

Abstract
The BarA-UvrY two-component system (TCS) in Escherichia coli is known to regulate a number of phenotypic traits. Both in vitro and in vivo assays, including the chicken embryo lethality assay, showed that this TCS regulates virulence in avian pathogenic E. coli (APEC) serotype O78:K80:H9. A number of virulence determinants, such as the abilities to adhere, invade, persist within tissues, survive within macrophages, and resist bactericidal effects of serum complement, were compromised in mutants lacking either the barA or uvrY gene. The reduced virulence was attributed to down regulation of type 1 and Pap fimbriae, reduced exopolysaccharide production, and increased susceptibility to oxidative stress. Our results indicate that BarA-UvrY regulates virulence properties in APEC and that the chicken embryo lethality assay can be used as a surrogate model to determine virulence determinants and their regulation in APEC strains.
Avian pathogenic Escherichia coli (APEC) infection causes avian colibacillosis, a complex syndrome characterized by air sacculitis, pericarditis, peritonitis, salpingitis, polyserositis, septicemia, synovitis, osteomyelitis, and yolk sac infection (19, 21). Cellulitis caused by APEC is the second leading cause of condemnation of broiler chickens and costs the U.S. poultry industry an estimated 40 million dollars per year (43). Most often, APEC strains infect chickens, turkeys, ducks, and other avian species through fecal dust via the respiratory tract. Isolates reported in some studies predominantly belong to the O1, O2, and O78 serogroups, other serotypes predominate in other studies, and often, untypeable isolates predominate (7, 19-22).
While many of the genes involved in APEC virulence have been identified, global regulation of the virulence of APEC, including large virulence plasmids, has only now begun to be characterized (6, 9, 10, 21, 26, 31). From attachment and colonization to the host cells to systemic invasion, a complex regulatory network in E. coli exists; this network senses the environment and activates genes that are required for each step in the infection process (18). The infection process requires rapid adaptation to the host environment by alteration of gene expression and, as a result, of bacterial structures and processes (15, 51, 62). In addition to strain-specific virulence determinants, it is highly likely that conserved global regulator(s) controlling adaptive metabolic systems are at play.
E. coli utilizes several evolutionarily conserved sensory systems to sense and adapt to their ever-changing surroundings. Common features in these signal transduction processes are conserved families of histidine-aspartate kinases that assist the bacteria in environmental adaptation (18, 52). The BarA protein is one such membrane-associated sensor protein conserved in certain gram-negative pathogens (23, 35, 39, 54). In uropathogenic E. coli, barA transcription is induced upon contact with the uroepithelial cell surface and has been implicated in the metabolic switching between glycolytic and gluconeogenic carbon sources (45, 70). Additionally, E. coli barA mutants show impaired catalase expression, which therefore renders cells sensitive to oxidative stress (36, 37). In Salmonella enterica serovar Typhimurium, barA or uvrY mutants exhibit a significant reduction in their ability to invade cultured epithelial cells, due in part to regulation of the type III secretion system required for modulating eukaryotic cellular physiology for uptake of bacteria (3, 4, 29).
The UvrY protein is the cognate response regulator for BarA in E. coli, and orthologs of UvrY are present in Pseudomonas (gacA), Erwinia (expA), Vibrio (varA), and Salmonella (sirA) species within evolutionarily conserved regions of their respective genomes (17, 44, 46, 59). The S. enterica UvrY ortholog, SirA, binds to the promoters of the hilA, hilC, and csrB genes, and thus, regulates bacterial motility and host cell invasion during infection (17, 56, 59).
In E. coli, the BarA-UvrY two-component system (TCS) affects the activity of CsrA RNA-binding protein by regulating the expression of csrB and csrC untranslated regulatory RNA. The csrB and csrC RNA binds to CsrA protein and prevents it from binding to the 5′ untranslated region of target mRNAs. CsrA controls carbon metabolism, flagellum biosynthesis, and biofilm formation (3, 57, 59, 66). These findings not only indicate an important, evolutionarily conserved role for the BarA-UvrY/SirA TCS in establishing early infection in pathogenic γ-proteobacteria but suggest that BarA-UvrY TCS could be an important regulator in the pathogenesis of E. coli.
The purpose of this investigation was to test the hypothesis that the BarA-UvrY TCS is critical to the virulence of APEC O78:K80:H9. To that end, isogenic barA and uvrY mutants of APEC O78:K80:H9 strain χ7122 were constructed using λ Red recombination as described previously (8) using the primers listed in Table 1. These mutants were tested for various attributes of virulence, including in vivo assays in developing chicken embryos and various in vitro assays to study the early steps in pathogenesis.
TABLE 1.
Primers used in this study
| Primer | Sequencea |
|---|---|
| barA knockout with kanamycin cassette | |
| OSM 58 | 5′-CATCGTCGCCATTCCGATATTGTTCGCGCGATTTCG TTGCATATGAATATCCTCC-3′ |
| OSM 59 | 5′-CGACATTATCCATCTCGTCCAACAGTTCCAGCAGCTACTTGTGTAGGCTGGAGC-3′ |
| uvrY knockout with chloramphenicol cassette | |
| OSM 43 | 5′-TGGTGCCGCCAGGGATACGACGCATTCTGGAAGTTGCATATGAATATCCTCCTTAGT-3′ |
| OSM 44 | 5′-CATTTGTTGAGCGATGTCAGAAGCAATGTAACGCTGACCGTGTAGGCTGGAGCTGCTTC-3′ |
| barA amplification and cloning primers | |
| OSM221 | 5′-CCCGAATTCATAGCATACGCCAAAATGAGGACAG-3′ |
| OSM222 | 5′-CCCGATATCATAACTCGACAAGACATCCATTA-3′ |
| uvrY amplification and cloning primers | |
| OSM 64 | 5′-CCCGAATTCATAATTTCATCGTAGGGCTTACTGTGA-3′ |
| OSM 74 | 5′-CCCCTGCAGATGCACGCCTGGCTGGTTAC-3′ |
| papA amplification primers | |
| OSM 309 | 5′-ACTCTGCGGACCACTTGGGA-3′ |
| OSM 310 | 5′-CCAACTATTCCTCAGGGGCA-3′ |
| fimA amplification primers | |
| OSM 260 | 5′-ACCGTTCAGTTAGGACAGGTTCGT-3′ |
| OSM 261 | 5′-TCTGCAGAGCCAGAACGTTTGTAT-3′ |
| 16S rrnA amplification primers | |
| OSM 17 | 5′-GTGCCCAGATGGGATTAGCTAGTAG-3′ |
| OSM 81 | 5′-GTTAGCCGGTGCTTCTTCTG-3′ |
The underlined sequences are restriction recognition sequences used for cloning.
Mutation in barA or uvrY reduces virulence of E. coli O78:K80:H9 (χ7122) in vivo.
The role of the BarA-UvrY TCS in the virulence of APEC strain χ7122 was determined using an chicken embryo lethality assay, in which live embryos were infected with a controlled amount of bacteria and were scored as alive or dead by movement of the embryo when held close to a bright light source (12, 16, 42, 69). Mutant strains of χ7122 were generated by λ Red-mediated recombination, with modifications suggested for clinical isolates of E. coli (8, 32, 38) (Table 2). Complementation of the mutants was accomplished by expressing wild-type copies of either the barA or uvrY gene in pBR322 (Tables 1 and 2).
TABLE 2.
