

Abstract
Using several approaches, we investigated the importance of clathrin-mediated endocytosis in the uptake of human rhinovirus serotype 2 (HRV2). By means of confocal immunofluorescence microscopy, we show that K+ depletion strongly reduces HRV2 internalization. Viral uptake was also substantially reduced by extraction of cholesterol from the plasma membrane with methyl-β-cyclodextrin, which can inhibit clathrin-mediated endocytosis. In accordance with these data, overexpression of dynamin K44A in HeLa cells prevented HRV2 internalization, as judged by confocal immunofluorescence microscopy, and strongly reduced infection. We also demonstrate that HRV2 bound to the surface of HeLa cells is localized in coated pits but not in caveolae. Finally, transient overexpression of the specific dominant-negative inhibitors of clathrin-mediated endocytosis, the SH3 domain of amphiphysin and the C-terminal domain of AP180, potently inhibited internalization of HRV2. Taken together, these results indicate that HRV2 uses clathrin-mediated endocytosis to infect cells.
Human rhinoviruses, members of the family Picornaviridae, use functionally and structurally unrelated membrane proteins for cell entry. Major-group viruses, represented by 91 serotypes, bind to intercellular adhesion molecule 1 (ICAM-1/CD54), a member of the immunoglobulin superfamily. Minor-group viruses (10 serotypes) gain access to the host cell via the low-density lipoprotein receptor (LDLR), the LDLR-related protein (LRP) (22), and possibly other members of the LDLR family such as the very-low-density lipoprotein receptor (VLDLR) (36). In their extracellular domains, these membrane proteins possess various numbers of ligand binding repeats, which are about 40 amino acids in length. Three disulfide bonds and a Ca2+ ion maintain the structure of each repeat; these are essential for ligand binding. LDLR binds and internalizes lipids assembled with apolipoprotein B and E, whereas VLDLR and LRP bind a large number of structurally and functionally unrelated macromolecules. A hallmark of these receptors is their high endocytosis rate. The ligands are released in endosomes due to the low pH environment and subsequently targeted to lysosomes for degradation (50). The receptors are recycled to the plasma membrane.
Upon uptake, the minor-group virus human rhinovirus type 2 (HRV2) and the major-group virus HRV14 progress into endosomal compartments where their RNA genomes are released into the cytosol through a pore in the endosomal membrane or by disruption of the endosome, respectively, to initiate viral replication (52).
The question of how viruses divert existing endocytic mechanisms used by the cells to carry out physiological functions has attracted considerable interest. All eukaryotic cells internalize a variety of ligands bound to their respective membrane receptors by clathrin-mediated endocytosis. This process is initiated by coated-pit nucleation and assembly involving the scaffolding protein clathrin, the adaptor complex protein AP-2, the linker proteins epsin and CALM/AP180, as well as the membrane lipid phosphatidylinositol-4,5-P2 and cargo receptors (62). Coated-pit maturation and fission then give rise to clathrin-coated vesicles by the action of endophilin, amphiphysin, and the GTPase dynamin, among others. Alternatively, clathrin-independent internalization also occurs. The best-characterized mechanism involves caveolae, small plasma membrane-derived vesicles containing the membrane protein caveolin. Recently, it has been found that this process is also dependent on functional dynamin. Apart from their involvement in cholesterol transport, the role of caveolae in constitutive endocytosis remains uncertain (26). Finally, clathrin- and caveolae-independent endocytosis has emerged as another pathway whose tracers are associated with detergent-insoluble microdomains or lipid rafts, but a precise description of the molecular mechanisms is still missing.
The cytoplasmic domains of receptors of the LDLR family contain NPXY and YXXL sequence motifs, which have been shown to direct them into clathrin-coated pits by associating with the clathrin terminal domain (28) or with AP-2 complexes (29). Because of the presence of these internalization signals in LDLRs, it was anticipated that uptake of minor-group HRVs would occur by clathrin-mediated endocytosis. A previous study by Huber et al. (23) showed that inhibition of clathrin-mediated endocytosis by K+ depletion or by expression of the dominant-negative dynamin mutant K44A (dynK44A) in HeLa cells (10), resulting in a defect in the severing of invaginated vesicles from the plasma membrane (53), has pleiotropic effects such as impairment of the acidification of the endosomal lumen. This increase of the pH, which might also alter the function of endosomal carrier vesicles (8, 23), complicated the interpretation of results on internalization of HRV2, which were based on infection, because the low pH environment of the endosomal compartment is absolutely required for uncoating of this serotype (39, 47). Since the contribution of clathrin-mediated endocytosis to HRV2 entry was not established in the previous study by Huber et al., we decided to reevaluate this issue by using in part approaches which have already been used before but also new methods to block coated-pit internalization, with a special focus on confocal immunofluorescence microscopy. We found that K+ depletion substantially impairs HRV2 uptake. We show that dynK44A expression and cholesterol depletion, treatments that inhibit clathrin-mediated endocytosis, strongly reduce HRV2 internalization. We also show that HRV2 bound to the surface of HeLa cells is localized in coated pits but not in caveolae. Finally, transient overexpression of the specific dominant-negative inhibitors of clathrin-mediated endocytosis, the SH3 domain of amphiphysin (Amph-SH3) and the C-terminal domain of AP-180 (AP180-C), resulted in a potent block of internalization of HRV2. These results indicate that HRV2 uses clathrin-mediated endocytosis for infection.
MATERIALS AND METHODS
Materials.
All chemicals were purchased from Sigma (St. Louis, Mo.) unless otherwise specified. Tissue culture media, supplements, and Lipofectamine were obtained from Invitrogen (Carlsbad, Calif.). Tissue culture plates and flasks were obtained from Costar (Cambridge, Mass.). Bafilomycin A1 (Alexis Biochemicals, San Diego, Calif.) was dissolved in dimethyl sulfoxide at 200 μM and stored at −20°C. The final concentration of dimethyl sulfoxide was kept below 1%. Alexa Fluor secondary antibodies and rhodamine-conjugated transferrin (dissolved in phosphate-buffered saline [PBS] at 5 mg/ml) were obtained from Molecular Probes (Eugene, Oreg.). Horseradish peroxidase-conjugated goat anti-chicken antibodies were obtained from Jackson Laboratories (West Baltimore, Pa.).
Vector pEGFP-C3 containing the clathrin light chain fused to green fluorescent protein (GFP-clathrin) (70) was a kind gift of Lois E. Greene (Laboratory of Cell Biology, Bethesda, Md.). Plasmid pCMV-Amph-SH3 was kindly provided by Harvey T. McMahon (Laboratory of Molecular Biology, Cambridge, United Kingdom).
Rabbit antiserum directed against the hemagglutinin (HA) tag (HA-probe, Y-11) and against the myc tag were obtained from Santa Cruz Biotechnology (Santa Cruz, Calif.) and Upstate Biotechnology (Lake Placid, N.Y.), respectively. Rabbit polyclonal antibody directed against caveolin-1 was obtained from Transduction Laboratories (Lexington, Ky.). Monoclonal antibody 8F5 (2.2 mg/ml) against HRV2 (57) was prepared in our laboratory.
Cells and viruses.
