

Abstract
Curli are extracellular surface fibers that are produced by many members of the Enterobacteriaceae and contribute to biofilm formation. The environmental cues that promote biofilm formation are poorly understood. We found that deletion of the N-acetylglucosamine-6-phosphate (GlcNAc-6P) deacetylase gene, nagA, resulted in decreased transcription from the curli-specific promoters csgBA and csgDEFG and a corresponding decrease in curli production in Escherichia coli. nagA is in an operon that contains nagB, nagC, nagD, and nagE, whose products are required for utilization of GlcNAc as a carbon source. NagC is a repressor of the nagBACD and nagE genes in the absence of intracellular GlcNAc-6P. We found that nagC mutants were also defective in curli production. Growth of a wild-type strain on media containing additional GlcNAc reduced curli gene transcription to a level similar to the level observed when nagA was deleted. The defect in curli production in nagA or nagC mutants was alleviated by deletion of the GlcNAc transporter gene, nagE. Curli-producing ΔnagA suppressor mutants whose cells were unable to take up GlcNAc were isolated. These results suggest that elevated levels of intracellular GlcNAc-6P signal cells to down-regulate curli gene expression.
Bacteria produce a wide variety of extracellular surface structures that help them adapt to their environments. These structures include curli, pili, flagella, and capsule among others. Such structures facilitate binding to host tissues, interactions with other bacteria, secretion of molecules, and motility. Curli are produced by many members of the Enterobacteriaceae (6, 56) and are associated with biofilm formation, host cell adhesion and invasion, and immune system activation (4, 5, 22, 27, 51, 53, 56). Curli fibers also share biochemical and structural characteristics with eukaryotic amyloid fibers and therefore provide an excellent model system for understanding amyloid fiber formation.
Two divergently transcribed operons, csgBA and csgDEFG, are required for curli production in Escherichia coli (24). Nearly identical operons have been identified in Salmonella spp. (12, 47). The csgBA operon encodes the major curli subunit CsgA and the nucleator protein, CsgB. The csgDEFG operon encodes four accessory proteins that are required for curli assembly. In the absence of CsgG, the curli subunits CsgA and CsgB are unstable, and no curli are formed (30). CsgG forms barrel-shaped homo-oligomers at the outer membrane and is hypothesized to be the pore through which CsgA and CsgB are secreted (45). csgF mutants secrete soluble, unpolymerized CsgA, suggesting that CsgB is nonfunctional in the absence of csgF (11). csgE mutants assemble ∼90% fewer fibers than wild-type (Wt) strains assemble, and the steady-state levels of CsgA and CsgB are reduced (11). Both CsgE and CsgF interact with CsgG at the outer membrane (45). CsgD is a positive transcriptional regulator of the csgBA promoter, but it does not regulate the csgDEFG promoter (24, 48). CsgD has also been shown to regulate genes required for the production of cellulose (57), which along with curli makes up the extracellular matrix produced by many enteric bacteria.
Curli gene expression is regulated in response to different environmental cues (for a review see reference 21). Regulators of csgD have an indirect effect on csgBA expression because CsgD is required for csgBA transcription. Curli genes are maximally expressed during stationary phase, and csgD expression is dependent on the stationary-phase sigma factor RpoS (2). Crl is a positive regulator of curli gene expression and many other stationary-phase induced genes (34, 43). Crl and RpoS interact to promote maximal curli gene expression (7). MlrA is a positive transcriptional regulator of csgD, and its expression is dependent on RpoS (8). Three two-component regulatory systems, Rcs, CpxA/R, and OmpR/EnvZ, regulate curli gene expression. The Rcs pathway positively regulates capsule synthesis in response to changes in the cell surface and negatively regulates both curli operons (19, 26, 31, 52). The CpxA/R system, which responds to envelope stress, negatively regulates both curli operons (16, 18). The OmpR/EnvZ system responds to changes in osmolarity and positively regulates csgD expression (47, 53). Two global regulatory factors, IHF and HN-S, influence curli gene expression. IHF positively regulates csgD expression (20), and HN-S positively or negatively regulates curli gene expression depending on the genetic background of the strain (2, 20, 34).
In this study we found that curli gene expression is responsive to high levels of intracellular N-acetylglucosamine-6-phosphate (GlcNAc-6P). When high levels of GlcNAc-6P accumulate in the cell, the nag genes (nagABCDE) are activated and support utilization of GlcNAc as a carbon source (Fig. 1) (35, 41). The nag genes are transcribed divergently as nagE and nagBACD transcripts. NagE is located in the inner membrane and transports GlcNAc into the cytoplasm, where it is immediately phosphorylated by the phosphotransferase system to form GlcNAc-6P, which is converted to glucosamine-6-phosphate (GlcN-6P) by the deacetylase NagA (25, 29, 41, 46, 55). NagB deaminates GlcN-6P to form fructose, which can be used in glycolysis (41, 46, 55). NagC acts as a transcriptional repressor of the nag genes when GlcNAc is not available (36, 38, 40). A role for NagD in GlcNAc metabolism and transport has not been identified. Regulation of surface fibers by GlcNAc appears to be common because type 1 pili produced by E. coli are also regulated by intracellular GlcNAc-6P levels (50).
FIG. 1.
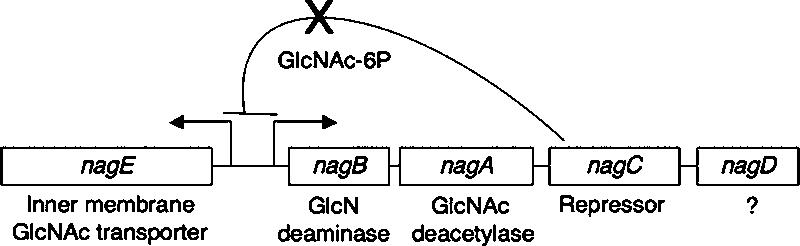
Schematic diagram the of the nag genes. nagBACD are expressed from a single transcript, and nagE is divergently transcribed. The intergenic region is 330 bp long. NagC represses transcription from the nagBACD and nagE promoters in the absence of GlcNAc-6P.
MATERIALS AND METHODS
Bacterial growth.
To induce curli production, bacteria were grown at 26°C for 48 h on YESCA plates (1 g yeast extract per liter, 10 g Casamino Acids per liter). Curli assembly was monitored by growing organisms on CR-YESCA plates (YESCA containing 50 μg/ml Congo red [CR] and 10 μg/ml Coomassie blue). To assess GlcNAc utilization, bacteria were grown on MacConkey agar plates containing 1% GlcNAc or W minimal medium plates (10.5 g K2HPO4 per liter, 4.5 g KH2PO4 per liter, 0.2 g MgSO4 · 7H2O per liter) supplemented with 0.2% NH4, 1% leucine, 1% threonine, 0.1% thiamine, and 0.4% GlcNAc or glucose. When needed, antibiotics were added to plates at the following concentrations: kanamycin, 50 μg/ml; chloramphenicol, 25 μg/ml; and ampicillin, 100 μg/ml.