E. coli strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotype | Source or reference |
|---|---|---|
| E. coli strains | ||
| DH5α | luxS supE44 Δ(φ80ΔlacZ M15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Invitrogen |
| K-12 MG1655 | F− λ−ilvG rbf50 rph-1 | Lab collection |
| AAEC072 | MG1655 ΔfimBEACDFGH | T. Romeo |
| LG1315 | Aerobactin producer | L. K. Nolan |
| LG1522 | Aerobactin indicator strain | L. K. Nolan |
| χ7122 | APEC O78:K80:H9 gyrA; Nalr | R. Curtiss III (50) |
| SM3000 | χ7122 barA::kan | This work |
| SM3001 | χ7122 uvrY::cm | This work |
| SM3002 | χ7122 barA::kan uvrY::cm | This work |
| SM3003 | SM3000 carrying pSM1; Apr | This work |
| SM3004 | SM3001 carrying pSM2; Apr | This work |
| Plasmids | ||
| pBR322 | Cloning vector; Ampr | Invitrogen |
| pSM1 | barA gene with EcoRI-EcoRV site of pBR322; Ampr | This work |
| pSM2 | uvrY within the EcoRV-BamHI site of pBR322; Ampr | This work |
| pType-1 | The fimACDFGH operon constitutively expressed in pACYC184 | J. Johnson |
| pPapG1 | The papG1 allele expressed constitutively in pACYC184 | J. Johnson |
Bacterial strains were grown for a total of 48 h in LB medium in two subcultures without shaking at 37°C to facilitate pilus formation. The barA or uvrY mutants did not exhibit a growth defect, as determined by generation time in LB and tryptone broth at 37°C. Cells were pelleted by centrifugation and gently resuspended in phosphate-buffered saline (PBS). A 10-fold serial dilution of bacterial suspension was made in PBS (9.0 to 1.0 log10 CFU in 0.1 ml). Groups of 20 12-day-old specific-pathogen-free (SPF) chicken embryos were infected with 0.1 ml of each dilution through the allantoic cavity using an 18-gauge needle. The needle hole was sealed with adhesive cement. The eggs were then incubated at 37°C and examined every 12 h for 7 days. The first time each embryo was observed to be dead was recorded (lack of movement on candling). The highest dilution at which half of all the embryos died was considered the minimum lethal dose (MLD50). The MLD50 of wild-type E. coli O78:K80:H9 strain χ7122 was determined to be 4 ± 0.5 log10 CFU, while the MLD50s of prototrophic E. coli K-12 strains DH5α and HB101 were >9 ± 0.5 log10 CFU, and they were considered to be avirulent. The effects of various mutations and their complement were determined by inoculating 0.1 ml of ∼5 × 103 CFU bacterial culture (final count in 0.1 ml determined by plating) into the allantoic cavities of a set of 20 12-day-old SPF chicken embryos. The experiment was repeated twice. Our results indicated that a mutation in either the barA or uvrY gene reduced the virulence of E. coli O78:K80:H9 strain χ7122 (Fig. 1). The reduction in virulence was significant (P < 0.05 between each set of eggs from two experiments by the paired t test), as 12 of 20 embryos and 16 of 20 embryos inoculated with the mutant strains were still alive after 5 days, while only 2 of 20 embryos survived for the wild-type strain. A plasmid-borne copy of the wild-type gene was capable of restoring virulence of the mutant strain; however, the complementation was not 100%. Tomenius et al. report that barA plasmid clones acquire mutations that result in poor complementation (60). It could also be due to the loss of plasmid from the bacterial strain in vivo, within the embryo, due to the absence of antibiotic selection. There was a 10% loss of plasmid-bearing colonies in the case of barA/p-barA complemented strain, as determined by dilution plating. A uvrY mutant was less virulent than a barA mutant, indicating that the transcription modulator, the UvrY protein, has a larger role in determining virulence. However, the reduction in virulence in the uvrY mutant was not similar to the level of avirulent E. coli K-12 strain, indicating as expected, that certain virulence determinants are independent of the BarA-UvrY regulatory system, and that the virulence of APEC is multifactorial in nature.
FIG. 1.

The BarA-UvrY two-component system regulates the virulence of E. coli O78:K80:H9 in chicken embryos. The lethality of E. coli K-12 DH5α (control), χ7122 (wild type [wt]), SM3000 (barA::kan) (▪), SM3001 (uvrY::cm) (▴), SM3002 (barA::kan uvrY::cm) (×), SM3003 (barA::kan) carrying pSM1 (□), and SM3004 (uvrY::cm) carrying pSM2 (▵) was determined by inoculating 0.1 ml of 5 × 10 3 CFU of bacteria into the allantoic cavity of a set of 20 12-day-old embryonated chicken eggs. Bacterial strains were grown in LB broth to an optical density at 600 nm of 1.5 at 37°C for 48 h without shaking. Cells were washed and gently resuspended in phosphate-buffered saline. Bacterial suspension in 0.1 ml of PBS was injected into the allantoic cavities of embryonated eggs using an 18-gauge needle, and the hole was subsequently sealed with glue. The eggs were incubated at 37°C and examined every 12 h for 7 days. The first time that each embryo was first observed to be dead (no movement on candling) was recorded. The results are the means of two such experiments.
The barA and uvrY mutants poorly colonize embryonic tissues and fail to persist within the liver and spleen.
To further investigate the reason for the reduced virulence in either a barA or uvrY mutant, a set of eight 12-day-old embryos were infected with various strains. At 24 h and 48 h, a set of four embryos was harvested, and the bacterial load was determined in various tissues (Table 3). Although barA mutants were able to colonize the chorioallantoic membrane (CAM) and the liver (∼3.0 ± 1.4 log10 CFU/mg of tissue), multiply in allantoic fluid (ALF) and amniotic fluid (∼2.0 ± 1.0 log10 CFU/ml of fluid), similar to the wild-type strain, they were unable to persist in the lungs or spleen. The persistence in liver and lungs decreased 10-fold (or more) after 48 h of infection. The uvrY mutant could initially replicate in ALF and colonize CAM, liver, and lungs, but it failed to persist in these organs, particularly in the liver (∼200-fold decrease) after 48 h of incubation. However, complementation of the uvrY mutant strain by a plasmid-borne copy of the wild-type uvrY gene (p-uvrY) (pSM2) restored colonization and persistence. These results, particularly the number of bacteria in the liver and spleen, indicate that UvrY may regulate virulence determinants required for systemic infection in the chicken embryo. Since the initial site of APEC infection is the lungs (air sacculitis), followed by a generalized infection (perihepatitis, pericarditis, or septicemia), our results indicate that a nonfunctional BarA-UvrY TCS may lead to poor colonization of the lung tissues and limit systemic invasion. Interestingly, unlike wild-type strains, embryos infected with mutants did not exhibit pericardial lesions, a characteristic of cellulitis-derived isolates (40). Our results showing the abilities of APEC strain χ7122 to colonize lungs, invade internal organs, and disseminate in allantoic and amniotic fluids of a 12-day-old embryos are essentially similar to that shown for the same APEC strain in 3.5-week-old chickens by Mellata et al. (33). Our results, therefore, suggest that 12-day-old SPF chicken embryos could serve as surrogate models for determining virulence.
TABLE 3.