HeLa-H1 cells (Flow Laboratories) were grown in monolayers in minimal essential medium (MEM) containing 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). For HRV2 internalization into HeLa cells, MEM supplemented with 2% FCS, l-glutamine, antibiotics, and 30 mM MgCl2 (MEM infection medium) was used.
HeLa cells stably expressing dynK44A and wild-type dynamin (dynwt) under the control of the tetracycline-regulated promoter were kindly supplied by S. L. Schmid. The cells were cultivated in Dulbecco's MEM with high glucose, l-glutamine, and sodium pyruvate supplemented with 10% heat-inactivated FCS, 400 μg of G418/ml, 200 ng of puromycin/ml, and 1 μg of tetracycline/ml. Cells were plated on coverslips in six-well plates in the absence of tetracycline for 48 h for induction. The cells were 60% confluent where used.
Constructs and transfections.
A rat brain marathon-ready cDNA library (Clontech, Palo Alto, Calif.) was used to amplify a DNA fragment encoding the C terminus of AP180 (amino acids 530 to 915 [18]). The primers used were GAATTCGCTGCCACCACCACCGCCGCCG and GAATTCTTACAAGAAATCCTTGATGTTAAGATCCG. This fragment was inserted into eukaryotic expression vector pCI (Promega, Madison, Wis.), which had been modified to allow fusion with a myc tag, using an EcoRI site. The same procedure was used to produce the myc-tagged N-terminal domain of AP180 (amino acids 1 to 289).
A cDNA corresponding to canine rab5 (7) was amplified from an MDCK cDNA library (a kind gift of I. Fialka and L. Huber, Institute of Molecular Pathology, Vienna, Austria) and cloned as a myc-tag fusion protein in the same modified pCI vector. The Ser34 → Asn mutation was introduced by PCR using primer elongation.
Internalization of HRV2 and of fluorescent-transferrin conjugates.
Cells were grown on coverslips and transfected with plasmids encoding the dominant-negative mutants 2 days before the experiment. They were then incubated with virus (∼30 PFU per cell) for 20 min at 34°C in MEM infection medium, washed with PBS, and prepared for immunofluorescence. For cointernalization of HRV2 and fluorescent transferrin, cells were preincubated with MEM without FCS at 34°C for 30 min. The medium was replaced by MEM infection medium without FCS and containing 5 μg of rhodamine-transferrin/ml together with HRV2. Incubation was at 34°C for 20 min, followed by three washes with ice-cold PBS prior to fixation.
Fluorescence microscopy.
Cells were fixed in 3% paraformaldehyde in PBS for 15 min at room temperature, quenched for 10 min in 50 mM NH4Cl in PBS, washed three times with PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min, washed three times, and blocked with 5% FCS in PBS for 15 min. Incubation with primary antibodies (8F5 or HA-probe) diluted 1:200 in PBS containing 1% FCS was performed for 1 h at room temperature. Cells were washed with PBS and incubated for 1 h at room temperature with secondary antibodies diluted 1:400 in PBS containing 1% FCS. The coverslips were washed three times with PBS and rinsed briefly in double-distilled H2O, and the cells were mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, Calif.). Samples were viewed under a Leica TCS NT confocal microscope (Heidelberg, Germany). Images were processed using Adobe Photoshop software.
Potassium depletion.
HeLa-H1 cells were washed with K+-free buffer (140 mM NaCl, 20 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, 1 mg of d-glucose/ml [pH 7.4]), incubated with K+-free buffer diluted 1:1 with water (hypotonic buffer) for 5 min, washed three times with K+-free buffer, and incubated with HRV2 for 20 min at 34°C in the same buffer prior to fixation and immunofluorescence. As a control, the same buffers containing 10 mM KCl were used.
Cholesterol depletion and virus-uncoating assay.
HeLa-H1 cells grown in 24-well plates were preincubated with 10 mM methyl-β-cyclodextrin (MβCD) in MEM infection medium at 34°C for 30 min. For control purposes, cells were also incubated without any addition of MβCD or in the presence of 400 nM bafilomycin A1. HRV2 (∼30 PFU per cell) was added, and the incubation was continued for 20 min in the absence or presence of the drugs. After being washed three times with PBS, samples were taken immediately (T = 0) or after further incubation for the specified time periods prior to determination of the virus titer.
Virus titer determination.
Infected cells were broken by three freeze-thaw cycles, debris was removed by a brief centrifugation, and serial 10-fold dilutions of the supernatants were prepared in MEM infection medium. Samples were transferred onto subconfluent monolayers of HeLa-H1 cells grown in 96-well culture plates containing 100 μl of MEM infection medium. After incubation at 34°C for 5 days, cells were stained with 0.1% crystal violet (in water) for 20 min. The tissue culture infective dose which infects 50% of the cells (TCID50) was calculated according to the method of Blake and O'Connell (5).
RESULTS
Because previous reports have suggested internalization of HRV2 via a clathrin-independent pathway (3, 23, 34) we initially decided to study the underlying mechanism by asking whether LDLRs lacking the coated-pit localization signal were able to mediate viral infection.
The full-length human LDLR or a mutant lacking 33 amino acids at the C terminus, including the clathrin-coated-pit-targeting signal NPVY, were stably transfected into a fibroblast cell line (M4) deficient in LDLR and LRP expression as a result of gene disruption (21, 24, 68). Unfortunately, the expression level of the truncated LDLR was much higher than that of wild-type LDLR in all clones examined, precluding a strict comparison of the infection efficiencies mediated by the two receptors. Nevertheless, we observed only small differences in infectivity by HRV2 between these clones, which did not match the very large difference in receptor expression (results not shown); this suggested that the full-length receptor is considerably more efficient in mediating infection.
Although these results did not allow the drawing of a formal conclusion, we were concerned that the clathrin-mediated endocytosis pathway might be more important for HRV2 infection than previously estimated and decided to proceed to a reevaluation of this topic.
Hypotonic shock followed by K+ depletion strongly inhibits HRV2 internalization.
Previous experiments using K+ depletion to dissociate clathrin coats from the plasma membrane (31, 34) have suggested that HRV2 infectivity was not diminished by this treatment. The authors of the latter report took the inhibition of total protein synthesis as an indication of virus-induced host cell shutoff. However, K+ depletion can have pleiotropic consequences on cell metabolism (3), and these results need not necessarily reflect an effect on viral entry. This prompted us to directly examine the effect of K+ depletion on HRV2 entry by using immunofluorescence and confocal microscopy.
HeLa-H1 cells were incubated in hypotonic K+-free buffer for 5 min prior to challenge with HRV2 at ∼30 PFU per cell in isotonic K+-free buffer for 20 min. As a control, cells were treated with the same buffers, but containing 10 mM KCl. Subsequently, cells were fixed, prepared for immunofluorescence detection of HRV2 by use of the monoclonal antibody 8F5, and examined by confocal microscopy. In all control cells, HRV2 accumulated in perinuclear vesicles, presumably early and late endosomes, whereas in K+-depleted cells, intracellular virus-specific fluorescence was dramatically reduced (Fig. 1). In ∼80% of the K+-depleted cells, staining was seen only on the plasma membrane, most probably representing surface-bound virus which failed to penetrate into the cells, while in the remainder of the cells, virus was found in peripheral or scattered vesicles, without perinuclear accumulation. We observed the same inhibition of viral entry when K+ depletion was performed on A431 carcinoma cells (data not shown).