Bacterial strains and plasmids.
Strains and plasmids used in this study are listed in Table 1. Deletion mutants were constructed by the method of Datsenko and Wanner (17). The primers used for deletion mutagenesis are listed in Table 2. To construct the deletion strains, the primer pairs indicated in Table 1 were used to PCR amplify the antibiotic resistance genes from pKD3 (chloramphenicol) or pKD4 (kanamycin). The PCR products were electroporated into strain MC4100 or C600 expressing the λ red recombination system from pKD46 (17). Strains MMB103 (ΔnagA), MMB173 (ΔnagAD), and SKM2 (ΔnagEA) were obtained by P1 transduction of the ΔnagA allele into strain C600, MMB161 (ΔnagD), or MMB152 (ΔnagE). Strain MMB152 (ΔnagE) was transformed with pKD46, and the ΔnagC:Knr PCR product was electroporated into it to create strain MMB185 (ΔnagEC). Strains MMB103 (ΔnagA), MMB171 (ΔnagC), and LSR6 (C600:ΔcsgBA ΔcsgDEFG) (11) were transformed with pKD46, and the ΔlacZ:Cmr PCR product was electroporated into these strains to create strains MMB1035 (ΔnagA ΔlacZ), MMB1714 (ΔnagC ΔlacZ), and MMB661 (ΔcsgBA ΔcsgDEFG ΔlacZ). All deletion mutants were checked by PCR.
TABLE 1.
Strains and plasmids used in this work
| Strain or plasmid | Relevant characteristicsa | Reference | Primersb |
|---|---|---|---|
| Strains | |||
| C600 | F−thr leu thi lac tonA | 9 | |
| MC4100 | F−araD139 Δ(argF-lac)U169 rspL150(strR) relA1 flbB5301 deoC1 ptsF25 rbsR | 10 | |
| MMB5 | MC4100 ΔnagA::Knr | This study | NagAH1P1, NagAH2P2 |
| MMB103 | C600 ΔnagA::Knr | This study | |
| MMB120 | C600 ΔnagB::Cmr | This study | NagBH1P1, NagBH1P1 |
| MMB171 | C600 ΔnagC::Knr | This study | NagCH1P1, NagCH2P2 |
| MMB161 | C600 ΔnagD::Cmr | This study | NagDH1P1, NagH1P1 |
| MMB152 | C600 ΔnagE::Cmr | This study | NagEH1P1, NagEH2P2 |
| MMB124 | C600 ΔnagAB::Cmr | This study | NagBH1P1, NagAH2P2 |
| MMB71 | C600 ΔnagAC::Cmr | This study | NagAH1P1, NagCH2P2 |
| MMB173 | MMB161 ΔnagA::Knr | This study | |
| SKM2 | MMB152 ΔnagA::Knr | This study | |
| MMB185 | MMB152 ΔnagC::Knr | This study | |
| MMB87 | C600 Δnag operon (ΔnagBACD ΔnagE::Cmr) | This study | NagH1P1, NagH2P2 |
| MMB200 | C600 ΔlacZ::Cmr | This study | LacZH1P1, LacZH2P2 |
| MMB1035 | MMB103 ΔlacZ::Cmr | This study | |
| MMB1714 | MMB171 ΔlacZ::Cmr | This study | |
| LSR6 | C600 ΔcsgBA ΔcsgDEFG::Knr | 11 | |
| MMB661 | LSR6 ΔlacZ::Cmr | This study | |
| Plasmids | |||
| pTrc99A | Expression vector | 1 | |
| pNagA | nagAhis in pTrc99A | This study | NagAEcoR1, NagAhisBamH1 |
| pNagC | nagC in pTrc99A | This study | NagCEcoR1, NagCBamH1 |
| pNagC4 | nagC4 (NagCG156R) in pTrc99A | This study | NagCEcoR1, NagCBamH1 |
| pRJ800 | Promoterless lacZ | 44 | |
| pBA-14 | csgBA promoter in pRJ800 | This study | |
| pD-1 | csgDEFG promoter in pRJ800 | This study | D1 top, D1 bot |
Knr, kanamycin resistance; Cmr, chloramphenicol resistance.
Primers listed in Table 2 that were used to construct the deletion strain or to PCR amplify the gene. If no primers are indicated, see the text for details concerning strain or plasmid construction.
TABLE 2.