Attributes of APEC strain χ7122 and various isogenic mutants to colonize 12-day-old chicken embryos, invade internal organs, and disseminate in allantoic and amniotic fluids after 24 h and 48 h of infection
| Strain | Genotype | Aerobactin productiona | Hemolysin productionb | Serum resistancec [log10 (CFU/ml)] at the following time after infection:
|
Tissued | Tissue infectivitye [log10 (CFU/ml or mg of tissue) at the following time after infection:
|
||
|---|---|---|---|---|---|---|---|---|
| 0 h | 4 h | 24 h | 48 h | |||||
| χ7122 | Wild type | + | + | 8.6 ± 0.9 | 8.9 ± 0.9 | CAM | 2.2 ± 1.2 | 3.1 ± 1.5 |
| ALF | 2.0 ± 1.2 | 3.4 ± 1.0 | ||||||
| AMF | 2.2 ± 1.2 | 3.1 ± 1.6 | ||||||
| Liver | 3.8 ± 1.9 | 3.9 ± 1.2 | ||||||
| Spleen | 2.0 ± 1.9 | 3.2 ± 1.8 | ||||||
| Lungs | 3.3 ± 1.9 | 3.8 ± 1.8 | ||||||
| SM3000 | barA | −/+ | −/+ | 8.5 ± 0.9 | 5.0 ± 0.4 | CAM | 2.1 ± 1.9 | 3.2 ± 1.5 |
| ALF | 2.0 ± 0.7 | 2.0 ± 1.3 | ||||||
| AMF | 2.0 ± 1.0 | 2.1 ± 1.5f,g | ||||||
| Liver | 3.1 ± 1.4f,g | 2.1 ± 1.0 | ||||||
| Spleen | <1 ± 0.1f,g | <1 ± 0.1f,g | ||||||
| Lungs | 1.8 ± 0.8f,g | <1 ± 0.1f,g | ||||||
| M3001 | uvrY | −/+ | −/+ | 8.1 ± 0.8 | 4.1 ± 0.9 | CAM | 2.2 ± 1.2 | <1 ± 0.1f,g |
| ALF | 1.8 ± 1.0f,g | <1 ± 0.1f,g | ||||||
| AMF | <1 ± 0.1f,g | <1 ± 0.1f,g | ||||||
| Liver | 3.3 ± 1.4f,g | 1.5 ± 1.2f,g | ||||||
| Spleen | <1 ± 0.1f,g | <1 ± 0.1f,g | ||||||
| Lungs | 2.1 ± 1.3f,g | <1 ± 0.1f,g | ||||||
| SM3004 | uvrY/p-uvrY | + | + | 8.4 ± 0.8 | 9.1 ± 0.9 | CAM | 2.7 ± 0.9 | 3.2 ± 1.3 |
| ALF | 2.1 ± 1.3 | 2.0 ± 0.9 | ||||||
| AMF | 3.6 ± 1.4 | 3.5 ± 1.0 | ||||||
| Liver | 3.5 ± 1.4 | 3.6 ± 1.5 | ||||||
| Spleen | 3.2 ± 1.4 | 3.5 ± 1.5 | ||||||
| Lungs | 3.2 ± 1.3 | 3.5 ± 1.5 | ||||||
Aerobactin production was determined by cross-feeding E. coli K-12 LG1522 (63). Lawns of LG1522 were plated onto M-9 minimal agar medium containing 40 μg/ml of tryptophan and 200μg/ml of 2,2′-dipyridyl. Fresh bacterial cultures in LB of 107 cells in 20 μl PBS were spotted onto the lawn of the aerobactin indicator strain LG1522 and incubated for 18 h at 37oC. Growth of the indicator organism around the spots in a halo indicated aerobactin production denoted by a + sign. E. coli LG1315 was used as a positive control (L. K. Nolan), and E. coli HB101 and DH5α were used as negative controls. A + sign indicates growth of reporter strain LG1522 comparable to that of LG1315; a −/+ sign indicates growth of reporter strain is less than that seen around the positive control, while a − sign indicates no growth of the reporter strain.
Hemolysin activity was measured by spotting 107 bacterial cells in 20 μl of PBS (and also by streaking) onto sheep blood agar plates and incubating the cells for 18 h at 37oC. The appearance of the zone of erythrocyte lysis around or under the bacterial colonies indicated hemolysis. E. coli K-12 DH5α was used as a negative control, and DH5α carrying plasmid pSF4000 (67) was used as a positive control. Two natural field isolates, VMRCVM-APEC isolate 10 (hly+) and isolate 11 (hly mutant), were used as APEC-specific controls. Marginal differences between the wild type and various mutants are denoted (−/+), but spectrophotometric measurement of heme release from sheep erythrocytes did not exhibit a statistically significant difference between the wild-type strain and various mutants.
Serum bactericidal assay was measured by the ability of 107 CFU/ml of freshly grown bacteria in 100-μl volume to survive in 300 μl of 10% fresh normal chicken serum (obtained from blood collected by wing vein puncture of 4- to 5-week-old chickens). Viable cell counts were done just before addition of bacteria to serum (time zero) and at 1 h and 3 h after incubation. Bacterial serum sensitivity was defined as a 2-log-unit decrease in the number of viable bacteria. Serum-sensitive (DH5α) and serum-resistant (VMRCVM-APEC isolate 20, a natural field isolate) E. coli strains were used as negative and positive controls, respectively. Results are presented as mean values of two independent experiments ± standard deviations of the means.
CAM, chorioallantoic membrane; ALF, allantoic fluid; AMF, amniotic fluid.
The persistence of wild-type APEC and various mutants was determined by the chicken embryo lethality assay followed by enumerating bacterial counts in various organs of 12-day-old embryos (42). The allantoic cavities of eight embryonated eggs were inoculated with 100 μl of 5 × 103 CFU of each bacterial culture in PBS. At 24 h and 48 h, the eggs were candled for viability. Half of the embryos inoculated with E. coli χ7122 (wild type) and SM3004 (uvrY/p-uvrY) had died by 24 h, and the other half showed morbidity. At 48 h, most of the morbid embryos died. At a given time, the dead embryos were chosen for dissection. Tissues or fluids were aseptically removed from each embryo, weighed, homogenized in 2-ml microcentrifuge tubes, and centrifuged at 1,000 rpm for 5 min at room temperature. Three serial dilutions of the supernatant were plated in triplicate onto LB plates with appropriate antibiotics and incubated at 37oC for 18 h, and the bacterial load was determined in log10 CFU per mg (tissue or organ) or ml (allantoic and amniotic fluids) ± standard deviation for four embryos from each group.
This value is significantly different (P < 0.05) from the value for the wild-type strain.
This value is significantly different (P < 0.05) from the value for the complemented strain.
Mutation in barA or uvrY reduces serum resistance, while aerobactin production is not altered.
The poor survival of the mutants in vivo could be due to a reduced ability to resist the bactericidal effects of serum complement (Table 3). Resistance to serum has been associated with E. coli causing infections in poultry and extraintestinal infections in other species (9, 14, 40). Serum resistance is a multifactorial characteristic involving outer membrane proteins, lipopolysaccharide, type 1 fimbriae, capsule, and O antigen and production of aerobactin (28). APEC strains more often contain ColV plasmids that encode serum resistance (41). APEC O78:K80:H9 carries three plasmids, of which one is of ColV origin. A similar plasmid from an O2:K2 serotype has recently been sequenced and found to carry serum resistance genes (24). One likely explanation is that BarA-UvrY TCS regulates a plasmid-borne pathogenic trait, directly or indirectly via other regulators. It is known that this TCS regulates stationary-phase sigma factor RpoS (36), which in turn regulates plasmid-borne pathogenicity genes, such as the spvR gene in Salmonella (68). Alternately, since the BarA-UvrY TCS affects carbon metabolism by regulating RpoS and CsrA, it is likely that the sugar substrates necessary to produce core O-78 antigen may be affected, leading to reduced serum resistance, as reported previously (33). Also, it is likely that this TCS may regulate pilus expression, contributing to serum resistance. Such an effect in production of exopolysaccharide (EPS) and pili was observed in E. coli K-12 and is reported for the strain under study (see Table 6; A. Mitra, S. Acharya, I. Patel, N. Chakraborty, G. Purrinton-Herren, D. Colley, T. Cebula, and S. Mukhopadhyay, unpublished observation).
TABLE 6.
Mutation in barA or uvrY reduces attachment and survival of E. coli O78:K80:H9 χ7122 in chicken macrophages
| Relevant genotype | No. of cells or bacteria (log10 CFU/ml)a
|
Attachment indexf | Survival indexg | |||
|---|---|---|---|---|---|---|
| Initial cellsb | Attached or invaded cells after 2 hc,d | Invaded fraction surviving after 8 hd | Calculated attached bacteriae | |||
| Wild type | 8.4 ± 0.9 | 7.0 ± 0.8 | 5.1 ± 0.7 | 7.0 ± 0.7 | 4.5 × 10−2 | 1.3 × 10−2 |
| barA | 8.9 ± 0.9 | 7.0 ± 0.8 | 3.8 ± 0.6 | 7.0 ± 0.7 | 1.4 × 10−2 | 7.0 × 10−4 |
| uvrY | 8.9 ± 0.9 | 7.0 ± 0.8 | 3.4 ± 0.5 | 7.0 ± 0.5 | 1.4 × 10−2 | 7.0 × 10−4 |
| uvrY/p-uvrY | 8.7 ± 0.9 | 7.1 ± 0.8 | 5.3 ± 0.7 | 7.1 ± 0.6 | 2.2 × 10−2 | 1.7 × 10−2 |
Results represent mean values ± standard deviations from three independent experiments.