FIG. 1.

K+ depletion inhibits HRV2 internalization. HeLa-H1 cells seeded on coverslips were subjected to hypotonic shock for 5 min, washed, and incubated with HRV2 in K+-free medium for 20 min at 34°C. Control cells were treated with the same buffers containing 10 mM K+. Cells were fixed, and HRV2 was visualized by using the anti-HRV2 monoclonal antibody 8F5. Preparations were photographed by using a confocal microscope. Noninfected cells did not show any staining with 8F5. Bar, 10 μm.
These results demonstrate that K+ depletion in fact strongly inhibits HRV2 endocytosis and underscores the importance of using confocal immunofluorescence microscopy to directly examine the effect of the treatment on viral entry. Measurement of infectivity might be misleading, as K+ depletion does not inhibit internalization completely and virus spreads rapidly from the infected cells to other cells. Moreover, virus particles bound to the plasma membrane cannot be removed entirely (3). Therefore, these are still able to infect after cessation of the treatment with K+-free buffer. Attempts to dissociate virus from the plasma membrane by exposure to low pH might even result in uncoating, penetration of the RNA into the cytosol, and infection (5a).
HRV2 internalization is inhibited by cholesterol depletion.
Next, we examined the effect of cholesterol depletion on the internalization of HRV2. Depletion of cholesterol from the plasma membrane by using MβCD has been shown to impair clathrin-mediated endocytosis (48, 60). MβCD strongly reduces endocytosis of transferrin and epidermal growth factor (EGF), whereas endocytosis of ricin, a general membrane marker, is less affected (48). Therefore, we anticipated that clathrin-mediated uptake of HRV2, as suggested by the K+ depletion experiment, would be inhibited by MβCD, keeping in mind that the effect of this treatment is not entirely specific because cholesterol depletion can also affect caveolae (63, 71).
In the next experiment, we assessed internalization by measuring the loss of infectivity of cell-associated virus as a result of uncoating in endosomes. The low endosomal pH results in inactivation of the infecting virus as a consequence of conformational change and expulsion of its RNA. The extent of this reaction depends on the incubation time (up to about 3 h); during this period, de novo-synthesized infectious particles were not detected.
HeLa-H1 cells were treated with MβCD for 30 min prior to incubation with HRV2 for 20 min at 34°C to allow internalization; cells were washed, and fresh medium was added (T = 0). After 0, 1, and 3 h of incubation at 34°C, cells were collected and cell-associated virus was released by freeze-thawing prior to determination of the number of infectious virions. In untreated cells, we consistently found that the virus titer dropped by about 2 orders of magnitude from 0 to 3 h of incubation, whereas in the presence of bafilomycin A1, a drug preventing acidification of endosomes (4, 47), the titer remained almost constant (Fig. 2A). When cells were pretreated with MβCD, the virus titer decreased much less with time compared with what was seen with untreated cells. This result is consistent with reduced internalization of the virus as a consequence of impaired clathrin-mediated endocytosis.
FIG. 2.

Cholesterol depletion inhibits internalization of HRV2. (A) HeLa-H1 cells in 24-well plates were incubated for 30 min at 34°C with 400 nM bafilomycin (white) or 10 mM MβCD (black) or left untreated (gray). Cells were challenged with HRV2 at 30 PFU/cell for 20 min, washed three times with PBS, and incubated in fresh medium containing the respective drugs for 0, 1, and 3 h at 34°C prior to determination of virus titer, as described in Materials and Methods. Shown are the means and standard errors of two independent experiments. (B) Cells were treated for 30 min with 10 mM MβCD or left untreated as indicated. HRV2 was added, and incubation was continued for 20 min prior to fixation, permeabilization, and immunofluorescence detection of virus. Representative confocal images are shown. Virus is retained on the plasma membrane of MβCD-treated cells. Note that the cells tend to round up as a consequence of the MβCD treatment. Bar, 10 μm.
To confirm that endocytosis of HRV2 is inhibited in MβCD-treated cells, the localization of cell-associated virus was examined by confocal fluorescence microscopy. As depicted in Fig. 2B, HRV2 was found predominantly at the plasma membrane of MβCD-treated cells, in contrast to untreated cells, where the virus was entirely intracellular and localized mainly to the perinuclear region. Membrane-associated virus was seen in most of the MβCD-treated cells; however, some cells also showed intracellular staining concomitant with plasma membrane staining. Plasma membrane staining was also observed upon incubation of MβCD-treated cells with fluorescent transferrin, as reported by Wu and colleagues (70), or with fluorescent LDL, indicating that the drug effectively impaired receptor-mediated endocytosis; these ligands, similar to HRV2, showed some intracellular staining in a small number of cells (data not shown). HRV2-associated fluorescence in the cytoplasm was then quantified by using TCNT Leica software. Using confocal pictures taken with identical parameters of more than 50 cells subjected to the various treatments, an area excluding the plasma membrane was delineated; the mean fluorescence intensity, corrected for background (areas without cells), of each pixel in these areas was recorded and was found to be 21.4 ± 6 intensity units for untreated cells and 7.8 ± 3 intensity units for MβCD-treated cells.
It is noteworthy that a reduction of internalization of Lipoplex transfection complexes upon cholesterol depletion has been reported; these complexes are taken up by clathrin-mediated endocytosis (71). The authors of this previous study observed plasma membrane staining by the labeled complexes in cholesterol-depleted cells by using confocal immunofluorescence, similar to our finding for HRV2 (Fig. 2B).
Taken together, the results of the cholesterol depletion experiments support the view that HRV2 is taken up via clathrin-coated pits; however, if we did not have additional evidence, this assay would not formally prove that clathrin-mediated endocytosis is used by the virus.
Internalization of HRV2 is markedly reduced in HeLa cells expressing dynK44A.
The results presented above, suggesting that HRV2 internalization is dependent on the clathrin-mediated pathway, imply that HRV2 internalization should also depend on functional dynamin. This protein is essential for clathrin-coated-pit scission from the plasma membrane, and a dominant-negative inhibitor, GTPase-deficient dynK44A, has been shown to effectively block endocytosis of ligands such as transferrin and EGF (10); however, dynK44A expression can also affect other pathways (20, 42).
HeLa cells stably transfected with dynK44A or dynwt under the control of the tTa-responsive promoter (12) were used for the following experiments. Induction takes place upon removal of tetracycline from the medium 2 days prior to the experiment. Yet, examining dynK44A or dynwt expression by immunofluorescence, we found that after induction only 60 to 80% of the cells express the recombinant dynamins; this allowed us to observe cells with no expression together with cells expressing dynK44A or dynwt. Using confocal immunofluorescence microscopy, we observed a strong inhibition of HRV2 internalization in the cells expressing dynK44A after 20 min of incubation with HRV2 (Fig. 3A). This block of internalization was observed in all (>95%) dynK44A-expressing cells examined. In these cells, virus was found to be exclusively associated with the plasma membrane, whereas it was entirely in the perinuclear region in nontransfected cells or cells expressing dynwt.
FIG.3.