Primers used in this work
| Primer | Sequence |
|---|---|
| NagAH1P1 | 5′-CCTGGCTCCTTGCTCAGGGCAATATTTTTTAAAATCGGGGGTCAGAGTGTAGGCTGGAGCTGCTTC-3′a |
| NagAH2P2 | 5′-GTTTTACGAGATCAACATTACCTATCTGAGCTTGTCCGCCTGGTGTCATATGAATATCCTCCTTAG-3′a |
| NagBH1P1 | 5′-CTTAATCCGCCAACGGCTTACATTTTACTTATTGAGGTGAATAGTGTAGGCTGGAGCTGCTTC-3′a |
| NagBH2P2 | 5′-ATTCGTGGCCGGTAAAGATCCGGCCCTGGGTTAATGCATACATATGAATATCCTCCTTAG-3′a |
| NagCH1P1 | 5′-CAAGACCATCGTTAACGGTAACGAGGTCGTAACTCAATAAGAGAAAGTGTGTAGGCTGGAGCTGCTTC-3′a |
| NagCH2P2 | 5′-CGTGCATCAGCACGCCGTCGATATCGCAAATTACATTTTTAATGGTCATATGAATATCCTCCTTAG-3′a |
| NagDH1P1 | 5′-AGTGGCGCTTATTGTTGTCAATATTCTGGGTAGTCCGTGTAGGCTGGAGCTGCTTC-3′a |
| NagEH1P1 | 5′-GCTTGAAACGAGCCAAATAGGGTTCTCGTAGGGGGAATAAGGTGTAGGCTGGAGCTGCTTC-3′a |
| NagEH2P2 | 5′-GTTGGATGCGACGCTCAAGCGTCGCATCAGGCATAAAGCAGACATATGAATATCCTCCTTAG-3′a |
| NagH1P1 | 5′-TTACTTTTTGATTTCATACAGCGGTGTTTGACCCGCCAGTGTAGGCTGGAGCTGCTTC-3′a |
| NagH2P2 | 5′-GTCATCAGAGCCAACCACGTCCGCAGACGTGGTTGCTATCATATGAATATCCTCCTTAG-3′a |
| LacZH1P1 | 5′-TGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCATGGTGTAGGCTGGAGCTGCTTC-3′a |
| LacZH2P2 | 5′-ACGCGAAATACGGGCAGACATGGCCTGCCCGGTTATTACATATGAATATCCTCCTTAG-3′a |
| NagAEcoR1 | 5′-CCGGAATTCCGGGGGTCAGAATGTATGCAT-3′b |
| NagAhisBamH1 | 5′-GGATCCTTAGTGATGGTGATGGTGATGTTGAGTTACGACCTCGGTACCG-3′b |
| NagCEcoR1 | 5′-CCGGAATTCGAGAAAGTATGACACCAGGCG-3′b |
| NagCBamH1 | 5′-CGCGGATCCTTAATTTTCCAGCAAATGCTG-3′b |
| NagEptop | 5′-CTAGCTAGCATTCACCTCAATAAGTAAAATG-3′b |
| NagE200B | 5′-CATCGTGGATTCCTCAAAGCG-3′b |
| NagESacI | 5′-CGCGAGCTCAGGGGGAATAAGATGAATATT-3′b |
| NagEmidtop | 5′-AACCGTCTGCTGATCCCAACC-3′b |
| NagEendtop | 5′-CTGGTATCGCCGATTACC-3′b |
| D1 top | 5′-GGATCCCCATGGGATGAAACCCCGCTTTTTTTATTG-3′b |
| D1 bot | 5′-CTGCAGTTAAGAAATTAAATCATTTCAACTTGGTTG-3′b |
The underlined sequence anneals to the P1 or P2 priming sites on pKD4 or pKD3. The sequence not underlined is homologous to the gene(s) that is deleted.
The underlined sequence anneals to the gene, and the sequence not underlined is restriction sites and/or a six-His tag that was included at the 5′ end of the primer.
nagA and nagC were PCR amplified from C600 genomic DNA, and the PCR products were cloned into the EcoRI and BamHI sites of pTrc99A (1), creating pNagA and pNagC (Tables 1 and 2). pNagC4 was constructed by PCR amplifying nagC from a CR+ ΔnagA suppressor strain and cloning the PCR product into the EcoRI and BamHI sites of pTrc99A, creating pNagC4 (Tables 1 and 2). The plasmids were sequenced at the University of Michigan sequencing center.
pBA-14 was constructed by subcloning the ScaI/BamHI fragment from pLR2 (45), which contained the csgBA promoter, into the SmaI and BamHI sites of pRJ800 (a pBR322 derivative containing a promoterless lacZ gene) (44). pD-1 was constructed by PCR amplifying the csgDEFG promoter from chromosomal DNA (Tables 1 and 2). This fragment was cloned into pCR2.1 (Invitrogen), creating pTopoIGR6. Following sequencing, the EcoRI and BamHI fragment was subcloned from pTopoIGR6 into pRJ800, creating pD-1. pBA-14 contained the csgBA promoter fused to lacZ, and pD-1 contained the csgDEFG promoter fused to lacZ.
Electron microscopy.
Bacteria were grown on YESCA plates for 48 h at 26°C. Samples were stained with uranyl acetate as described previously (11) and were viewed using a Phillips CM10 microscope.
SDS-polyacrylamide gel electrophoresis and Western blotting.
Bacteria were grown on YESCA plates for 48 h at 26°C. Bacteria were scraped off plates and resuspended in 10 mM Tris (pH 7.4), which was followed by normalizaton by optical density at 600 nm. To monitor CsgA stability, samples were briefly treated with formic acid as described previously (14). Whole-cell lysates were electrophoresed on 13% sodium dodecyl sulfate (SDS)-polyacrylamide and blotted onto polyvinylidene difluoride using standard techniques. CsgA and CsgG polyclonal antibodies were raised in rabbits with the purified proteins (Proteintech, Chicago, IL) and were used at dilutions of 1:10,000 and 1:100,000, respectively. The secondary antibodies were anti-rabbit antibodies conjugated to horseradish peroxidase (Sigma, St. Louis, MO) and were used at a dilution of 1:5,000. Western blots were developed using the Pierce super signal detection system.
β-Galactosidase assays.
Bacteria were grown on YESCA plates or buffered YESCA plates with and without 10 mM GlcNAc. YESCA plates were buffered at pH 8.1 with 0.1 M N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES) (Sigma, St. Louis, MO). Samples were taken after 48 h of growth at 26°C. Bacteria were scraped off the plates and resuspended in 1 ml of 50 mM Tris (pH 8). β-Galactosidase assays were performed as described previously (33).
Isolation of CR+ ΔnagA suppressor strains.
Overnight cultures of Wt strain C600 or ΔnagA mutant MMB103 were grown in LB, and 10 μl of each culture was plated onto a CR-YESCA plate. The bacteria were grown at 26°C for 96 h. After 96 h of growth CR+ ΔnagA suppressors appeared. These CR+ ΔnagA suppressors were struck out and isolated as single colonies to obtain a pure population of CR+ ΔnagA suppressors.
Sequencing nagE and nagC from CR+ ΔnagA suppressor strains.
nagE and nagC were PCR amplified from genomic DNA from 11 CR+ ΔnagA suppressor strains using primers NagEptop and NagE200B and primers NagCEcoR1 and NagCBamH1, respectively (Table 2). The PCR products were sequenced at the University of Michigan sequencing center. nagC was sequenced using the same primers that were used for PCR. nagE was sequenced using primers NagEptop, NagESacI, NagEmidtop, and NagEendtop (Table 2).
RESULTS
High levels of GlcNAc-6P in the cell cause a defect in curli production.
Curli-producing bacteria stain red on plates amended with CR, while noncurliated strains remain white (13). The nagA gene was identified in a transposon mutagenesis screen by assaying for mutants that were defective in CR binding (23). In order to verify this observation and further characterize this phenomenon, we constructed nonpolar nagA deletions in E. coli strains C600 and MC4100 and found that they were defective in CR binding (Fig. 2A). CR binding was restored in the nagA deletion strains by expression of nagA in trans (Fig. 2A). Deletion of nagA in C600 resulted in a more dramatic reduction in CR binding compared to the reduction with the same deletion in MC4100 (Fig. 2A). Electron microscopy analysis demonstrated that while only a few ΔnagA bacteria produced curli, the fibers that were observed were structurally similar to those produced by Wt strains (Fig. 2C and D).
