated with the cells at an MOI of 10 or 1 for 1 h in the presence of bafilomycin (in pH 7.4 medium), no de novo viral synthesis was observed (TCID50 values not shown). Therefore, the conformational modification has to take place when the virus is still attached to the plasma membrane to allow for RNA translocation. This probably occurs concomitantly with the conformational change. This demonstrates that HRV2 uncoating and RNA transfer into the cytosol can be artificially induced at the plasma membrane by exposure to pH 5.3.
DISCUSSION
In vivo uncoating of HRV2 is accompanied by a structural change from D-antigen to C-antigen, which takes place in late endosomes in HeLa cells (30). The virus remains bound to its receptors when it starts to traverse the endocytic compartments at decreasing pH. For vectorial RNA release, the capsid must come close to the lipid bilayer: it is believed that the hydrophobic N terminus of VP1, which becomes externalized upon the transition of native virus to subviral particles, inserts into the membrane and aids in opening a pore for transit of the viral RNA to the cytosol (12, 31). However, due to the dimension of the rod-like receptors (17), the bound capsid might be held at a distance that prevents it from contacting the lipid bilayer. Furthermore, most of the natural ligands of the LDLR family dissociate in the early endosome at a pH of about 5.9 to 6.0 and are further shuttled to lysosomes, whereas the receptors are recycled to the plasma membrane (26). Therefore, the question arises of whether HRV2 stays with any of the receptors (LDLR, VLDLR, and LRP) or is released in early compartments and subsequently progresses as part of the fluid phase to later compartments.
We first showed that low-pH-induced structural changes occur to a similar extent at 34 and 4°C (Fig. 1A). This allowed us to investigate, with plasma membrane-bound virus but in the absence of internalization, whether the low pH and any of the receptors acted synergistically in catalyzing the structural changes. Thus, when mimicking physiological conditions, we found no evidence for this (Fig. 1B). We then asked whether HRV2 behaved similarly to natural ligands in dissociating from the receptors at the pH prevailing in early endosomes. Indeed we found that bound native virus was readily released when the pH was decreased to 6.0 (Fig. 3A) and that binding of the virus to the plasma membrane was substantially reduced at this pH (Fig. 3B). Deletion of the epidermal growth factor (EGF) precursor region and of the YWTD β-propeller domain (ΔEGF) in LDLR (7) and VLDLR (25) suggested that these domains were responsible for the acid-dependent dissociation of natural ligands from the receptors. However, ligand binding and receptor trafficking were differentially affected. ΔEGF LDLR was unable to bind LDL, but it bound and internalized β-VLDL. In this case, the ligand was not released even below a pH of ≤4.5, and the ligand-receptor complex was transported to and degraded in lysosomes. On the other hand, deletion of these domains in the VLDLR resulted in recycling of receptor-associated protein (RAP) together with the truncated receptor. Although RAP binding was unaffected, low-pH-dependent dissociation in endosomes did not occur. It is assumed that conformational changes in the EGF precursor region and YWTD β-propeller domain of the receptors within the acidic endosomal environment are responsible for ligand dissociation. Because surface-bound β-VLDL is not released from the plasma membrane when bound to wild-type LDLR to any significant extent at pH 6.0 (7), presumably acidic pH and low-calcium ions act synergistically to allow ligand dissociation in early, sorting endosomes (8). It will be interesting to determine how deletions of the EGF precursor region and YWTD β-propeller domain in the LDLR and VLDR affect endocytic trafficking of the receptor in the presence of the ligand HRV2 and whether they have any influence on uncoating and infection. In any case, our data strongly suggest that, in vivo, similar to other ligands, the virus progresses to late endocytic compartments with the bulk of the fluid phase but not in association with its receptors.