Overexpression of dynK44A inhibits HRV2 uptake in stably transformed HeLa cells. (A) dynK44A or dynwt expression was induced in HeLa cells by incubation without tetracycline. After 48 h, cells were incubated with HRV2 (∼30 PFU/cell) for 20 min at 34°C, fixed, and stained for confocal immunofluorescence detection of virus and dynamin mutants. Cells expressing dynK44A show strongly reduced virus uptake. In these cells, virus clearly accumulates on the plasma membrane. Cells expressing dynwt internalize virus-like untransfected cells. Bar, 10 μm. (B) Cells induced to express HA-tagged dynK44A were incubated with HRV2 and rhodamine-transferrin for 20 min at 34°C, washed, fixed, and stained for immunofluorescence by using anti-HRV2 and anti-HA antibodies. HRV2 (green) and transferrin (red) internalization are blocked in cells overexpressing dynK44A. Note the colocalization (yellow) of virus and transferrin, presumably in early endosomes of the cell that does not express dynK44A. Bar, 10 μm. (C) Cells induced to express HA-dynK44A or HA-dynwt were infected with HRV2 (∼15 PFU/cell) for 20 min, washed, and incubated overnight at 34°C before immunofluorescence detection of virus-producing cells and of the HA tag. For each experiment, a total of ∼250 cells expressing the respective dynamins was observed and the fraction of cells synthesizing virus was determined (black bars). This was divided by the fraction of virus-producing cells that did not express dynamin (white bars).
These data contrast with the results of previous reports indicating dynamin-independent endocytosis of HRV2 (3, 23). Reasons for the apparent discrepancy may include subtle differences in methodology, differences in the expression levels achieved, or the concentration of the virus used. HRV2 particles bound to the plasma membrane of dynK44A-expressing cells may have also become trapped in clathrin-coated tubes induced by the mutant (11), as has been shown for influenza virus (49); this might be taken for internalized virus.
We also performed cointernalization of HRV2 and fluorescent transferrin in HeLa cells induced to express dynK44A. As shown in Fig. 3B, there was a block of transferrin as well as of HRV2 endocytosis in dynK44A-positive cells, indicating that clathrin-mediated endocytosis was effectively impaired in these cells. Identical results were obtained when fluorescent LDL and HRV2 were coincubated with the cells (data not shown). In addition, throughout this study, we observed colocalization of transferrin and HRV2 in endosomes each time both markers were used simultaneously, as, for instance, in the cell that does not express dynK44A shown in Fig. 3B.
Next, using immunofluorescence microscopy, we compared infection efficiencies for cells expressing dynK44A and dynwt by counting cells that produced virus 1 day after infection. Figure 3C shows that there was a significantly smaller number of virus-producing cells among dynK44A-positive cells than among nontransfected cells or cells expressing dynwt. The values obtained in this experiment are very similar to those reported for Hantaan virus using the same cells; this virus enters cells by clathrin-dependent receptor-mediated endocytosis (25).
In conclusion, our results demonstrate that HRV2 endocytosis is fully dynamin dependent. Other dynamin-dependent pathways include caveolae (20, 42) and a recently described lipid raft-dependent pathway (30); lipid rafts are very unlikely to mediate HRV2 uptake for several reasons, which are delineated below. Therefore, we believe that the block of HRV2 internalization by dynK44A most probably reflects the effect of this mutant on clathrin-mediated endocytosis.
Colocalization of HRV2 and GFP-clathrin light-chain A on the plasma membrane.
To examine whether membrane-bound virus would be localized in clathrin-coated pits, we labeled these structures by using a GFP-tagged clathrin light-chain A. This fusion protein has been shown to specifically target coated pits in the plasma membrane (70). Two days after transfection, cells were incubated with HRV2 for 1 h at 4°C, fixed, and processed for immunofluorescence detection of viral particles. Cells expressing GFP-clathrin were examined by confocal microscopy (Fig. 4A). A large proportion of fluorescent dots corresponding to virus on the plasma membrane was found to colocalize with GFP-clathrin.
FIG. 4.

Membrane-bound HRV2 is concentrated in coated pits. (A) HeLa-H1 cells were transfected with a GFP-clathrin light-chain A expression plasmid to label coated pits. Two days after transfection, they were incubated with about 30 PFU of HRV2/cell at 4°C for 1 h and processed for confocal immunofluorescence detection of HRV2. Images show the colocalization (yellow) of HRV2 (red) and GFP-clathrin (green) at the plasma membrane of a cell. (B) Same as in panel A, except that cells were incubated for 30 min with 10 mM MβCD prior to incubation with HRV2 for 20 min at 34°C. Note that cells appear rounded due to the action of MβCD. (C) Nontransfected HeLa-H1 cells were incubated with HRV2 for 1 h at 4°C, fixed, permeabilized, and stained for immunofluorescence detection of HRV2 (red) and caveolin-1 (green). Bars, 10 μm.
Colocalization of HRV2 and GFP-clathrin could be observed even after cholesterol depletion by MβCD (Fig. 4B), which flattens the pits but does not remove them from the membrane (48, 60, 70). In about 15 fields examined, we consistently found that 60 to 80% of plasma membrane-bound HRV2 colocalized with GFP-clathrin, suggesting association with coated pits. When plasma membrane-bound HRV2 was compared with endogenous caveolin-1 in HeLa cells, there was a complete lack of correlation between the two markers, indicating that an involvement of caveolae in HRV2 internalization is very unlikely (Fig. 4C). A further indication that caveolae (or lipid rafts) were not involved was the lack of association of HRV2 with detergent-insoluble fractions containing the lipid domains in a sucrose density gradient flotation assay used to isolate Triton X-100-resistant membranes (results not shown). It is also notable that LDLR family members are not present in these domains (19, 64).
HRV2 internalization is blocked by transient overexpression of two dominant-negative inhibitors of clathrin-mediated endocytosis.
Because MβCD and dynK44A are not entirely specific inhibitors of clathrin-mediated endocytosis, we also used two mutant proteins with known specific dominant-negative effect on the clathrin-mediated pathway.
The first mutant is myc-tagged Amph-SH3, which was shown to induce a potent blockade of receptor-mediated endocytosis of transferrin and EGF (43, 56, 67). Amph-SH3 was transiently expressed in HeLa-H1 cells. Two days after transfection, cells were incubated for 20 min with HRV2, fixed, and processed for immunofluorescence detection of virus and the myc-tag epitope. Examination of cells by using confocal microscopy revealed the strong inhibitory effect of Amph-SH3 overexpression on HRV2 endocytosis (Fig. 5). As expected for a dominant-negative mutant, which acts by overwhelming the wild-type protein to prevent its function, the effect was dependent on the expression level; a full inhibition of virus uptake indicated by the absence of perinuclear and cytoplasmic staining was usually observed at moderate and high expression levels of Amph-SH3. In the latter case, plasma membrane staining indicated retention of the virus on the surface of the cells (Fig. 5).
FIG. 5.

HRV2 internalization is blocked by transient overexpression of Amph-SH3. HeLa-H1 cells seeded on coverslips were transfected with an expression plasmid encoding myc-tagged Amph-SH3. Two days after transfection, they were incubated with HRV2, fixed, and permeabilized. HRV2 and Amph-SH3 were detected by using anti-HRV2 monoclonal antibody 8F5 and anti-myc antiserum, respectively, followed by fluorescent conjugates. Cells were examined and photographed by using a confocal microscope. In cells overexpressing Amph-SH3, HRV2 does not accumulate in the perinuclear compartment or in the cytoplasm but remains attached to the plasma membrane. Bar, 10 μM.