FIG. 2.

ΔnagA and ΔnagC strains are defective in curli production. (A) CR-YESCA plate containing Wt strains (Wtc, C600; Wtm, MC4100) and ΔnagA strains (ΔnagAc, MMB103; ΔnagAm, MMB5) transformed with pTrc99A or pNagA after 48 h of growth at 26°C. (B) CR-YESCA plate containing Wt (C600), ΔnagBACD ΔnagE (MMB87) (Δnag), ΔnagA (MMB103), ΔnagB (MMB120), ΔnagC (MMB171), ΔnagD (MMB161), ΔnagE (MMB152), ΔnagEA (SKM2), and ΔnagEC (MMB185) strains after 48 h of growth at 26°C. (C and D) Electron micrographs of Wt (MC4100) (C) and ΔnagA (MMB5) (D) strains. Scale bars = 400 nm.
Nonpolar deletions of nagB, nagD, and nagE had no effect on CR binding (Fig. 2B), while deletion of nagC resulted in modestly reduced CR binding (Fig. 2B). The defect in CR binding in the ΔnagC strain was complemented by expression of nagC in trans (see Fig. 5B). Deletion of all the nag genes (ΔnagBACD ΔnagE) resulted in a CR+ phenotype (Fig. 2B), demonstrating that nagA and nagC were not required for curli production in the absence of other nag genes. One plausible explanation for why the ΔnagC and ΔnagA strains were defective in curli production was that these strains still import GlcNAc through NagE, resulting in high intracellular levels of GlcNAc-6P. In nagA mutants, GlcNAc-6P is not converted to GlcN-6P (55), causing GlcNAc-6P to accumulate in the cell. In nagC mutants, the nag genes are constitutively active, resulting in high levels of both nagE transcription and nagA transcription. The higher levels of nagE transcription in nagC mutants likely result in more GlcNAc transported into the cell and therefore higher intracellular GlcNAc-6P levels. To test the hypothesis that NagE-mediated transport of GlcNAc into the cell was required for the reduction in curli production in nagA and nagC mutants, nagE/nagA and nagE/nagC double deletion mutants were constructed. Both ΔnagEA and ΔnagEC strains bound CR with efficiency similar to that of the Wt (Fig. 2B). ΔnagAB, ΔnagAC, and ΔnagAD strains were defective in CR binding (data not shown).
FIG. 5.

CR+ ΔnagA suppressors map to nagE or nagC. (A) Intergenic region between nagBACD and nagE and part of the nagE open reading frame. The start codons for nagB and nagE are indicated by boldface type and arrows. The arrowheads indicate places where IS1 or IS5 elements are inserted into the nagE promoter or gene in the CR+ ΔnagA suppressors. In one suppressor the underlined sequence is duplicated, and in another suppressor the sequence indicated by gray type is deleted; both result in premature stop codons in nagE. (B) CR-YESCA plate containing Wt (C600), ΔnagC (MMB171), and ΔnagA (MMB103) strains transformed with pTrc99A, pNagC, or pNagC4 after 48 h of growth at 26°C. (C) Plate containing MacConkey agar with 1% GlcNAc and the Wt (C600) and ΔnagC (MMB171) strains transformed with pTrc99A, pNagC, or pNagC4 after overnight growth at 37°C.
CsgG and CsgA steady-state levels are reduced in ΔnagA and ΔnagC strains.
The stability of CsgA and CsgG was tested using different nag mutants. CsgA polymerizes into an SDS-insoluble fiber on the cell surface (15). Therefore, whole cells were treated with formic acid to liberate CsgA monomers for Western analysis. In strains that were CR− (ΔnagA, ΔnagC, ΔnagAB, ΔnagAC, and ΔnagAD) (Fig. 2B and data not shown), the steady-state levels of CsgA and CsgG were greatly reduced compared to the levels in the Wt (Fig. 3A, lanes 2, 4, 8, 14, 16, and 18, and Fig. 3B, lanes 1, 2, 4, 8, 9, and 10). The ΔnagC strain, which had an intermediate CR binding phenotype, consistently produced more CsgA and CsgG than the ΔnagA strain produced (Fig. 2B; Fig. 3A, lanes 4 and 8; and Fig. 3B, lanes 2 and 4). In strains that were CR+ (ΔnagB, ΔnagD, ΔnagE, ΔnagEA, ΔnagEC, and ΔnagBACD ΔnagE) (Fig. 2B) the CsgA and CsgG levels were similar to those in the Wt (Fig. 3A, lanes 2, 6, 10, 12, 20, 22, and 24, and Fig. 3B, lanes 1, 3, 5, 6, 7, 11, and 12).
FIG. 3.

Steady-state levels of CsgA and CsgG are reduced when intracellular levels of GlcNAc-6P are high. (A and B) Western blots of whole-cell lysates from different strains in a C600 background after 48 h of growth at 26°C on YESCA plates developed with anti-CsgA antibodies (A) or with anti-CsgG antibodies (B). (C) Whole-cell Western blot of Wt strain MMB200 transformed with pRJ800 after 48 h of growth at 26°C on YESCA plates or YESCA (pH 8.1) plates with or without 10 mM GlcNAc developed with anti-CsgA antibodies. The samples in even-numbered lanes in panels A and C were treated with formic acid (FA) prior to electrophoresis.
Because CsgG is required for the stability and secretion of CsgA (30), we examined whether overexpression of CsgG could restore CR binding to a ΔnagA strain. Expression of CsgG from the trc promoter in a ΔnagA strain resulted in amounts of outer membrane-localized CsgG similar to the amounts in the Wt (data not shown). However, CR binding was not restored in this strain (data not shown), suggesting that CsgG mislocalization or stability was not solely responsible for the curli deficiency in the ΔnagA strain.
Transcription from the csgBA and csgDEFG promoters is reduced in ΔnagA and ΔnagC strains.