In this context, it is of note that the affinity of the major group virus HRV3 for its receptor ICAM-1 also decreases by 50-fold upon lowering the pH from 8 to 6 as shown by Biacore measurements. The virus was stable under these conditions, which were used to regenerate the surface (6). This suggests that the dissociation is not due to a reversible modification of the virion and might be taken to indicate that HRV3 and possibly other major group HRVs also detach from their receptors in early endosomes. However, major group HRVs are strongly dependent on ICAM-1 to induce the conformational change. Thus, virus modification has to precede receptor dissociation. In agreement with this hypothesis, uncoating and endosomal penetration of the major group virus HRV14 take place in early endosomes (1, 36).
When membrane-bound HRV2 was exposed to pH 5.3 for 20 min at 4°C, it was entirely converted to C-antigen (Fig. 1). However, the release of these subviral particles into the supernatant was retarded (Fig. 2). Because neither A nor B particles exhibit affinity for the receptor, this raises the question of how they remain attached for these extended time periods. Only A particles are hydrophobic and bind to liposomes (20). It is therefore conceivable that primarily A particles are formed under these conditions, which may subsequently be partially converted to B particles. This prompted us to investigate whether exposure of the virus to low pH (5.3) at the plasma membrane would result in infection. This was indeed the case (Fig. 6). Of note is the complete absence of residual infectious virus after low-pH incubation at time 0 and 4°C. Since bafilomycin was also present when the cells were warmed to 34°C to allow new virus synthesis, infection by native virus through the endocytic route can be excluded. By the same token, it is highly unlikely that subviral A particles became internalized and released their RNA, since the great majority of viral protein was released into the medium upon exposure to low pH after 60 min (see Fig. 2 and 6). This indicates that infection can occur from the plasma membrane, provided that the virus is exposed to low pH. So far, this has not been demonstrated for a nonenveloped virus that requires low endosomal pH for infectivity. Madhus and colleagues carried out similar experiments earlier, but failed to detect productive uncoating (23). However, they used inhibition of cellular protein synthesis after 36 h as a measure of viral infection. Since no clear experimental details are otherwise given, the difference in technique might have resulted in a lack of detection of HRV2 infection.
As depicted in Fig. 7, we propose the following mechanism for HRV2 uncoating at the plasma membrane, taking into account that subviral A but not B particles are hydrophobic. The function of the receptors is to concentrate and hold the virus in a position close to the membrane. When receptor-bound virus is exposed to a pH of ≤5.3, the resulting C-antigenic particles (A particles) no longer bind to their receptors, but due to their proximity to the membrane, the probability that newly exposed hydrophobic sequences in VP1 (together with VP4?) can now insert into the plasma membrane is enhanced. These hydrophobic domains together with VP4 can form a pore through which the RNA is then transferred into the cytoplasm. The interaction appears to be transient, since all C-antigenic particles (now presumably B particles) that had been generated are subsequently released into the supernatant medium. Nevertheless, the time allowed for virus-membrane interaction is sufficient to result in productive uncoating and infection. According to this model, during its natural entry route, HRV2 is present in the fluid phase of the endosome after receptor dissociation. Once the pH has dropped below 5.6, the multivesicular nature of endosomal carrier vesicles or late endosomes might greatly enhance the probability of contact between the now hydrophobic virus particle and the membrane even in the absence of receptors.
FIG. 7.

Potential mechanism of HRV2 uncoating at the plasma membrane upon exposure to low pH. HRV2 binds to its receptors close to the top of the fivefold axis. Upon low-pH treatment, hydrophobic domains in the N terminus of VP1 become externalized, giving rise to C-antigenic particles that are subsequently released from the receptors. Insertion of these domains into the plasma membrane can now occur, resulting in pore formation and RNA release.
Acknowledgments
This work was supported by grants from Virologiefonds to D.B and R.F. and Jubiläumsfonds der Österreichischen Nationalbank no. 7511 to R.F.