Amphiphysin interacts with the AP-2 adaptor complex at its N terminus and with the proline-rich domain of dynamin at its C-terminal SH3 domain. Amph-SH3 presumably prevents recruitment of dynamin to clathrin-coated pits and also interferes with dynamin ring formation (43). Therefore, we cannot exclude the possibility that Amph-SH3 also affects other dynamin-dependent but clathrin-independent endocytosis pathways, although to our knowledge no data indicating this have been presented.
To target even more specifically the clathrin-mediated endocytosis pathway, we used another, recently described, dominant-negative inhibitor, AP180-C, which has also been used in other studies to block this pathway (40). AP180 has a phosphatidylinositol-4,5-P2 binding domain at its N terminus and interacts with the clathrin heavy chain at its C terminus through several clathrin-binding motifs (13, 18). It is believed to induce the nucleation of clathrin lattices on the plasma membrane. This process is impaired by overexpression of AP180-C, and as a consequence, clathrin is redistributed in the cells and the number of coated pits is drastically reduced (18).
The cDNA corresponding to AP180-C was amplified from a rat brain cDNA library and cloned as an N terminally myc-tagged fusion protein in a eukaryotic expression vector. AP180-C was then expressed transiently in HeLa-H1 cells. As a control, 2 days after transfection, cells were incubated with fluorescent transferrin for 20 min prior to fixation and immunofluorescence detection of myc-tagged AP180-C. As expected, expression of the truncated protein effectively blocked transferrin uptake (Fig. 6A). We then examined internalization of HRV2 (Fig. 6B). Like transferrin internalization, HRV2 endocytosis was strongly inhibited in cells expressing AP180-C, and as observed for Amph-SH3, inhibition of HRV2 internalization in many cells was accompanied by retention of the virus on the plasma membrane (Fig. 6B); this membrane staining demonstrates that viral receptors are still present on the cells. Therefore, although virus can bind to the surface of cells expressing AP180-C, there is no clathrin-independent mechanism that would promote its access to the endocytic compartment. Next, we transfected cells with the N-terminal domain of AP180 (AP180-N), which, as previously reported, does not inhibit transferrin internalization (18) (results not shown). Accordingly, even a strong expression of AP180-N did not affect HRV2 uptake (Fig. 6C), showing that the inhibition observed for AP180-C is specific for this mutant and also ruling out a general blockade of endocytosis caused by the overexpression of a recombinant protein in HeLa-H1 cells.
FIG. 6.

HRV2 internalization is blocked by transient overexpression of AP180-C. (A to C) HeLa-H1 cells seeded on coverslips were transfected with an expression plasmid encoding myc-tagged AP180-C (A and B) or AP180-N (C). Two days after transfection, they were incubated with fluorescent transferrin (A) or HRV2 (B and C), fixed, and permeabilized. HRV2 and AP180-C or AP180-N were detected with anti-HRV2 monoclonal antibody 8F5 and anti-myc antiserum, respectively, followed by fluorescent conjugates. Cells were examined and photographed by using a confocal microscope. In cells overexpressing AP180-C, transferrin and HRV2 internalization are blocked. In cells overexpressing AP180-N, HRV2 internalization is not blocked. Bar, 10 μM. (D) AP180-C expression was rated by visual inspection as being low, medium, or high in fluorescence intensity. The percentage of cells in each group without any cytoplasmic (perinuclear as well as peripheral) fluorescence is given for transferrin and HRV2 (>75 cells/group; mean of three experiments ± standard deviation).
For both virus and transferrin, the extent of inhibition of internalization was dependent on the expression level of AP180-C. To quantify this effect, we visually evaluated the degree of AP180-C expression in transfected cells by comparing the fluorescence intensities in the cytoplasm. Then, cells were divided into three groups according to low, medium, or high expression, and in each group, the percentage of cells that did not show any perinuclear or cytoplasmic fluorescence from internalized transferrin or HRV2 was determined. Figure 6D shows that the values obtained for virus and for transferrin depend almost identically on the expression level of AP180-C. Importantly, practically all cells with medium or high AP180-C expression lacked the characteristic perinuclear accumulation of HRV2 in HeLa cells.
Finally, we examined the dependence of HRV2 endocytosis on functional Rab5. This small GTPase is broadly involved in the regulation of vesicular trafficking of early endosomes and probably also of receptor-mediated endocytosis (37, 69). HeLa-H1 cells were transfected with the dominant-negative mutant of Rab5, Rab5 S34N, which preferentially binds GDP and inhibits endosome fusion (58). HRV2 internalization was examined by immunofluorescence. In all cells expressing Rab5 S34N (Fig. 7), we observed a substantial reduction of virus internalization compared with what was seen with nontransfected cells, where fluorescence was stronger and perinuclear. There was also some accumulation of virus on the plasma membrane of some transfected cells. HRV2 internalization was reduced but not abolished; in general, very small vesicles containing the virus were detected. In accord with the data reported by Stenmark and colleagues (58), control experiments with transferrin showed a reduced diffuse staining and occasional accumulation of the ligand at the plasma membrane. Overexpression of wild-type Rab5 resulted in expanded early endosomes in which HRV2 as well as transferrin were found to accumulate (data not shown). The influence of the Rab5 S34N mutant on HRV2 endocytosis indicates trafficking of the virus through a Rab5-dependent early endosomal compartment such as that involved in the receptor-mediated endocytosis pathway.
FIG. 7.
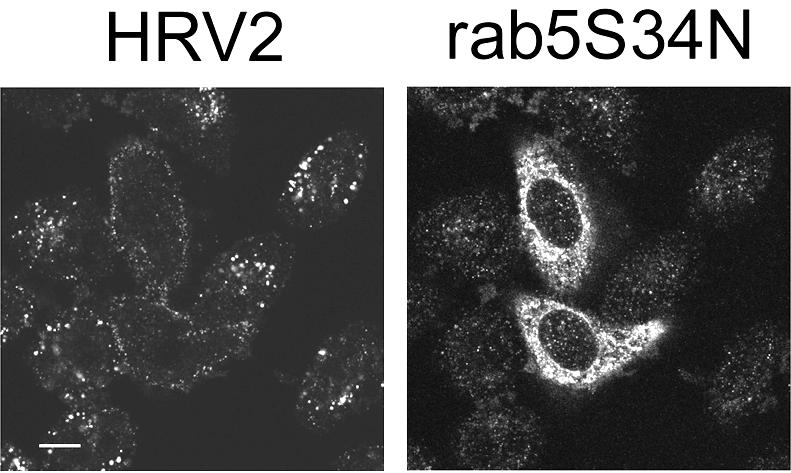
Rab5 S34N expression reduces uptake of HRV2. HeLa-H1 cells seeded on coverslips were transfected with an expression plasmid encoding myc-tagged Rab5 S34N. Two days after transfection, they were incubated with HRV2, fixed, and permeabilized. HRV2 and Rab5 S34N were detected by using anti-HRV2 monoclonal antibody and anti-myc antiserum, respectively, and fluorescent conjugates. They were examined and photographed by using a confocal microscope. In cells expressing Rab5 S34N, HRV2 does not accumulate in the perinuclear compartment but is retained, at least in part, on the plasma membrane. Bar, 10 μM.