The decreases in the steady-state levels of CsgA and CsgG in the ΔnagA and ΔnagC strains could have been due to a defect in csg transcription. Transcription from the csgBA and csgDEFG promoters was monitored in ΔnagA and ΔnagC strains. Two plasmids (pBA-14 and pD-1) in which the csgBA or csgDEFG promoter region was fused to lacZ were constructed. Wt (MMB200), ΔnagA (MMB1035), ΔnagC (MMB1714), and ΔcsgBA ΔcsgDEFG (MMB661) strains were transformed with pBA-14 and pD1, and the β-galactosidase activity in each of the strains was monitored after 48 h of growth on YESCA plates. In the ΔnagA strain, the csgBA promoter activity was reduced sevenfold compared to the activity in the Wt strain and the csgDEFG promoter activity was reduced almost twofold (Table 3). In the ΔnagC strain the csgBA promoter activity was reduced almost twofold, while the csgDEFG promoter activity was modestly affected (Table 3). csgBA promoter activity was eliminated in a ΔcsgBA ΔcsgDEFG strain (Table 3), which is consistent with previously described work that demonstrated that CsgD is required for csgBA promoter activity (24). csgDEFG promoter activity was slightly reduced in the ΔcsgBA ΔcsgDEFG strain (Table 3). Previous work with Salmonella enterica serovar Typhimurium demonstrated that csgDEFG promoter activity was not affected in a csgD mutant strain (48). Both csgBA and csgDEFG promoter activities were reduced when the Wt strain was grown on YESCA containing 300 mM NaCl, which is consistent with previously published reports (data not shown) (49).
TABLE 3.
PcsgBA and PcsgDEFG activities in Wt, ΔnagA, ΔnagC, and ΔcsgBA ΔcsgDEFG strains after 48 h of growth on YESCA plates
| Strain | Activities (Miller units) of the following promoter fusions:
|
||
|---|---|---|---|
| Nonea | PcsgBAb | PcsgDEFGc | |
| Wt (MMB200) | 7.18 ± 0.85d | 260 ± 31.8 | 1,281 ± 115 |
| ΔnagA (MMB1035) | 8.67 ± 0.82 | 35.4 ± 2.37 | 720 ± 69 |
| ΔnagC (MMB1714) | 6.16 ± 0.61 | 149 ± 11.2 | 944 ± 103 |
| ΔcsgBA ΔcsgDEFG (MMB661) | 4.29 ± 0.38 | 25.5 ± 2.48 | 836 ± 45.5 |
The strains were transformed with pRJ800, which contains a promoterless lacZ fusion.
pBA-14, which contains the csgBA promoter fused to lacZ.
pD-1, which contains the csgDEFG promoter fused to lacZ.
The values are means ± standard deviations from one experiment performed in triplicate. Each value is representative of the values from at least three independent experiments.
One plausible explanation for the decrease in csgBA and csgDEFG promoter activity in ΔnagA and ΔnagC strains is that there was an artificially high level of GlcNAc-6P inside the cells. To test this possibility, the Wt strain containing the csgBA or csgDEFG promoter fusion was grown on buffered YESCA plates in the presence and absence of 10 mM GlcNAc. The plates were buffered to prevent pH fluctuations caused by addition of GlcNAc, since pH changes could potentially complicate the analysis. Transcription from the csgBA promoter was reduced approximately 6-fold and csgDEFG promoter activity was reduced 2.5-fold after growth on YESCA plates containing 10 mM GlcNAc (Table 4). Western blots confirmed that there were reductions in the steady-state levels of CsgA when Wt cells were grown in the presence of 10 mM GlcNAc (Fig. 3C, lanes 2 and 4). The CsgA protein levels and transcription from the csgBA promoter were reproducibly lower on buffered YESCA plates (pH 8.1, 7.0, and 5.5) than on unbuffered YESCA plates (Fig. 3C, lanes 2 and 6, Tables 3 and 4, and data not shown). Although we are not sure why growth on buffered plates reduced CsgA stability and csgBA transcription, this effect was specific to the csgBA promoter as the csgDEFG promoter was not affected by growth on buffered plates (Tables 3 and 4).
TABLE 4.
PcsgBA and PcsgDEFG activities in a Wt strain after growth on YESCA with or without 10 mM GlcNAc
| Strain | Activity (Miller units)
|
|
|---|---|---|
| Without GlcNAca | With 10 mM GlcNAca | |
| Wt (MMB200)/pRJ800 | 9.52 ± 1.1b | 7.94 ± 1.56 |
| Wt (MMB200)/pBA-14 | 125 ± 6 | 19.5 ± 0.87 |
| Wt (MMB200)/pD-1 | 1,244 ± 51 | 495 ± 45.8 |
Strains were grown on YESCA (pH 8.1) with or without 10 mM GlcNAc for 48 h at 26°C.
The values are means ± standard deviations from one experiment performed in triplicate and are representative of the values from at least three independent experiments.
CR+ ΔnagA suppressors develop after extended growth on YESCA plates.
We found that CR+ ΔnagA suppressors developed after 96 h of growth on CR-YESCA plates. CR+ ΔnagA suppressors were isolated as single colonies (Fig. 4A) and were analyzed to determine whether they retained the nagA deletion. The CR+ ΔnagA suppressors were unable to utilize GlcNAc as the sole carbon source, indicating that they retained the nagA deletion (Fig. 4B and C). PCR was used to confirm the presence of the nagA deletion in the suppressors (data not shown). We reasoned that CR+ ΔnagA suppressors may have reduced the capacity to take up GlcNAc and that NagE was nonfunctional or not expressed. Therefore, we sequenced nagE in 11 ΔnagA CR+ suppressor mutants, and 10 of these 11 suppressor mutants had changes in nagE (Fig. 5A). We found that IS1 and IS5 elements had inserted into the nagE promoter or shortly after the nagE start codon (Fig. 5A). Two suppressors had a duplication or a deletion of 14-bp sequences in nagE that resulted in premature stop codons in nagE (Fig. 5A).
FIG. 4.

CR+ ΔnagA suppressors develop after extended growth on YESCA plates. After 96 h of growth on CR-YESCA plates, CR+ ΔnagA suppressors arose. These suppressors were mixed with CR− ΔnagA isolates, and the two populations were isolated. (A) CR-YESCA plate containing the Wt (C600), original ΔnagA (MMB103), CR+ ΔnagA suppressor, and CR− ΔnagA (isolate mixed with the CR+ ΔnagA suppressor) strains after growth for 48 h at 26°C. (B and C) The strains were streaked onto W minimal medium plates with glucose (B) or GlcNAc (C) as the sole carbon source and incubated at 37°C for 48 h.
We hypothesized that the 11th suppressor might have a mutation in nagC that resulted in reduced expression of nagE, thereby reducing GlcNAc transport into the cell. We sequenced nagC in the last suppressor and found a point mutation that caused a single amino acid substitution at position 156 that changed glycine to arginine. We refer to this allele of nagC below as nagC4. nagC4 was cloned into plasmid pTrc99A, and its ability to complement a ΔnagA strain for CR binding was assessed. nagC4 restored CR binding and curli production to a ΔnagA strain, while nagC did not complement a ΔnagA strain for CR binding (Fig. 5B). Since nagC4 could complement a ΔnagA strain for CR binding, we predicted that overexpression of this allele in a Wt strain might prevent the strain from utilizing GlcNAc as a carbon source by constitutively repressing the nag genes. Bacteria that can utilize GlcNAc turn pink when they are grown on MacConkey agar plates containing GlcNAc, and strains that do not utilize GlcNAc remain white. When Wt nagC was overexpressed in a Wt or ΔnagC strain, GlcNAc was utilized as a carbon source (Fig. 5C). However, overexpression of nagC4 in a Wt or ΔnagC strain resulted in an inability of the strain to utilize GlcNAc (Fig. 5C), suggesting that nagC4 constitutively represses the nag genes.