REFERENCES
- 1.Bayer, N., E. Prchla, M. Schwab, D. Blaas, and R. Fuchs. 1999. Human rhinovirus HRV14 uncoats from early endosomes in the presence of bafilomycin. FEBS Lett. 463:175-178. [DOI] [PubMed] [Google Scholar]
- 2.Bayer, N., D. Schober, E. Prchla, R. F. Murphy, D. Blaas, and R. Fuchs. 1998. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J. Virol. 72:9645-9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belnap, D. M., B. M. McDermott, Jr., D. J. Filman, N. Cheng, B. L. Trus, H. J. Zuccola, V. R. Racaniello, J. M. Hogle, and A. C. Steven. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA 97:73-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blake, K., and S. O'Connell. 1993. Virus culture, p. 81-122. In D. R. Harper (ed.), Virology Labfax. Blackwell Scientific Publications, London, United Kingdom.
- 5.Brown, M. S., J. Herz, and J. L. Goldstein. 1997. LDL receptor structure: calcium cages, acid baths and recycling receptors. Nature 388:629-630. [DOI] [PubMed] [Google Scholar]
- 6.Casasnovas, M., and T. A. Springer. 1995. Kinetics and thermodynamics of virus binding to receptor. Studies with rhinovirus, intercellular adhesion molecule-1 (ICAM-1), and surface plasmon resonance. J. Biol. Chem. 270:13216-13224. [DOI] [PubMed] [Google Scholar]
- 7.Davis, C. G., J. L. Goldstein, T. C. Sudhof, R. G. Anderson, D. W. Russell, and M. S. Brown. 1987. Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature 326:760-765. [DOI] [PubMed] [Google Scholar]
- 8.Dirlam-Schatz, K. A., and A. D. Attie. 1998. Calcium induces a conformational change in the ligand binding domain of the low density lipoprotein receptor. J. Lipid Res. 39:402-411. [PubMed] [Google Scholar]
- 9.Greve, J. M., C. P. Forte, C. W. Marlor, A. M. Meyer, H. Hoover-Litty, D. Wunderlich, and A. McClelland. 1991. Mechanisms of receptor-mediated rhinovirus neutralization defined by two soluble forms of ICAM-1. J. Virol. 65:6015-6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruenberger, M., D. Pevear, G. D. Diana, E. Kuechler, and D. Blaas. 1991. Stabilization of human rhinovirus serotype-2 against pH-induced conformational change by antiviral compounds. J. Gen. Virol. 72:431-433. [DOI] [PubMed] [Google Scholar]
- 11.Helenius, A., J. Kartenbeck, K. Simons, and E. Fries. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84:404-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hewat, E., E. Neumann, and D. Blaas. 2002. The concerted conformational changes during human rhinovirus 2 uncoating. Mol. Cell 10:317-326. [DOI] [PubMed] [Google Scholar]
- 13.Hewat, E. A., and D. Blaas. 1996. Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J. 15:1515-1523. [PMC free article] [PubMed] [Google Scholar]
- 14.Hewat, E. A., E. Neumann, J. F. Conway, R. Moser, B. Ronacher, T. C. Marlovits, and D. Blaas. 2000. The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: a structural view. EMBO J. 19:6317-6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofer, F., M. Gruenberger, H. Kowalski, H. Machat, M. Huettinger, E. Kuechler, and D. Blass. 1994. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 91:1839-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoover-Litty, H., and J. M. Greve. 1993. Formation of rhinovirus-soluble ICAM-1 complexes and conformational changes in the virion. J. Virol. 67:390-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon, H., and G. G. Shipley. 2000. Vesicle-reconstituted low density lipoprotein receptor. Visualization by cryoelectron microscopy. J. Biol. Chem. 275:30458-30464. [DOI] [PubMed] [Google Scholar]
- 18.Kolatkar, P. R., J. Bella, N. H. Olson, C. M. Bator, T. S. Baker, and M. G. Rossmann. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18:6249-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korant, B. D., K. Lonberg-Holm, J. Noble, and J. T. Stasny. 1972. Naturally occurring and artificially produced components of three rhinoviruses. Virology 48:71-86. [DOI] [PubMed] [Google Scholar]