DISCUSSION
All the data presented in this study strongly suggest that HRV2 penetrates via clathrin-mediated endocytosis, although this assertion is very difficult to prove formally because inhibitors or dominant-negative mutant proteins do not always completely block coated-pit formation or internalization. Nevertheless, the complete lack of HRV2-associated cytoplasmic staining in most of the K+-depleted cells or in cells overexpressing AP180-C or Amph-SH3 indicates that viral particles cannot enter the cells in the absence of functional clathrin-mediated endocytosis, at least during an incubation time of 20 to 30 min. Under control conditions, this time is largely sufficient for the virus to initiate infection. On the other hand, virus is clearly detectable on the surface of the cells, indicating that binding to the cells still occurs.
The very short time needed for HRV2 to be internalized and to infect cells is fully consistent with the rapid kinetics of coated-pit endocytosis; this is in marked contrast to the slower internalization process of ligands and viruses using caveolae. Notably, simian virus 40 (1), echovirus 1 (35), and filoviruses (17), which infect cells via the caveolar system, have a slow kinetics of internalization and require a minimum of 1.5 to 2 h to initiate infection. Our uncoating assay (Fig. 2) indicated that most of the HRV2 accomplished the pH-dependent uncoating within less than 1 h at 34°C. Along the same line, the inhibition of entry by cholesterol depletion using MβCD was already apparent after 20 min of incubation with the virus (Fig. 2, T = 0); this is fully consistent with an inhibition of clathrin-mediated endocytosis by the drug. However, because depletion of an essential membrane component is a rather harsh treatment, as seen from the changes in the cellular morphology, we cannot rule out pleiotropic effects.
Another convincing element which suggests the prominence of clathrin-mediated endocytosis in HRV2 internalization is the block of internalization by expression of dynK44A. This mutant, which was previously thought to specifically block clathrin-coated-pit fission from the membrane, is now considered a rather nonspecific inhibitor of endocytosis; nevertheless, we observed a strong inhibition of HRV2 endocytosis by dynK44A, which is expected for a clathrin-dependent ligand.
It is noteworthy that all markers of the clathrin-independent endocytosis pathways described so far are intimately linked to detergent-resistant membrane fractions (40, 54). These comprise glycosylphosphatidylinositol (GPI)-anchored proteins, which are delivered either to recycling endosomes (51) or to the Golgi complex (40, 41), and the interleukin-2 receptor (30). Interestingly, direct interaction of the GPI-anchored urokinase receptor and LRP promotes internalization of this complex by clathrin-coated vesicles (9), suggesting that clathrin-mediated endocytosis becomes preponderant as soon as a receptor containing a coated pit targeting motif is involved.
In the case of HRV2, a role for lipid rafts in internalization can be excluded because virus and LDLR (and most probably also LRP) do not associate with these domains. On the other hand, a decay-accelerating factor-using, lipid raft-dependent, enterovirus 11 variant cannot infect nocodazole-treated cells (59), and this drug does not impair HRV2 infection (4).
It was recently demonstrated that LRP is internalized more rapidly than LDLR (32). This raises the question of which receptor, LDLR or LRP, is predominantly involved in HRV2 entry. Indeed, fibroblasts derived from patients genetically deficient in LDLR expression are infected almost as efficiently as wild-type cells with both receptors present (22). Both receptors possess coated-pit-targeting motifs and are internalized by clathrin-mediated endocytosis (33). Because our experiments with HeLa cells point to a clathrin-dependent entry, we believe that both receptors internalize the virus via the same route.
Data on the internalization pathway of a number of viruses have been reported. The great majority was found to enter the cells by clathrin-mediated endocytosis. These include vesicular stomatitis virus (61), Semliki Forest virus (15), Sindbis virus (6, 14), adenovirus (65), adeno-associated virus type 2 (16), canine parvovirus (45), influenza virus (49), Hantaan virus (25), foot-and-mouth disease virus (38), parechovirus 1 (27), and JC virus (46). Nevertheless, several of these studies relied only on pharmacological inhibitors or dominant-negative dynK44A, which are not entirely specific for clathrin endocytosis, and a recent report has indicated that influenza virus also enters cells via clathrin-independent pathways (55). Interestingly, canine parvovirus enters the cells by clathrin-mediated endocytosis via the transferrin receptor, a prototype receptor of this pathway (44). This is similar to the internalization of HRV2 through coated pits via LDLR family members that is described in the present study. Clathrin-coated vesicles mediate fast and efficient transfer of receptor-bound particles to an acidic endosomal compartment, a property that is highly favorable for the initiation of infection by pH-dependent viruses (2, 47). Simian virus 40, echovirus 1, filoviruses, and respiratory syncytial virus have been found to be internalized via caveolae (1, 17, 35, 66), a specificity which might be related to a requirement of these viruses to follow the intracellular routes of these organelles.
A recent review on virus entry by S. B. Sieczkarski and G. R. Whittaker underscores the great care with which data obtained using nonspecific inhibitors of clathrin-mediated endocytosis should be interpreted (54). Although we have to acknowledge some contradictions between the results of the present report and those of our previous collaborative studies (e.g., regarding the effect of dynamin mutants on HRV2 uptake), these investigations have paved the way for the characterization of HRV2 internalization (3, 23, 47); our results now establish definitely that this virus utilizes clathrin to penetrate into the host cell. Measuring the internalization of radioactive HRV2 is difficult, and interpretation of the results is complicated because it is impossible to quantitatively remove plasma membrane-bound virus; this appears to represent a substantial proportion of the total cell-associated virus when coated-pit internalization is inhibited (3; this study). Therefore, our data emphasize the importance of analyzing virus endocytosis by direct examination with a confocal microscope; this method is particularly appropriate in the case of HRV2 internalization because the rapid translocation of viral proteins to the perinuclear compartment allows easy differentiation between internalized and surface-bound virus. Finally, we complete and refine our data with the dominant-negative protein domains Amph-SH3 and AP180-C, which specifically inhibit the clathrin-dependent pathway.
In summary, the results of the present study indicate that the normal endocytosis pathway of HRV2 is clathrin dependent. This property might be related to the strict dependence of minor-group rhinoviruses on low pH for uncoating; therefore, penetration in endosomes by a very efficient pathway is likely to represent a strong benefit in the infection cycle of these viruses.
Acknowledgments
We thank Joachim Herz for the kind gift of receptor-negative M4 cells; Harvey T. McMahon and Lois Green for the plasmids encoding the Amph-SH3 domain and the clathrin-GFP fusion protein, respectively; and Sandra Schmid and Hanna Damke for the cells stably expressing dynK44A.
This work was supported by the Austrian Virology Foundation and by Austrian Science Foundation grant no. 14503-MOB.