DISCUSSION
E. coli can use GlcNAc-6P as a carbon source and/or as a precursor for peptidoglycan and lipopolysaccharide biosynthesis. Recently, GlcNAc-6P was also shown to be a signaling molecule that regulates the expression of type 1 pili (51). Curli gene expression is under the control of many environmental factors, including oxygen tension, temperature, and ionic strength (for a review see reference 20). Here we show that GlcNAc-6P modulates curli gene expression. Mutations that disrupt GlcNAc metabolism result in reduced levels of csgBA and csgDEFG transcription and curli production (Fig. 2 and 3 and Table 3). Addition of excess GlcNAc to media reduces csgBA and csgDEFG transcription in a Wt strain (Table 4). Curli production is restored in nagA or nagC mutants by deletion of the GlcNAc transporter gene, nagE (Fig. 2 and 3). Curli production is also restored in nagA mutants by suppressor mutations that map to nagE or nagC (Fig. 4 and 5). Collectively, these results suggest that excess GlcNAc-6P inside the cell results in reduced curli gene expression. However, the molecular details of curli gene repression by GlcNAc-6P remain unknown.
NagC is thought to be a functional regulator only in the absence of GlcNAc since NagC-GlcNAc-6P binds DNA poorly (40). In the absence of nagA, GlcNAc-6P accumulates in the cell (55) and binds to NagC, preventing NagC-mediated repression at the nag promoters (40). Given our observation that nagA mutants have lower csgBA and csgDEFG promoter activities, it is possible that NagC directly activates the csgBA promoter in the absence of GlcNAc-6P. However, NagC is not required for curli expression in certain genetic backgrounds. Both ΔnagEC and ΔnagBACD ΔnagE strains produce curli (Fig. 2B and 3), and a search for NagC binding sites in the intergenic region between the two curli operons did not reveal any putative sites (data not shown). Furthermore, the curli defect in the ΔnagA strain, which is phenotypically a nagC mutant (40), is much more pronounced than the defect in the ΔnagC strain (Fig. 2B and 3 and Table 3).
Instead, we hypothesized that the ΔnagC strain is defective in curli production because of an increase in nagE expression, which results in more intracellular GlcNAc-6P. In the case of the ΔnagA strain, not only is nagE up-regulated, resulting in an increase in the intracellular GlcNAc-6P levels, but once GlcNAc-6P enters the cell, there is no way to degrade it (40, 55). When the GlcNAc-6P levels in the cell are lower, as they are in ΔnagEC, ΔnagEA, ΔnagBACD ΔnagE, or CR+ ΔnagA suppressor strains, curli production is restored (Fig. 2B, 3, 4, and 5).
The mutation of one of the CR+ ΔnagA suppressor mutants that we isolated mapped to nagC. The allele of nagC, nagC4, was dominant over Wt nagC (Fig. 5B and C) and did not exhibit GlcNAc sugar sensitivity (Barnhart and Chapman, unpublished results), which is the inability to grow in rich media in the presence of exogenous GlcNAc (3, 55). nagC4 likely results in reduced GlcNAc transport into the cell due to a decrease in nagE expression. Therefore, when nagC4 is expressed in a ΔnagA strain, CR binding is restored because GlcNAc-6P does not accumulate in the cells.
nagC4 encodes a protein that differs from Wt NagC only at amino acid 156. This amino acid is not within the helix-turn-helix motif of NagC, which is amino acids 33 to 56 (37), suggesting that the mutation does not affect DNA binding. Previous work identified nagA suppressor mutants that allow bacteria to grow in the presence of GlcNAc while they remain unable to utilize GlcNAc as a carbon source (55). One such suppressor mutation was mapped to nagC (39). The nagC mutant did not respond to GlcNAc-6P, constitutively repressed the nag genes, and was dominant over Wt nagC. Plumbridge (39) postulated that this nagC allele, which results in a leucine-to-proline substitution at amino acid 125, did not bind to GlcNAc-6P. We predict that amino acid 156 and amino acid 125 form part of the GlcNAc-6P-responsive domain of NagC.
Here, we provide genetic evidence that nagA suppressor mutations also occur in nagE. It was postulated that disruption of nagE would also allow suppression of GlcNAc sugar sensitivity in nagA mutants (39). Although we isolated nagA suppressors by screening for reconstitution of curli production, the CR+ ΔnagA suppressors were not sugar sensitive (Barnhart and Chapman, unpublished results). Since the CR+ ΔnagA suppressor mutants block GlcNAc-6P uptake by rendering nagE nonfunctional, these results support the hypothesis that high intracellular levels of GlcNAc-6P down-regulate csg expression.
Curli are not the only extracellular surface fibers regulated in response to GlcNAc. The type 1 pilus genes in E. coli are subject to phase variation, which is controlled by an invertible element in the promoter that switches the promoter between the on and off orientations. FimB is the recombinase that switches the promoter from the off orientation to the on orientation (28). High levels of GlcNAc turn off fimB transcription in a NagC-dependent fashion (50), which should fix the type 1 pilus promoter in the off position. Like curli, type 1 pili are required for biofilm formation (42). E. coli also produces an extracellular GlcNAc polymer important for biofilm formation (54). However, it is not known whether the nag genes are required for assembly of this polymer. Additionally, in Vibrio cholerae a novel type IV pilus is positively regulated in response to chitin, which is a GlcNAc polymer (32). Therefore, intracellular levels of GlcNAc appear to be an emerging regulator of extracellular fiber production and community behavior.
Acknowledgments
We thank Elisabeth Ashman for construction of pD-1. We thank Robert Bender and Christopher Rosario for reading the manuscript. We also thank members of the Chapman and Hultgren labs and Staffan Normark for helpful discussions.
This work was supported by NIH grant AI54967-01 to M.R.C.