HD-11 macrophages were obtained from the laboratory of S. K. Samal. The cells were grown in RPMI 1640 supplemented with 2mM l-glutamine, sodium pyruvate, and nonessential amino acids, without antibiotics, in 10% fetal bovine serum at a volume of 3 ml in six-well tissue culture plates at 37oC in 5% CO2. All bacterial cultures were grown with two passages of 48 h each time in static LB with appropriate antibiotics prior to infection for type 1 pilus induction. P-pilus expression was induced by growing bacteria on tryptic soy agar plates with the appropriate antibiotics. Experiments were performed with bacteria grown under both condition with essentially similar results.
Adherence assays were performed essentially by the method of Elsinghorst (13) as described in Table 5, footnote c.
After the initial 2-h incubation, an additional set of wells was washed with PBS (Mg2+/Ca2+) and then incubated for another 6 h in medium containing 100 μg ml−1 gentamicin before enumerating internalized bacteria to determine invasion frequencies.
These are the calculated values for only adherent bacteria obtained by the following formula: log10 CFU/ml of attached or invaded cells − log10 CFU/ml of invaded cells surviving gentamicin treatment.
Attachment index is calculated as follows: (CFU/ml of only adherent bacteria)/(total bacterial inoculum [CFU/ml]).
Intracellular survival fraction (survival index) is calculated as follows: (CFU/ml surviving gentamicin treatment)/(CFU/ml of only adherent population).
The ability to scavenge iron is critical for survival and pathogenesis in vivo, under conditions of low free iron that limit bacterial growth. The aerobactin iron acquisition system of APEC appeared not to be affected by the BarA-UvrY TCS (Table 3). Thus, the inability of barA or uvrY mutants to persist in the internal organs of 12-day-old chicken embryos appears to be independent of iron acquisition.
Mutation in barA or uvrY reduces MRHA.
To further understand the mechanistic basis for reduced embryo lethality in barA and uvrY mutants, we examined the effect(s) of these mutations on attachment. APEC strains adhere to chicken epithelial cells of the pharynx and trachea by type 1 fimbriae via d-mannose residues, but not in the deeper tissues (48). P fimbriae, which recognize globoseries and glycolipids, are responsible for colonization of lungs, air sacs, and internal organs but not peripheral tissues, such as trachea (48). Hemagglutination (HA) assays with chicken red blood cells under conditions that induce type 1 and P fimbriae indicated that the BarA-UvrY TCS may be regulating a mannose-resistant adherence (Table 4), either via P fimbriae or through other novel adhesins. There was a more than sixfold decrease in hemagglutination in the mutants (Table 4, log2 4 in the wild type versus log2 1 in mutants). We found similar results with human O+ P+ and guinea pig red blood cells (data not reported). The total HA could be restored by expressing p-uvrY in a uvrY strain, but not the mannose-resistant hemagglutination (MRHA) phenotype, suggesting that type 1 fimbriae also had a subordinate role in adhesion of these strains as reported earlier (12, 34). It is possible that the observed MRHA phenotype could also be a function of adhesins other than P fimbriae.
TABLE 4.
Mutation in barA and uvrY exhibits a reduction in mannose-resistant hemagglutination to chicken blood
| Strain | Genotype | Hemagglutination of 1% chicken erythrocytea [log2 (1/dilution)]
|
|
|---|---|---|---|
| Without 50 mM mannose | With 50 mM mannose | ||
| χ7122 | Wild type | 4 | 4 |
| AAEC072 | Δfim | 1 | <1 |
| AAEC072/p-Type-1 | Δfim/p-fimACDFGH clusterb | 5 | <1 |
| AAEC072/p-PapG1 | Δfim/p-pap clusterc | 4 | 4 |
| AAEC072/pSM1 | Δfim/p-barA | 4 | 2 |
| AAEC072/pSM2 | Δfim/p-uvrY | 5 | 5 |
| SM3000 | barA | 1 | 1 |
| SM3001 | uvrY | 1 | <1 |
| SM3002 | barA uvrY | 1 | <1 |
| SM3003 | barA/p-barA | 3 | 2 |
| SM3004 | uvrY/ p-uvrY | 4 | 3 |
The values are mean log2 of inverse dilution at which hemagglutination (HA) was observed with chicken blood. The standard deviation was <0.05 in all cases. The bacterial cultures were grown with two passages of 48 h each in static LB broth with appropriate antibiotics at 37oC to maximize type 1 fimbria expression. The assay was done on ice in duplicate in 96-well microtiter plates. Each bacterial culture was diluted twofold, before blood was added to study agglutination. The experiment was repeated twice with essentially similar results. The highest reciprocal of the dilution at which 50% of the erythrocytes sedimented to the bottom of the plate is taken as the HA titer.
Plasmid constitutively overexpressing type 1 fimbriae. It is a known positive control for the mannose-sensitive phenotype.
Plasmid overexpressing Pap fimbriae. It is a known positive control for the mannose-resistant phenotype.
The BarA-UvrY TCS is known to regulate the production of a basic polysaccharide, unbranched β-1,6-N-acetyl-d-glucosamine (PGA), via CsrA (64). PGA has been shown to regulate adhesion to abiotic surfaces, but there is no report of its role in adhesion to biotic surfaces. Curli is another adhesin that has been implicated in binding to various biotic surfaces and is a known virulence determinant of APEC strains (50). Expression of curli is regulated by stationary-phase sigma factor RpoS (49), which, in turn, is regulated by BarA-UvrY TCS (36). Other factors that could contribute to the HA phenotype are capsular polysaccharides and outer membrane proteins (11). Thus, the reduction in HA in the absence of the BarA-UvrY TCS could be a reflection of global down regulation of several of these factors.
Mutation in barA or uvrY reduces adherence and invasion to cultured chicken embryo fibroblasts.
To understand why mutant strains of χ7122 cause a decrease in embryo lethality and reduction in HA, we assayed for the possible effects barA and uvrY might have on the initial attachment phase of bacteria to a cultured chicken embryo fibroblast line, DF-1 (55). For APEC to cause colibacillosis, bacterial cells must be able to invade epithelial cells and move through the host fibroblasts that make up the connective tissue. In vivo, the uvrY mutant colonized the chorioallantoic membrane, an epithelium, 100-fold more poorly than the wild-type strain did (Table 3). Since we did not have a transformed chicken epithelial cell line, we assayed the abilities of barA and uvrY mutant strains and uvrY-complemented strain of APEC to adhere to and invade chicken fibroblasts (Table 5). Deletion of either the barA or uvrY gene in strain χ7122 reduced bacterial attachment to fibroblasts by 100-fold (∼2 log10 CFU/ml difference) of the wild type, respectively (Table 5). These less adherent phenotypes could be complemented to wild-type χ7122 levels when the respective gene was provided in trans. Complementation was best achieved in the uvrY/p-uvrY complemented strain (Table 5 and Fig. 2A to D). About 16% of the adherent APEC could invade DF-1 cells, as indicated by their ability to resist gentamicin treatment after 8 h of initial infection (Table 5). However, a mutation in either barA or uvrY (especially uvrY), lead to almost 100-fold reduction in invasiveness of these mutants that were adhering to DF-1 cells. The invasiveness could be restored to near wild-type levels in the uvrY/p-uvrY complemented strain. These results indicate that the BarA-UvrY TCS, either directly or indirectly, regulates a number of bacterial determinants responsible for attachment and invasion of APEC strains. The BarA-SirA TCS has been shown to be required for full virulence in S. enterica because of its effects on the type III secretion system (4, 30). This TCS has also been implicated in regulating invasiveness in Salmonella by regulating pathogenicity island I genes through the master regulator HilA (30). Whether such regulation of yet unknown APEC-specific pathogenicity island/type III secretion system operates in E. coli O78:K80:H9 strain is currently under investigation.
TABLE 5.