REFERENCES
- 1.Anderson, H. A., Y. Chen, and L. C. Norkin. 1996. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 7:1825-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basak, S., and H. Turner. 1992. Infectious entry pathway for canine parvovirus. Virology 186:368-376. [DOI] [PubMed] [Google Scholar]
- 3.Bayer, N., D. Schober, M. Huttinger, D. Blaas, and R. Fuchs. 2001. Inhibition of clathrin-dependent endocytosis has multiple effects on human rhinovirus serotype 2 cell entry. J. Biol. Chem. 276:3952-3962. [DOI] [PubMed] [Google Scholar]
- 4.Bayer, N., D. Schober, E. Prchla, R. F. Murphy, D. Blaas, and R. Fuchs. 1998. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J. Virol. 72:9645-9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blake, K., and S. O'Connell. 1993. Virus culture, p. 81-122. In D. R. Harper (ed.), Virology labfax. Blackwell Scientific Publications, West Smithfield, London, United Kingdom.
- 5a.Brabec, M., G. Baravalle, D. Blaas, and R. Fuchs. 2003. Conformational changes, plasma membrane penetration, and infection by human rhinovirus type 2: role of receptors and low pH. J. Virol. 77:5370-5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carbone, R., S. Fre, G. Iannolo, F. Belleudi, P. Mancini, P. G. Pelicci, M. R. Torrisi, and P. P. DiFiore. 1997. eps15 and eps15r are essential components of the endocytic pathway. Cancer Res. 57:5498-5504. [PubMed] [Google Scholar]
- 7.Chavrier, P., R. G. Parton, H. P. Hauri, K. Simons, and M. Zerial. 1990. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 62:317-329. [DOI] [PubMed] [Google Scholar]
- 8.Clague, M. J., S. Urbe, F. Aniento, and J. Gruenberg. 1994. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J. Biol. Chem. 269:21-24. [PubMed] [Google Scholar]
- 9.Czekay, R. P., T. A. Kuemmel, R. A. Orlando, and M. G. Farquhar. 2001. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol. Biol. Cell 12:1467-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damke, H., T. Baba, D. Warnock, and S. Schmid. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127:915-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damke, H., D. D. Binns, H. Ueda, S. L. Schmid, and T. Baba. 2001. Dynamin GTPase domain mutants block endocytic vesicle formation at morphologically distinct stages. Mol. Biol. Cell 12:2578-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damke, H., M. Gossen, S. Freundlieb, H. Bujard, and S. L. Schmid. 1995. Tightly regulated and inducible expression of dominant interfering dynamin mutant in stably transformed HeLa cells. Methods Enzymol. 257:209-220. [DOI] [PubMed] [Google Scholar]
- 13.Dell'Angelica, E. C. 2001. Clathrin-binding proteins: got a motif? Join the network! Trends Cell Biol. 11:315-318. [DOI] [PubMed] [Google Scholar]
- 14.DeTulleo, L., and T. Kirchhausen. 1998. The clathrin endocytic pathway in viral infection. EMBO J. 17:4585-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doxsey, S. J., F. M. Brodsky, G. S. Blank, and A. Helenius. 1987. Inhibition of endocytosis by anti-clathrin antibodies. Cell 50:453-463. [DOI] [PubMed] [Google Scholar]
- 16.Duan, D. S., Q. Li, A. W. Kao, Y. P. Yue, J. E. Pessin, and J. F. Engelhardt. 1999. Dynamin is required for recombinant adeno-associated virus type 2 infection. J. Virol. 73:10371-10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Empig, C. J., and M. A. Goldsmith. 2002. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 76:5266-5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ford, M. G., B. M. Pearse, M. K. Higgins, Y. Vallis, D. J. Owen, A. Gibson, C. R. Hopkins, P. R. Evans, and H. T. McMahon. 2001. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291:1051-1055. [DOI] [PubMed] [Google Scholar]
- 19.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henley, J. R., E. W. A. Krueger, B. J. Oswald, and M. A. McNiven. 1998. Dynamin-mediated internalization of caveolae. J. Cell Biol. 141:85-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herz, J., D. E. Clouthier, and R. E. Hammer. 1992. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71:411-421. [DOI] [PubMed] [Google Scholar]
- 22.Hofer, F., M. Gruenberger, H. Kowalski, H. Machat, M. Huettinger, E. Kuechler, and D. Blaas. 1994. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 91:1839-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber, M., M. Brabec, N. Bayer, D. Blaas, and R. Fuchs. 2001. Elevated endosomal pH in HeLa cells overexpressing mutant dynamin can affect infection by pH-sensitive viruses. Traffic 2:727-736. [DOI] [PubMed] [Google Scholar]
- 24.Ishibashi, S., M. S. Brown, J. L. Goldstein, R. D. Gerard, R. E. Hammer, and J. Herz. 1993. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 92:883-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin, M., J. Park, S. Lee, B. Park, J. Shin, K. J. Song, T. I. Ahn, S. Y. Hwang, B. Y. Ahn, and K. Ahn. 2002. Hantaan virus enters cells by clathrin-dependent receptor-mediated endocytosis. Virology 294:60-69. [DOI] [PubMed] [Google Scholar]
- 26.Johannes, L., and C. Lamaze. 2002. Clathrin-dependent or not: is it still the question? Traffic 3:443-451. [DOI] [PubMed] [Google Scholar]
- 27.Joki-Korpela, P., V. Marjomaki, C. Krogerus, J. Heino, and T. Hyypia. 2001. Entry of human parechovirus 1. J. Virol. 75:1958-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kibbey, R. G., J. Rizo, L. M. Gierasch, and R. G. W. Anderson. 1998. The LDL receptor clustering motif interacts with the clathrin terminal domain in a reverse turn conformation. J. Cell Biol. 142:59-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirchhausen, T. 1999. Adaptors for clathrin-mediated traffic. Ann. Rev. Cell Dev. Biol. 15:709-732. [DOI] [PubMed] [Google Scholar]
- 30.Lamaze, C., A. Dujeancourt, T. Baba, C. G. Lo, A. Benmerah, and A. Dautry-Varsat. 2001. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell 7:661-671. [DOI] [PubMed] [Google Scholar]
- 31.Larkin, J. M., M. S. Brown, J. L. Goldstein, and R. G. Anderson. 1983. Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell 33:273-285. [DOI] [PubMed] [Google Scholar]
- 32.Li, Y., W. Lu, M. P. Marzolo, and G. Bu. 2001. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J. Biol. Chem. 276:18000-18006. [DOI] [PubMed] [Google Scholar]
- 33.Li, Y., M. P. Marzolo, P. van Kerkhof, G. J. Strous, and G. J. Bu. 2000. The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J. Biol. Chem. 275:17187-17194. [DOI] [PubMed] [Google Scholar]
- 34.Madshus, I. H., K. Sandvig, S. Olsnes, and B. van Deurs. 1987. Effect of reduced endocytosis induced by hypotonic shock and potassium depletion on the infection of Hep 2 cells by picornaviruses. J. Cell. Physiol. 131:14-22. [DOI] [PubMed] [Google Scholar]
- 35.Marjomaki, V., V. Pietiainen, H. Matilainen, P. Upla, J. Ivaska, L. Nissinen, H. Reunanen, P. Huttunen, T. Hyypia, and J. Heino. 2002. Internalization of echovirus 1 in caveolae. J. Virol. 76:1856-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marlovits, T. C., C. Abrahamsberg, and D. Blaas. 1998. Very-low-density lipoprotein receptor fragment shed from HeLa cells inhibits human rhinovirus infection. J. Virol. 72:10246-10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLauchlan, H., J. Newell, N. Morrice, A. Osborne, M. West, and E. Smythe. 1998. A novel role for Rab5-GDI in ligand sequestration into clathrin-coated pits. Curr. Biol. 8:34-45. [DOI] [PubMed] [Google Scholar]