REFERENCES
- 1.Amann, E., B. Ochs, and K. J. Abel. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301-315. [DOI] [PubMed] [Google Scholar]
- 2.Arnqvist, A., A. Olsen, and S. Normark. 1994. Sigma S-dependent growth-phase induction of the csgBA promoter in Escherichia coli can be achieved in vivo by sigma 70 in the absence of the nucleoid-associated protein H-NS. Mol. Microbiol. 13:1021-1032. [DOI] [PubMed] [Google Scholar]
- 3.Bernheim, N. J., and W. J. Dobrogosz. 1970. Amino sugar sensitivity in Escherichia coli mutants unable to grow on N-acetylglucosamine. J. Bacteriol. 101:384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bian, Z., A. Brauner, Y. Li, and S. Normark. 2000. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J. Infect. Dis. 181:602-612. [DOI] [PubMed] [Google Scholar]
- 5.Bian, Z., Z. Q. Yan, G. K. Hansson, P. Thoren, and S. Normark. 2001. Activation of inducible nitric oxide synthase/nitric oxide by curli fibers leads to a fall in blood pressure during systemic Escherichia coli infection in mice. J. Infect. Dis. 183:612-619. [DOI] [PubMed] [Google Scholar]
- 6.Bokranz, W., X. Wang, H. Tschape, and U. Romling. 2005. Expression of cellulose and curli fimbriae by Escherichia coli isolated from the gastrointestinal tract. J. Med. Microbiol. 54:1171-1182. [DOI] [PubMed] [Google Scholar]
- 7.Bougdour, A., C. Lelong, and J. Geiselmann. 2004. Crl, a low temperature-induced protein in Escherichia coli that binds directly to the stationary phase sigma subunit of RNA polymerase. J. Biol. Chem. 279:19540-19550. [DOI] [PubMed] [Google Scholar]
- 8.Brown, P. K., C. M. Dozois, C. A. Nickerson, A. Zuppardo, J. Terlonge, and R. Curtiss III. 2001. MlrA, a novel regulator of curli (AgF) and extracellular matrix synthesis by Escherichia coli and Salmonella enterica serovar Typhimurium. Mol. Microbiol. 41:349-363. [DOI] [PubMed] [Google Scholar]
- 9.Campbell, A. 1961. Sensitive mutants of bacteriophage lambda. Virology 14:22-32. [DOI] [PubMed] [Google Scholar]
- 10.Casadaban, M. J. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541-555. [DOI] [PubMed] [Google Scholar]
- 11.Chapman, M. R., L. S. Robinson, J. S. Pinkner, R. Roth, J. Heuser, M. Hammar, S. Normark, and S. J. Hultgren. 2002. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295:851-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collinson, S. K., S. C. Clouthier, J. L. Doran, P. A. Banser, and W. W. Kay. 1996. Salmonella enteritidis agfBAC operon encoding thin, aggregative fimbriae. J. Bacteriol. 178:662-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collinson, S. K., P. C. Doig, J. L. Doran, S. Clouthier, T. J. Trust, and W. W. Kay. 1993. Thin, aggregative fimbriae mediate binding of Salmonella enteritidis to fibronectin. J. Bacteriol. 175:12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collinson, S. K., L. Emody, K. H. Muller, T. J. Trust, and W. W. Kay. 1991. Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J. Bacteriol. 173:4773-4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collinson, S. K., J. M. Parker, R. S. Hodges, and W. W. Kay. 1999. Structural predictions of AgfA, the insoluble fimbrial subunit of Salmonella thin aggregative fimbriae. J. Mol. Biol. 290:741-756. [DOI] [PubMed] [Google Scholar]
- 16.Danese, P. N., and T. J. Silhavy. 1998. CpxP, a stress-combative member of the Cpx regulon. J. Bacteriol. 180:831-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorel, C., O. Vidal, C. Prigent-Combaret, I. Vallet, and P. Lejeune. 1999. Involvement of the Cpx signal transduction pathway of E. coli in biofilm formation. FEMS Microbiol. Lett. 178:169-175. [DOI] [PubMed] [Google Scholar]
- 19.Ferrieres, L., and D. J. Clarke. 2003. The RcsC sensor kinase is required for normal biofilm formation in Escherichia coli K-12 and controls the expression of a regulon in response to growth on a solid surface. Mol. Microbiol. 50:1665-1682. [DOI] [PubMed] [Google Scholar]
- 20.Gerstel, U., C. Park, and U. Romling. 2003. Complex regulation of csgD promoter activity by global regulatory proteins. Mol. Microbiol. 49:639-654. [DOI] [PubMed] [Google Scholar]
- 21.Gerstel, U., and U. Romling. 2003. The csgD promoter, a control unit for biofilm formation in Salmonella typhimurium. Res. Microbiol. 154:659-667. [DOI] [PubMed] [Google Scholar]
- 22.Gophna, U., M. Barlev, R. Seijffers, T. A. Oelschlager, J. Hacker, and E. Z. Ron. 2001. Curli fibers mediate internalization of Escherichia coli by eukaryotic cells. Infect. Immun. 69:2659-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammar, M. 1997. Assembly and adhesive properties of curli. Ph.D. thesis. Karolinska Institute, Stockholm, Sweden.