Mutation in barA or uvrY reduces attachment and invasion of E. coli O78:K80:H9 strain χ7122 to chicken embryo fibroblasts
| Relevant genotype | No. of cells or bacteria (log10 CFU/ml)a
|
Attachment indexf | Invasion indexg | |||
|---|---|---|---|---|---|---|
| Initial cellsb | Attached and invaded bacteria after 2 hc | Invaded bacterial fraction surviving after 4 hd | Calculated attached bacteriae | |||
| Wild type | 8.6 ± 0.9 | 7.1 ± 0.9 | 6.4 ± 0.8 | 7.1 ± 0.9 | 2.7 × 10−2 | 2.0 × 10−1 |
| barA | 8.6 ± 0.9 | 6.7 ± 0.8 | 4.8 ± 0.7 | 6.6 ± 0.8 | 1.2 × 10−2 | 1.5 × 10−2 |
| uvrY | 8.6 ± 0.9 | 6.3 ± 0.8 | 4.0 ± 0.6 | 6.3 ± 0.8 | 5.0 × 10−3 | 5.0 × 10−3 |
| uvrY/p-uvrY | 8.6 ± 0.9 | 7.2 ± 0.9 | 6.3 ± 0.8 | 7.1 ± 0.8 | 4.5 × 10−2 | 1.3 × 10−2 |
Results represent mean values ± standard deviations from three independent experiments.
DF-1 chicken embryo fibroblasts were obtained from the laboratory of S. K. Samal (Virginia-Maryland Regional College of Veterinary Medicine, College Park, Md.). The cells were grown in Dulbecco's modified Eagle's medium with 4 mM l-glutamine, sodium bicarbonate (1.5 g liter−1), glucose (4.5 g liter−1), and 10% fetal bovine serum. Cells were seeded at 4 × 106 in 3 ml medium per well in 24-well tissue culture plates and incubated at 37oC with 5% CO2 for 2 days (∼70% confluence) prior to infection. Each well was infected with a bacterium/DF-1 cell ratio of 10:1. For type 1 pilus induction, bacterial cultures were grown with two passages of 48 h each time in static LB with the appropriate antibiotics prior to infection. Growing bacteria on tryptic soy agar plates with the appropriate antibiotics induced P-type pilus expression. The experiment was performed with bacteria grown under both conditions with essentially similar results.
Adherence assays were performed essentially by the method of Elsinghorst (13). Just before infection, the medium in each well was replaced with 3 ml of fresh prewarmed cell culture medium. Twenty microliters of bacterial suspension was added to each well to attain a multiplicity of infection of 10. The plates were centrifuged at 600 × g for 5 min and incubated at 37oC for 2 h. After 2 h, one set of triplicate wells was lysed by the addition of 20 μl of 5% Triton X-100. Bacteria present in the lysates, representing the total number of bacteria (intra- and extracellular), were enumerated by plating.
To determine invasion frequencies, after the initial 2-h incubation, an additional set of wells was washed with PBS (Mg2+/Ca2+) and incubated for another 4 h in medium containing 100 μg ml−1 of the membrane-impermeable bactericidal antibiotic gentamicin to kill adhered extracellular bacteria. Wells were washed three times with PBS, lysed with 1 ml 0.1% Triton X-100, and plated in various dilutions.
To measure adherence, a second set of wells was washed five times with PBS supplemented with 2 mM MgCl2 and CaCl2, lysed with 1 ml of 0.1% Triton X-100, and plated in various dilutions. "Only adherence" was calculated as the numbers of bacteria recovered after PBS wash subtracted from the total number of bacteria present in each well.
Attachment index is calculated as follows: (CFU/ml of only adherent bacteria)/(total bacterial inoculum [CFU/ml]).
Invasion index was determined as the number of bacteria surviving incubation with gentamicin divided by the total number of bacteria present just before the addition of gentamicin, i.e., the only adherent population.
FIG. 2.

Mutation in uvrY reduces attachment to cultured chicken fibroblasts and macrophages. The monolayers of cultured cells over glass coverslips were incubated with bacteria (grown under static conditions) with a multiplicity of infection of 10. As described in Table 5, footnote c, the unattached bacteria were washed, stained with Hema-3 stain (Fischer Scientific, Middletown, VA), and visualized using a Spot RT camera attached to a Olympus BH-2 microscope with a 100× objective lens. Attachment of E. coli χ7122 (wild type [wt]), uvrY mutant, and uvrY mutant complemented with a plasmid-borne copy of the wild-type uvrY gene (p-uvrY). (A to D) Attachment to DF-1 chicken fibroblasts. Arrows indicate surface-attached bacteria. (E to H) Attachment to HD11 chicken macrophages. Panels F and H show attached bacteria, and panel G shows bacteria that are mostly engulfed (indicated by arrows).
Mutation in barA or uvrY reduces survival within chicken macrophages.
The most common form of APEC infection in poultry is characterized by initial respiratory tract colonization followed by a systemic spread to other parts of the body. Avian air sacs do not have cellular defense mechanisms and depend initially on the influx of heterophils followed by macrophages as a cellular defense (34, 61). APEC strains are known to be associated in vivo with macrophages of the air sacs and lungs, while nonpathogenic strains were observed to lack these attributes (34). Moreover, the pathogenic APEC strains are more resistant to killing by chicken macrophages in vitro than the less-pathogenic strains are (33, 47). Therefore, using HD-11 chicken macrophage line, we examined the effects of mutations of barA and uvrY on bacterial survival within cultured macrophages.
Internalized fractions of bacteria surviving within cultured HD-11 macrophages were enumerated by the standard gentamicin protection assay (13). Briefly, HD-11 cells were infected with the APEC strain(s) and incubated for a total of 2 h for adhesion prior to treatment of the infected cells with gentamicin to kill any external bacteria. Subsequently, the gentamicin-treated mixture was incubated for another 6 hours, before internalized bacteria were enumerated by dilution plating (13). There appeared to be no differences in adhesion (Table 6). However, mutation in barA reduced APEC survival within chicken macrophages by 1,000-fold compared to that of the wild type, and the uvrY mutant survived but at a level 104-fold less than that of the wild type (Table 6). Although there was not much of an adherence defect, the difference in survival of internalized bacteria, compared to wild type, is significant enough (P < 0.05 in all experiments) to be independent of adhesion. The mutant bacteria appeared to be engulfed quickly by the macrophages, while the wild-type or uvrY/p-uvrY complemented bacteria appeared to resist engulfment (Fig. 2F, H versus G, and the arrows indicating engulfed bacteria). A probable reason for this phenotype could be the lack of catalase activity in the mutant strains to counteract the oxidative onslaught of hydrogen peroxide within the macrophages. As expected, mutant colonies produced less catalase as determined by bubbling upon addition of 10 μl of 1% hydrogen\ peroxide on individual colonies grown on a LB plate. Exopolysaccharide and pili are also known to help APEC better survive within macrophages. It is also possible that mutation in uvrY leads to enhanced engulfment and destruction of the bacterium due to a reduction in pili and reduced EPS expression.
Mutations in barA or uvrY increase susceptibility to oxidative stress and reduce expression of exopolysaccharide and pili.
E. coli K-12, deficient in BarA and UvrY, has been shown to be impaired in catalase production and very sensitive to oxidative stress (36, 46). This phenotype is caused by the effect the BarA-UvrY TCS has on the expression of the stationary-phase sigma factor, RpoS, and the resultant regulation of stress responses (36). Indeed, this was true for the APEC strain χ7122 strain (Table 7). The barA or uvrY mutants exhibited a greater zone of inhibition of bacterial growth in the presence of 1% hydrogen peroxide (42 mm for the uvrY mutant compared to 32 mm for the wild-type strain [Table 7]).
TABLE 7.
Mutation in barA and uvrY genes in APEC strain χ7122 leads to lower pilus expression, exopolysaccharide production, and increased susceptibility to oxidative stressa
| Relevant genotype | Fold change in mRNA levels detected by quantitative real-time PCRb
|
Hydrogen peroxide sensitivity (zone of inhibition [mm])c | Exopolysaccharide (uronic acid) (U/mg protein)d | |
|---|---|---|---|---|
| fimA | papA | |||
| Wild type | 1.0 | 1.0 | 32.3 ± 0.2 | 2.0 ± 0.5 |
| barA | 1.8 ± 0.2 (↓) | 3.1 ± 0.2 (↓) | 35.3 ± 0.3 | 0.9 ± 0.3 |
| uvrY | 1.8 ± 0.3 (↓) | 3.2 ± 0.5 (↓) | 42.0 ± 0.4 | 0.8 ± 0.3 |
Bacterial strains were grown in LB under static conditions with the appropriate antibiotics. The same 24-h culture (in triplicate) was used for all the assays.