- 38.Miller, L. C., W. Blakemore, D. Sheppard, A. Atakilit, A. M. King, and T. Jackson. 2001. Role of the cytoplasmic domain of the β-subunit of integrin α5β6 in infection by foot-and-mouth disease virus. J. Virol. 75:4158-4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neubauer, C., L. Frasel, E. Kuechler, and D. Blaas. 1987. Mechanism of entry of human rhinovirus 2 into HeLa cells. Virology 158:255-258. [DOI] [PubMed] [Google Scholar]
- 40.Nichols, B. J. 2002. A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 4:374-378. [DOI] [PubMed] [Google Scholar]
- 41.Nichols, B. J., and J. Lippincott-Schwartz. 2001. Endocytosis without clathrin coats. Trends Cell Biol. 11:406-412. [DOI] [PubMed] [Google Scholar]
- 42.Oh, P., D. P. McIntosh, and J. E. Schnitzer. 1998. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J. Cell Biol. 141:101-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Owen, D. J., P. Wigge, Y. Vallis, J. D. A. Moore, P. R. Evans, and H. T. McMahon. 1998. Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 17:5273-5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parker, J. S., W. J. Murphy, D. Wang, S. J. O'Brien, and C. R. Parrish. 2001. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 75:3896-3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker, J. S., and C. R. Parrish. 2000. Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J. Virol. 74:1919-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pho, M. T., A. Ashok, and W. J. Atwood. 2000. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 74:2288-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prchla, E., E. Kuechler, D. Blaas, and R. Fuchs. 1994. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 68:3713-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodal, S. K., G. Skretting, O. Garred, F. Vilhardt, B. van Deurs, and K. Sandvig. 1999. Extraction of cholesterol with methyl-β-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell 10:961-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy, A. M. M., J. S. Parker, C. R. Parrish, and G. R. Whittaker. 2000. Early stages of influenza virus entry into Mv-1 lung cells: involvement of dynamin. Virology 267:17-28. [DOI] [PubMed] [Google Scholar]
- 50.Rudenko, G., L. Henry, K. Henderson, K. Ichtchenko, M. S. Brown, J. L. Goldstein, and J. Deisenhofer. 2002. Structure of the LDL receptor extracellular domain at endosomal pH. Science 298:2353-2358. [DOI] [PubMed] [Google Scholar]
- 51.Sabharanjak, S., P. Sharma, R. G. Parton, and S. Mayor. 2002. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2:411-423. [DOI] [PubMed] [Google Scholar]
- 52.Schober, D., P. Kronenberger, E. Prchla, D. Blaas, and R. Fuchs. 1998. Major and minor-receptor group human rhinoviruses penetrate from endosomes by different mechanisms. J. Virol. 72:1354-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sever, S., H. Damke, and S. L. Schmid. 2000. Dynamin:GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol. 150:1137-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sieczkarski, S. B., and G. R. Whittaker. 2002. Dissecting virus entry via endocytosis. J. Gen. Virol. 83:1535-1545. [DOI] [PubMed] [Google Scholar]
- 55.Sieczkarski, S. B., and G. R. Whittaker. 2002. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 76:10455-10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson, F., N. K. Hussain, B. Qualmann, R. B. Kelly, B. K. Kay, P. S. McPherson, and S. L. Schmid. 1999. SH3-domain-containing proteins function at distinct steps in clathrin-coated vesicle formation. Nat. Cell Biol. 1:119-124. [DOI] [PubMed] [Google Scholar]
- 57.Skern, T., C. Neubauer, L. Frasel, P. Gruendler, W. Sommergruber, W. Zorn, E. Kuechler, and D. Blaas. 1987. A neutralizing epitope on human rhinovirus type 2 includes amino acid residues between 153 and 164 of virus capsid protein VP2. J. Gen. Virol. 68:315-323. [DOI] [PubMed] [Google Scholar]
- 58.Stenmark, H., R. G. Parton, O. Steelemortimer, A. Lutcke, J. Gruenberg, and M. Zerial. 1994. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 13:1287-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stuart, A. D., H. E. Eustace, T. A. McKee, and T. D. Brown. 2002. A novel cell entry pathway for a DAF-using human enterovirus is dependent on lipid rafts. J. Virol. 76:9307-9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subtil, A., I. Gaidarov, K. Kobylarz, M. A. Lampson, J. H. Keen, and T. E. McGraw. 1999. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. USA 96:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Superti, F., L. Seganti, F. M. Ruggeri, A. Tinari, G. Donelli, and N. Orsi. 1987. Entry pathway of vesicular stomatitis virus into different host cells. J. Gen. Virol. 68:387-399. [DOI] [PubMed] [Google Scholar]
- 62.Takei, K., and V. Haucke. 2001. Clathrin-mediated endocytosis: membrane factors pull the trigger. Trends Cell Biol. 11:385-391. [DOI] [PubMed] [Google Scholar]
- 63.Torgersen, M. L., G. Skretting, B. van Deurs, and K. Sandvig. 2001. Internalization of cholera toxin by different endocytic mechanisms. J. Cell Sci. 114:3737-3747. [DOI] [PubMed] [Google Scholar]
- 64.Verkade, P., T. Harder, F. Lafont, and K. Simons. 2000. Induction of caveolae in the apical plasma membrane of Madin-Darby canine kidney cells. J. Cell Biol. 148:727-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang, K. N., S. Huang, A. KapoorMunshi, and G. Nemerow. 1998. Adenovirus internalization and infection require dynamin. J. Virol. 72:3455-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Werling, D., J. C. Hope, P. Chaplin, R. A. Collins, G. Taylor, and C. J. Howard. 1999. Involvement of caveolae in the uptake of respiratory syncytial virus antigen by dendritic cells. J. Leukoc. Biol. 66:50-58. [DOI] [PubMed] [Google Scholar]
- 67.Wigge, P., K. Kohler, Y. Vallis, C. A. Doyle, D. Owen, S. P. Hunt, and H. T. McMahon. 1997. Amphiphysin heterodimers: potential role in clathrin mediated endocytosis. Mol. Biol. Cell 8:2003-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willnow, T. E., and J. Herz. 1994. Genetic deficiency in low density lipoprotein receptor-related protein confers cellular resistance to Pseudomonas exotoxin A. Evidence that this protein is required for uptake and degradation of multiple ligands. J. Cell Sci. 107:719-726. [PubMed] [Google Scholar]
- 69.Woodman, P. G. 2000. Biogenesis of the sorting endosome: the role of Rab5. Traffic 1:695-701. [DOI] [PubMed] [Google Scholar]
- 70.Wu, X., X. Zhao, L. Baylor, S. Kaushal, E. Eisenberg, and L. E. Greene. 2001. Clathrin exchange during clathrin-mediated endocytosis. J. Cell Biol. 155:291-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zuhorn, I. S., R. Kalicharan, and D. Hoekstra. 2002. Lipoplex-mediated transfection of mammalian cells occurs through the cholesterol-dependent clathrin-mediated pathway of endocytosis. J. Biol. Chem. 277:18021-18028. [DOI] [PubMed] [Google Scholar]