- 20.Lonberg-Holm, K., L. B. Gosser, and E. J. Shimshick. 1976. Interaction of liposomes with subviral particles of poliovirus type 2 and rhinovirus type 2. J. Virol. 19:746-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lonberg-Holm, K., and J. Noble-Harvey. 1973. Comparison of in vitro and cell-mediated alteration of a human rhinovirus and its inhibition by sodium dodecyl sulfate. J. Virol. 12:819-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lonberg-Holm, K., and F. H. Yin. 1973. Antigenic determinants of infective and inactivated human rhinovirus type 2. J. Virol. 12:114-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madshus, I., S. Olsnes, and K. Sandvig. 1984. Different pH requirements for entry of the two picornaviruses, human rhinovirus 2 and murine encephalomyocarditis virus. Virology 139:346-357. [DOI] [PubMed] [Google Scholar]
- 24.Marlovits, T. C., C. Abrahamsberg, and D. Blaas. 1998. Very-low-density lipoprotein receptor fragment shed from HeLa cells inhibits human rhinovirus infection. J. Virol. 72:10246-10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikhailenko, I., W. Considine, K. M. Argraves, D. Loukinov, B. T. Hyman, and D. K. Strickland. 1999. Functional domains of the very low density lipoprotein receptor: molecular analysis of ligand binding and acid-dependent ligand dissociation mechanisms. J. Cell Sci. 112:3269-3281. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee, S., R. N. Ghosh, and F. Maxfield. 1997. Endocytosis. Physiol. Rev. 77:753-803. [DOI] [PubMed] [Google Scholar]
- 27.Neubauer, C., L. Frasel, E. Kuechler, and D. Blaas. 1987. Mechanism of entry of human rhinovirus 2 into HeLa cells. Virology 158:255-258. [DOI] [PubMed] [Google Scholar]
- 28.Noble, J. N., and K. Lonberg-Holm. 1973. Interactions of components of human rhinovirus type 2 with HeLa cells. Virology 51:270-278. [DOI] [PubMed] [Google Scholar]
- 29.Olson, N. H., P. R. Kolatkar, M. A. Oliveira, R. H. Cheng, J. M. Greve, A. McClelland, T. S. Baker, and M. G. Rossmann. 1993. Structure of a human rhinovirus complexed with its receptor molecule. Proc. Natl. Acad. Sci. USA 90:507-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prchla, E., E. Kuechler, D. Blaas, and R. Fuchs. 1994. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 68:3713-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prchla, E., C. Plank, E. Wagner, D. Blaas, and R. Fuchs. 1995. Virus-mediated release of endosomal content in vitro: different behavior of adenovirus and rhinovirus serotype 2. J. Cell Biol. 131:111-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rueckert, R. R. 1996. Picornaviridae: the viruses and their replication, p. 609-654. In D. M. Knipe, B. N. Fields, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 33.Rust, R. C., L. Landmann, R. Gosert, B. L. Tang, W. Hong, H.-P. Hauri, D. Egger, and K. Bienz. 2001. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 75:9808-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savolainen, C., S. Blomqvist, M. N. Mulders, and T. Hovi. 2002. Genetic clustering of all 102 human rhinovirus prototype strains: serotype 87 is close to human enterovirus 70. J. Gen. Virol. 83:333-340. [DOI] [PubMed] [Google Scholar]
- 35.Schlegel, A., T. H. Giddings, Jr., M. S. Ladinsky, and K. Kirkegaard. 1996. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70:6576-6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schober, D., P. Kronenberger, E. Prchla, D. Blaas, and R. Fuchs. 1998. Major and minor receptor group human rhinoviruses penetrate from endosomes by different mechanisms. J. Virol. 72:1354-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uncapher, C. R., C. M. Dewitt, and R. J. Colonno. 1991. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 180:814-817. [DOI] [PubMed] [Google Scholar]
- 38.White, J., J. Kartenbeck, and A. Helenius. 1980. Fusion of Semliki Forest virus with the plasma membrane can be induced by low pH. J. Cell Biol. 87:264-272. [DOI] [PMC free article] [PubMed] [Google Scholar]