Total RNA was isolated from 20-ml cultures using the TRIzol reagent (Invitrogen, Carlsbad, CA). The RNA was treated with DNase I and purified using Qiagen RNeasy midi columns (Qiagen, Valencia, CA). For quantitative real-time PCR, first-strand cDNA was synthesized from 5 μg of total RNA using Superscript II (Invitrogen, Carlsbad, CA) and 50 ng of random hexamers (Invitrogen, Carlsbad, CA). Internal gene-specific primer pairs were used to amplify papA (OSM 309 and OSM 310), fimA (OSM 260 and OSM 261), and 16S rrnA (OSM 17 and OSM 81). For real-time PCR, the first 10 ng of first-strand cDNA was amplified separately with 10 μM each of gene-specific primer and 16S rrnA-specific primers (OSM 17 and OSM 81) in a 25-μl total volume using a PCT-200 Opticon Cycler (Biorad, Hercules, CA) and SYBR green 1 PCR master mix. Threshold cycle (CT) values were determined for various amplification products. The ΔCT values between samples were normalized to those for the rrnA product, as ΔCT = (CT of mutant − CT of rrnA) − (CT of wild type − CT of rrnA). Because PCR products double with each amplification cycle, the fold difference in the initial concentration of each transcript is determined as 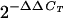 . The values are the means ± standard deviations of the mean for two independent experiment with triplicate samples. The wild type was assigned a value of 1.0. The downwards arrow indicates down regulation or lower regulation compared to the wild type.
. The values are the means ± standard deviations of the mean for two independent experiment with triplicate samples. The wild type was assigned a value of 1.0. The downwards arrow indicates down regulation or lower regulation compared to the wild type.
The hydrogen peroxide sensitivity was measured by putting a sterile filter paper disc soaked with 1% hydrogen peroxide on top of freshly overlaid bacteria (5 log10 CFU bacteria) in soft agar. The results are mean diameters of inhibition after 18 h of incubation at 37°C ± standard deviations of the means.
For EPS determination, bacteria were grown on LB agar overnight at 37°C, harvested by scraping, and resuspended in 2.5 ml of PBS. The cell number was determined from the turbidity at 600 nm. EPS was separated from the bacteria by vortexing each sample for 10 min, followed by ultracentrifugation of the bacterial suspension at 160,000 × g for 60 min at 4°C. The supernatant was removed and dialyzed in double-distilled water for 3 h in a membrane with a 6-kDa cutoff (Pierce). Uronic acids are common constituents of bacterial EPS. Uronic acid produced by various bacterial strains was determined by a colorimetric method, using pure uronic acid (Sigma, St. Louis, MO) as a standard, and expressed as units per milligram of protein (5).
Increased serum resistance, failure to persist in the liver and macrophages, and poor attachment to cultured cells indicated a possible role of exopolysaccharide and pili (27, 28). The BarA-UvrY TCS regulates carbon metabolism that provides substrates for synthesis of capsules, EPS, and surface antigens (A. Mitra and S. Mukhopadhyay, unpublished observation). The system is also known to indirectly regulate synthesis of a neutral unbranched polymer β-1,6-N-acetyl-d-glucosamine, encoded by the pga operon of E. coli (65). However, our studies indicate that BarA-UvrY also has an effect on the production of acidic EPS and capsules in E. coli K-12 (Mitra et al., submitted). Capsules contribute to the observed resistance to oxidative stress, survival within a phagosome, and inflammatory response. We found that disruption of BarA-UvrY led to a reduced uronic acid production (approximately twofold [Table 7]). Uronic acids are common constituents of bacterial EPS and a much more specific indicator of EPS (5). This may partly explain why an uvrY mutant fails to establish a systemic infection as observed in Table 3 or shows accelerated engulfment by macrophages.
Mellata et al. have demonstrated that possible regulation of type 1 and P fimbriae can provide protection for APEC from the bactericidal effects of phagocytes (34). As stated previously, type 1 pili are responsible for the initial stages of infection, while Pap contributes to tissue invasion in APEC (48). To further understand whether the defect in adhesion and bactericidal effect was due to decreased pilus expression, we examined the mRNA levels of pilus genes by quantitative real-time PCR in 24-h static cultures (Table 7). Since the genome of strain χ7122 has not yet been sequenced, we used published sequence from uropathogenic E. coli CFT073 to design primers for papA and fimA genes encoding the major Pap and type 1 structural proteins. The level of fimA expression was approximately twofold down regulated, and the level of papA expression was approximately threefold down regulated in either a barA or uvrY mutant (Table 7). These results support our phenotypic observations and suggest a new role of regulation of surface adhesins by the BarA-UvrY TCS. However, whether this regulation is direct or indirect is yet to be determined.
Conclusions.
This and other studies continue to dissect the virulence of APEC and to distinguish them from other extraintestinal pathogenic E. coli strains (26, 31, 53). While considerable work has been done to identify the virulence factors, little is known of their regulation. Large-scale genomic screenings have identified potential regulators (6, 10, 31), but only the Pst system has been recently examined in detail and shown to affect virulence in APEC (26). We now show that in addition to the Pst system/Pho regulon, there is also a BarA-UvrY regulon that controls virulence in APEC. It is interesting to note that a barA or uvrY mutant negatively affects the transcription of the rpoS gene, encoding the stationary-phase sigma factor RpoS (36). Decreased levels of RpoS result in down regulation of pstS transcription (58), which in turn governs the expression of the entire pst operon (1). In S. enterica, barA-sirA and pstS are known to affect expression of hilA, which is a regulator of the Salmonella pathogenicity island I-encoded type III secretion apparatus involved in bacterial invasion of epithelial cells (2, 25). However, there appears to be other factors that are, directly or indirectly, regulated by the BarA-UvrY TCS, including pili (type 1 and P) and exopolysaccharide that contribute to virulence in APEC. Whether this regulation is direct or indirect or a combination remains to be determined.
Using a chicken embryo lethality assay, we have shown that BarA-UvrY TCS regulates virulence factors in APEC serotype O78:K80:H9. A combination of virulence determinants, such as the abilities to adhere, invade, and survive within antigen-presenting cells, such as macrophages, and the ability to resist the bactericidal effect of serum complement are compromised in mutants lacking either barA and uvrY genes. The ability to resist the bactericidal effects of complement and persist within macrophages provides a survival advantage to APEC strains by potentiating efficient replication while abrogating elimination by the host immune responses. This was evident in our chicken embryo lethality assay, where isogenic mutant strains were rapidly eliminated from the livers and spleens of the infected embryos, while the wild-type APEC strain persisted within tissues, causing mortality. Our results also indicate that 12-day-old SPF chicken embryos can be used as a model to determine the initial virulence properties of APEC strains conveniently, since the mortality and colonization results of the wild-type strain are similar to those of 3.5-week-old chickens (33). Our results, therefore, suggest that the BarA-UvrY TCS may be a global regulator of APEC virulence.
Acknowledgments
We thank Roy Curtis III for providing the parent APEC strains; T. Romeo, J. Johnson, and L. K. Nolan for various strains and plasmids; and I. Dryburgh-Barry for assistance throughout the project. We are grateful to Siba Samal for his advice and critical analysis of our experimental design.
This work was supported by USDA-NRI-CSREES Competitive Grant 2004-35204-14749, USDA-Animal Health 2002-1106-0195318, and a Maryland Agriculture Experimental Station grant to S.M.