- 24.Hammar, M., A. Arnqvist, Z. Bian, A. Olsen, and S. Normark. 1995. Expression of two csg operons is required for production of fibronectin- and Congo red-binding curli polymers in Escherichia coli K-12. Mol. Microbiol. 18:661-670. [DOI] [PubMed] [Google Scholar]
- 25.Jones-Mortimer, M. C., and H. L. Kornberg. 1980. Amino-sugar transport systems of Escherichia coli K12. J. Gen. Microbiol. 117:369-376. [DOI] [PubMed] [Google Scholar]
- 26.Jubelin, G., A. Vianney, C. Beloin, J. M. Ghigo, J. C. Lazzaroni, P. Lejeune, and C. Dorel. 2005. CpxR/OmpR interplay regulates curli gene expression in response to osmolarity in Escherichia coli. J. Bacteriol. 187:2038-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kikuchi, T., Y. Mizunoe, A. Takade, S. Naito, and S. Yoshida. 2005. Curli fibers are required for development of biofilm architecture in Escherichia coli K-12 and enhance bacterial adherence to human uroepithelial cells. Microbiol. Immunol. 49:875-884. [DOI] [PubMed] [Google Scholar]
- 28.Klemm, P. 1986. Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli. EMBO J. 5:1389-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lengeler, J. 1980. Characterisation of mutants of Escherichia coli K12, selected by resistance to streptozotocin. Mol. Gen. Genet. 179:49-54. [DOI] [PubMed] [Google Scholar]
- 30.Loferer, H., M. Hammar, and S. Normark. 1997. Availability of the fibre subunit CsgA and the nucleator protein CsgB during assembly of fibronectin-binding curli is limited by the intracellular concentration of the novel lipoprotein CsgG. Mol. Microbiol. 26:11-23. [DOI] [PubMed] [Google Scholar]
- 31.Majdalani, N., and S. Gottesman. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59:379-405. [DOI] [PubMed] [Google Scholar]
- 32.Meibom, K. L., X. B. Li, A. T. Nielsen, C. Y. Wu, S. Roseman, and G. K. Schoolnik. 2004. The Vibrio cholerae chitin utilization program. Proc. Natl. Acad. Sci. USA 101:2524-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller, J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 34.Olsen, A., A. Arnqvist, M. Hammar, S. Sukupolvi, and S. Normark. 1993. The RpoS sigma factor relieves H-NS-mediated transcriptional repression of csgA, the subunit gene of fibronectin-binding curli in Escherichia coli. Mol. Microbiol. 7:523-536. [DOI] [PubMed] [Google Scholar]
- 35.Peri, K. G., H. Goldie, and E. B. Waygood. 1990. Cloning and characterization of the N-acetylglucosamine operon of Escherichia coli. Biochem. Cell Biol. 68:123-137. [DOI] [PubMed] [Google Scholar]
- 36.Plumbridge, J. 1995. Co-ordinated regulation of amino sugar biosynthesis and degradation: the NagC repressor acts as both an activator and a repressor for the transcription of the glmUS operon and requires two separated NagC binding sites. EMBO J. 14:3958-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plumbridge, J. 2001. DNA binding sites for the Mlc and NagC proteins: regulation of nagE, encoding the N-acetylglucosamine-specific transporter in Escherichia coli. Nucleic Acids Res. 29:506-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plumbridge, J. 1996. How to achieve constitutive expression of a gene within an inducible operon: the example of the nagC gene of Escherichia coli. J. Bacteriol. 178:2629-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plumbridge, J. A. 1992. A dominant mutation in the gene for the Nag repressor of Escherichia coli that renders the nag regulon uninducible. J. Gen. Microbiol. 138:1011-1017. [DOI] [PubMed] [Google Scholar]
- 40.Plumbridge, J. A. 1991. Repression and induction of the nag regulon of Escherichia coli K-12: the roles of nagC and nagA in maintenance of the uninduced state. Mol. Microbiol. 5:2053-2062. [DOI] [PubMed] [Google Scholar]
- 41.Plumbridge, J. A. 1989. Sequence of the nagBACD operon in Escherichia coli K12 and pattern of transcription within the nag regulon. Mol. Microbiol. 3:505-515. [DOI] [PubMed] [Google Scholar]
- 42.Pratt, L. A., and R. Kolter. 1998. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type 1 pili. Mol. Microbiol. 30:285-293. [DOI] [PubMed] [Google Scholar]
- 43.Pratt, L. A., and T. J. Silhavy. 1998. Crl stimulates RpoS activity during stationary phase. Mol. Microbiol. 29:1225-1236. [DOI] [PubMed] [Google Scholar]
- 44.Reznikoff, W., and W. McClure. 1986. E. coli promoters, p. 1-33. In W. Reznikoff and E. Gold (ed.), Maximizing gene expression. Butterworths Publishers, Boston, Mass.
- 45.Robinson, L. S., E. M. Ashman, S. J. Hultgren, and M. R. Chapman. 2006. Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol. Microbiol. 59:870-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rogers, M. J., T. Ohgi, J. Plumbridge, and D. Soll. 1988. Nucleotide sequences of the Escherichia coli nagE and nagB genes: the structural genes for the N-acetylglucosamine transport protein of the bacterial phosphoenolpyruvate:sugar phosphotransferase system and for glucosamine-6-phosphate deaminase. Gene 62:197-207. [DOI] [PubMed] [Google Scholar]
- 47.Romling, U., Z. Bian, M. Hammar, W. D. Sierralta, and S. Normark. 1998. Curli fibers are highly conserved between Salmonella typhimurium and Escherichia coli with respect to operon structure and regulation. J. Bacteriol. 180:722-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romling, U., M. Rohde, A. Olsen, S. Normark, and J. Reinkoster. 2000. AgfD, the checkpoint of multicellular and aggregative behaviour in Salmonella typhimurium regulates at least two independent pathways. Mol. Microbiol. 36:10-23. [DOI] [PubMed] [Google Scholar]
- 49.Romling, U., W. D. Sierralta, K. Eriksson, and S. Normark. 1998. Multicellular and aggregative behaviour of Salmonella typhimurium strains is controlled by mutations in the agfD promoter. Mol. Microbiol. 28:249-264. [DOI] [PubMed] [Google Scholar]
- 50.Sohanpal, B. K., S. El-Labany, M. Lahooti, J. A. Plumbridge, and I. C. Blomfield. 2004. Integrated regulatory responses of fimB to N-acetylneuraminic (sialic) acid and GlcNAc in Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 101:16322-16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tukel, C., M. Raffatellu, A. D. Humphries, R. P. Wilson, H. L. Andrews-Polymenis, T. Gull, J. F. Figueiredo, M. H. Wong, K. S. Michelsen, M. Akcelik, L. G. Adams, and A. J. Baumler. 2005. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol. Microbiol. 58:289-304. [DOI] [PubMed] [Google Scholar]
- 52.Vianney, A., G. Jubelin, S. Renault, C. Dorel, P. Lejeune, and J. C. Lazzaroni. 2005. Escherichia coli tol and rcs genes participate in the complex network affecting curli synthesis. Microbiology 151:2487-2497. [DOI] [PubMed] [Google Scholar]
- 53.Vidal, O., R. Longin, C. Prigent-Combaret, C. Dorel, M. Hooreman, and P. Lejeune. 1998. Isolation of an Escherichia coli K-12 mutant strain able to form biofilms on inert surfaces: involvement of a new ompR allele that increases curli expression. J. Bacteriol. 180:2442-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang, X., J. F. Preston III, and T. Romeo. 2004. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 186:2724-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.White, R. J. 1968. Control of amino sugar metabolism in Escherichia coli and isolation of mutants unable to degrade amino sugars. Biochem. J. 106:847-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zogaj, X., W. Bokranz, M. Nimtz, and U. Romling. 2003. Production of cellulose and curli fimbriae by members of the family Enterobacteriaceae isolated from the human gastrointestinal tract. Infect. Immun. 71:4151-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zogaj, X., M. Nimtz, M. Rohde, W. Bokranz, and U. Romling. 2001. The multicellular morphotypes of Salmonella typhimurium and Escherichia coli produce cellulose as the second component of the extracellular matrix. Mol. Microbiol. 39:1452-1463. [DOI] [PubMed] [Google Scholar]