Editor: F. C. Fang
REFERENCES
- 1.Aguena, M., E. Yagil, and B. Spira. 2002. Transcriptional analysis of the pst operon of Escherichia coli. Mol. Genet. Genomics 268:518-524. [DOI] [PubMed] [Google Scholar]
- 2.Altier, C. 2005. Genetic and environmental control of Salmonella invasion. J. Microbiol. 43:85-92. [PubMed] [Google Scholar]
- 3.Altier, C., M. Suyemoto, and S. D. Lawhon. 2000. Regulation of Salmonella enterica serovar Typhimurium invasion genes by csrA. Infect. Immun. 68:6790-6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altier, C., M. Suyemoto, A. I. Ruiz, K. D. Burnham, and R. Maurer. 2000. Characterization of two novel regulatory genes affecting Salmonella invasion gene expression. Mol. Microbiol. 35:635-646. [DOI] [PubMed] [Google Scholar]
- 5.Blumenkrantz, N., and G. Asboe-Hansen. 1973. New method for quantitative determination of uronic acids. Anal. Biochem. 54:484-489. [DOI] [PubMed] [Google Scholar]
- 6.Brown, P. K., and R. Curtiss. 1996. Unique chromosomal regions associated with virulence of an avian pathogenic Escherichia coli strain. Proc. Natl. Acad. Sci. USA 93:11149-11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheville, N. F., and L. H. Arp. 1978. Comparative pathologic findings of Escherichia coli infection in birds. J. Am. Vet. Med. Assoc. 173:584-587. [PubMed] [Google Scholar]
- 8.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dho-Moulin, M., and J. M. Fairbrother. 1999. Avian pathogenic Escherichia coli (APEC). Vet. Res. 30:299-316. [PubMed] [Google Scholar]
- 10.Dozois, C. M., F. Daigle, and R. Curtiss III. 2003. Identification of pathogen-specific and conserved genes expressed in vivo by an avian pathogenic Escherichia coli strain. Proc. Natl. Acad. Sci. USA 100:247-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dozois, C. M., M. Dho-Moulin, A. Bree, J. M. Fairbrother, C. Desautels, and R. Curtiss III. 2000. Relationship between the Tsh autotransporter and pathogenicity of avian Escherichia coli and localization and analysis of the Tsh genetic region. Infect. Immun. 68:4145-4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elankumaran, S., and A. T. Venugopalan. 1998. Adhesion to tracheal epithelium and adherence associated characteristics of avian Escherichia coli, p. 106-121. In Proceedings of the International Symposium on Infectious Bronchitis and Pneumovirus Infections in Poultry, Rauischholzhausen, Germany, 15 to 18 June 1998. Institut für Geflügelkrankheiten, Giessen, Germany.
- 13.Elsinghorst, E. A. 1994. Measurement of invasion by gentamicin resistance. Methods Enzymol. 236:405-420. [DOI] [PubMed] [Google Scholar]
- 14.Fairbrother, J. M., E. Nadeau, and C. L. Gyles. 2005. Escherichia coli in postweaning diarrhea in pigs: an update on bacterial types, pathogenesis, and prevention strategies. Anim. Health Res. Rev. 6:17-39. [DOI] [PubMed] [Google Scholar]
- 15.Falkow, S. 1997. Invasion and intracellular sorting of bacteria: searching for bacterial genes expressed during host/pathogen interactions. J. Clin. Investig. 100:239-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibbs, P. S., J. J. Maurer, L. K. Nolan, and R. E. Wooley. 2003. Prediction of chicken embryo lethality with the avian Escherichia coli traits complement resistance, colicin V production, and presence of the increased serum survival gene cluster (iss). Avian Dis. 47:370-379. [DOI] [PubMed] [Google Scholar]
- 17.Goodier, R. I., and B. M. Ahmer. 2001. SirA orthologs affect both motility and virulence. J. Bacteriol. 183:2249-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grebe, T. W., and J. B. Stock. 1999. The histidine protein kinase superfamily. Adv. Microb. Physiol. 41:139-227. [DOI] [PubMed] [Google Scholar]
- 19.Gross, W. B. 1991. Coliobacillosis, p. 138-144. In B. W. Calnek, H. J. Barnes, C. W. Beard, W. M. Reid, and H. W. Yoder, Jr. (ed.), Diseases of poultry, 9th ed. Iowa State University Press, Ames, Iowa.
- 20.Gross, W. B. 1961. Development of airsac disease. Avian Dis. 5:431-439. [Google Scholar]
- 21.Gross, W. B. 1994. Disease due to Escherichia coli in poultry, p. 237-259. In C. L. Gyles (ed.), Escherichia coli in domestic animals and man. CAB International, Wallingford, United Kingdom.
- 22.Harry, E. G., and L. A. Hemsley. 1965. The association between the presence of septicaemia strains of Escherichia coli in the respiratory and intestinal tracts of chickens and the occurrence of colisepticaemia. Vet. Rec. 77:35-40. [PubMed] [Google Scholar]
- 23.Ishige, K., S. Nagasawa, S. Tokishita, and T. Mizuno. 1994. A novel device of bacterial signal transducers. EMBO J. 13:5195-5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, T. J., K. E. Siek, S. J. Johnson, and L. K. Nolan. 2006. DNA sequence of a ColV plasmid and prevalence of selected plasmid-encoded virulence genes among avian Escherichia coli strains. J. Bacteriol. 188:745-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, B. D. 2005. Salmonella invasion gene regulation: a story of environmental awareness. J. Microbiol. 43:110-117. [PubMed] [Google Scholar]
- 26.Lamarche, M. G., C. M. Dozois, F. Daigle, M. Caza, R. Curtiss III, J. D. Dubreuil, and J. Harel. 2005. Inactivation of the Pst system reduces the virulence of an avian pathogenic Escherichia coli O78 strain. Infect. Immun. 73:4138-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.La Ragione, R. M., W. A. Cooley, and M. J. Woodward. 2000. The role of fimbriae and flagella in the adherence of avian strains of Escherichia coli O78:K80 to tissue culture cells and tracheal and gut explants. J. Med. Microbiol. 49:327-338. [DOI] [PubMed] [Google Scholar]
- 28.La Ragione, R. M., and M. J. Woodward. 2002. Virulence factors of Escherichia coli serotypes associated with avian colisepticaemia. Res. Vet. Sci. 73:27-35. [DOI] [PubMed] [Google Scholar]
- 29.Lawhon, S. D., J. G. Frye, M. Suyemoto, S. Porwollik, M. McClelland, and C. Altier. 2003. Global regulation by CsrA in Salmonella typhimurium. Mol. Microbiol. 48:1633-1645. [DOI] [PubMed] [Google Scholar]
- 30.Lawhon, S. D., R. Maurer, M. Suyemoto, and C. Altier. 2002. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol. Microbiol. 46:1451-1464. [DOI] [PubMed] [Google Scholar]
- 31.Li, G., C. Laturnus, C. Ewers, and L. H. Wieler. 2005. Identification of genes required for avian Escherichia coli septicemia by signature-tagged mutagenesis. Infect. Immun. 73:2818-2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Link, A. J., D. Phillips, and G. M. Church. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 179:6228-6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mellata, M., M. Dho-Moulin, C. M. Dozois, R. Curtiss III, P. K. Brown, P. Arne, A. Bree, C. Desautels, and J. M. Fairbrother. 2003. Role of virulence factors in resistance of avian pathogenic Escherichia coli to serum and in pathogenicity. Infect. Immun. 71:536-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellata, M., M. Dho-Moulin, C. M. Dozois, R. Curtiss III, B. Lehoux, and J. M. Fairbrother. 2003. Role of avian pathogenic Escherichia coli virulence factors in bacterial interaction with chicken heterophils and macrophages. Infect. Immun. 71:494-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizuno, T. 1997. Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of Escherichia coli. DNA Res. 4:161-168. [DOI] [PubMed] [Google Scholar]
- 36.Mukhopadhyay, S., J. P. Audia, R. N. Roy, and H. E. Schellhorn. 2000. Transcriptional induction of the conserved alternative sigma factor RpoS in Escherichia coli is dependent on BarA, a probable two-component regulator. Mol. Microbiol. 37:371-381. [DOI] [PubMed] [Google Scholar]
- 37.Mukhopadhyay, S., and H. E. Schellhorn. 1997. Identification and characterization of hydrogen peroxide-sensitive mutants of Escherichia coli: genes that require OxyR for expression. J. Bacteriol. 179:330-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy, K. C., and K. G. Campellone. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagasawa, S., S. Tokishita, H. Aiba, and T. Mizuno. 1992. A novel sensor-regulator protein that belongs to the homologous family of signal-transduction proteins involved in adaptive responses in Escherichia coli. Mol. Microbiol. 6:799-807. [DOI] [PubMed] [Google Scholar]
- 40.Ngeleka, M., J. K. Kwaga, D. G. White, T. S. Whittam, C. Riddell, R. Goodhope, A. A. Potter, and B. Allan. 1996. Escherichia coli cellulitis in broiler chickens: clonal relationships among strains and analysis of virulence-associated factors of isolates from diseased birds. Infect. Immun. 64:3118-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nolan, L. K., S. M. Horne, C. W. Giddings, S. L. Foley, T. J. Johnson, A. M. Lynne, and J. Skyberg. 2003. Resistance to serum complement, iss, and virulence of avian Escherichia coli. Vet. Res. Commun. 27:101-110. [DOI] [PubMed] [Google Scholar]
- 42.Nolan, L. K., R. E. Wooley, J. Brown, K. R. Spears, H. W. Dickerson, and M. Dekich. 1992. Comparison of a complement resistance test, a chicken embryo lethality test, and the chicken lethality test for determining virulence of avian Escherichia coli. Avian Dis. 36:395-397. [PubMed] [Google Scholar]
- 43.Norton, R. A. 1997. Avian cellulitis. World's Poult. Sci. J. 53:337-349. [Google Scholar]
- 44.Parkins, M. D., H. Ceri, and D. G. Storey. 2001. Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol. Microbiol. 40:1215-1226. [DOI] [PubMed] [Google Scholar]
- 45.Pernestig, A. K., D. Georgellis, T. Romeo, K. Suzuki, H. Tomenius, S. Normark, and O. Melefors. 2003. The Escherichia coli BarA-UvrY two-component system is needed for efficient switching between glycolytic and gluconeogenic carbon sources. J. Bacteriol. 185:843-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pernestig, A. K., O. Melefors, and D. Georgellis. 2001. Identification of UvrY as the cognate response regulator for the BarA sensor kinase in Escherichia coli. J. Biol. Chem. 276:225-231. [DOI] [PubMed] [Google Scholar]
- 47.Pourbakhsh, S. A., M. Boulianne, B. Martineau-Doize, and J. M. Fairbrother. 1997. Virulence mechanisms of avian fimbriated Escherichia coli in experimentally inoculated chickens. Vet. Microbiol. 58:195-213. [DOI] [PubMed] [Google Scholar]
- 48.Pourbakhsh, S. A., M. Dho-Moulin, A. Bree, C. Desautels, B. Martineau-Doize, and J. M. Fairbrother. 1997. Localization of the in vivo expression of P and F1 fimbriae in chickens experimentally inoculated with pathogenic Escherichia coli. Microb. Pathog. 22:331-341. [DOI] [PubMed] [Google Scholar]
- 49.Pratt, L. A., and T. J. Silhavy. 1998. Crl stimulates RpoS activity during stationary phase. Mol. Microbiol. 29:1225-1236. [DOI] [PubMed] [Google Scholar]
- 50.Provence, D. L., and R. Curtiss III. 1992. Role of crl in avian pathogenic Escherichia coli: a knockout mutation of crl does not affect hemagglutination activity, fibronectin binding, or Curli production. Infect. Immun. 60:4460-4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rathman, M., L. P. Barker, and S. Falkow. 1997. The unique trafficking pattern of Salmonella typhimurium-containing phagosomes in murine macrophages is independent of the mechanism of bacterial entry. Infect. Immun. 65:1475-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson, V. L., D. R. Buckler, and A. M. Stock. 2000. A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 7:626-633. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez-Siek, K. E., C. W. Giddings, C. Doetkott, T. J. Johnson, M. K. Fakhr, and L. K. Nolan. 2005. Comparison of Escherichia coli isolates implicated in human urinary tract infection and avian colibacillosis. Microbiology 151:2097-2110. [DOI] [PubMed] [Google Scholar]
- 54.Sahu, S. N., S. Acharya, H. Tuminaro, I. Patel, K. Dudley, J. E. LeClerc, T. A. Cebula, and S. Mukhopadhyay. 2003. The bacterial adaptive response gene, barA, encodes a novel conserved histidine kinase regulatory switch for adaptation and modulation of metabolism in Escherichia coli. Mol. Cell. Biochem. 253:167-177. [DOI] [PubMed] [Google Scholar]
- 55.Schaefer-Klein, J., I. Givol, E. V. Barsov, J. M. Whitcomb, M. VanBrocklin, D. N. Foster, M. J. Federspiel, and S. H. Hughes. 1998. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology 248:305-311. [DOI] [PubMed] [Google Scholar]
- 56.Stocki, S. L., L. A. Babiuk, N. A. Rawlyk, A. A. Potter, and B. J. Allan. 2002. Identification of genomic differences between Escherichia coli strains pathogenic for poultry and E. coli K-12 MG1655 using suppression subtractive hybridization analysis. Microb. Pathog. 33:289-298. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki, K., X. Wang, T. Weilbacher, A. K. Pernestig, O. Melefors, D. Georgellis, P. Babitzke, and T. Romeo. 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J. Bacteriol. 184:5130-5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taschner, N. P., E. Yagil, and B. Spira. 2004. A differential effect of sigmaS on the expression of the Pho regulon genes of Escherichia coli. Microbiology 150:2985-2992. [DOI] [PubMed] [Google Scholar]
- 59.Teplitski, M., R. I. Goodier, and B. M. Ahmer. 2003. Pathways leading from BarA/SirA to motility and virulence gene expression in Salmonella. J. Bacteriol. 185:7257-7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tomenius, H., A. K. Pernestig, C. F. Mendez-Catala, D. Georgellis, S. Normark, and O. Melefors. 2005. Genetic and functional characterization of the Escherichia coli BarA-UvrY two-component system: point mutations in the HAMP linker of the BarA sensor give a dominant-negative phenotype. J. Bacteriol. 187:7317-7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toth, T. E., P. Siegel, and H. Veit. 1987. Cellular defense of the avian respiratory system. Influx of phagocytes: elicitation versus activation. Avian Dis. 31:861-867. [PubMed] [Google Scholar]
- 62.Vazquez-Torres, A., and F. C. Fang. 2000. Cellular routes of invasion by enteropathogens. Curr. Opin. Microbiol. 3:54-59. [DOI] [PubMed] [Google Scholar]
- 63.Vidotto, M. C., E. E. Muller, J. C. de Freitas, A. A. Alfieri, I. G. Guimaraes, and D. S. Santos. 1990. Virulence factors of avian Escherichia coli. Avian Dis. 34:531-538. [PubMed] [Google Scholar]
- 64.Wang, X., A. K. Dubey, K. Suzuki, C. S. Baker, P. Babitzke, and T. Romeo. 2005. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol. Microbiol. 56:1648-1663. [DOI] [PubMed] [Google Scholar]
- 65.Wang, X., J. F. Preston III, and T. Romeo. 2004. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 186:2724-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei, B. L., A. M. Brun-Zinkernagel, J. W. Simecka, B. M. Pruss, P. Babitzke, and T. Romeo. 2001. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol. Microbiol. 40:245-256. [DOI] [PubMed] [Google Scholar]
- 67.Welch, R. A., and S. Falkow. 1984. Characterization of Escherichia coli hemolysins conferring quantitative differences in virulence. Infect. Immun. 43:156-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilson, J. A., T. J. Doyle, and P. A. Gulig. 1997. Exponential-phase expression of spvA of the Salmonella typhimurium virulence plasmid: induction in intracellular salts medium and intracellularly in mice and cultured mammalian cells. Microbiology 143:3827-3839. [DOI] [PubMed] [Google Scholar]
- 69.Wooley, R. E., P. S. Gibbs, T. P. Brown, and J. J. Maurer. 2000. Chicken embryo lethality assay for determining the virulence of avian Escherichia coli isolates. Avian Dis. 44:318-324. [PubMed] [Google Scholar]
- 70.Zhang, J. P., and S. Normark. 1996. Induction of gene expression in Escherichia coli after pilus-mediated adherence. Science 273:1234-1236. [DOI] [PubMed] [Google Scholar]