

Abstract
The first and key step in alkane metabolism is the terminal hydroxylation of alkanes to 1-alkanols, a reaction catalyzed by a family of integral-membrane diiron enzymes related to Pseudomonas putida GPo1 AlkB, by a diverse group of methane, propane, and butane monooxygenases and by some membrane-bound cytochrome P450s. Recently, a family of cytoplasmic P450 enzymes was identified in prokaryotes that allow their host to grow on aliphatic alkanes. One member of this family, CYP153A6 from Mycobacterium sp. HXN-1500, hydroxylates medium-chain-length alkanes (C6 to C11) to 1-alkanols with a maximal turnover number of 70 min−1 and has a regiospecificity of ≥95% for the terminal carbon atom position. Spectroscopic binding studies showed that C6-to-C11 aliphatic alkanes bind in the active site with Kd values varying from ∼20 nM to 3.7 μM. Longer alkanes bind more strongly than shorter alkanes, while the introduction of sterically hindering groups reduces the affinity. This suggests that the substrate-binding pocket is shaped such that linear alkanes are preferred. Electron paramagnetic resonance spectroscopy in the presence of the substrate showed the formation of an enzyme-substrate complex, which confirmed the binding of substrates observed in optical titrations. To rationalize the experimental observations on a molecular scale, homology modeling of CYP153A6 and docking of substrates were used to provide the first insight into structural features required for terminal alkane hydroxylation.
Various microorganisms utilize aliphatic alkanes as their sole carbon and energy source. They contain enzyme systems like the diiron- and copper-containing methane monooxygenases found in many methanotrophs, such as Methylococcus capsulatus (Bath) (24), integral membrane-bound diiron AlkB proteins present in many α, β, and γ-proteobacteria and high-G+C-content gram-positive bacteria (40), and membrane-bound heme-containing cytochrome P450 enzymes (CYP) found in many alkane-degrading yeasts and fungi (3, 9, 36).
CYPs have been identified in all kingdoms of life (http://drnelson.utmem.edu/cytochromep450.html). In the prokaryotes, CYPs are involved in the synthesis of secondary metabolites and in the degradation of alternative—typically hydrophobic—carbon sources. Well-studied examples are P450cam, P450cin, P450bioI, and the thermophilic CYP119 (13, 15, 23). Recently, we found that many strains able to metabolize linear alkanes (40) contain CYP enzymes (37) related to CYP153A1 (the first example of this CYP family, which was purified and cloned from Acinetobacter sp. EB104) (2, 21). These strains include Alcanivorax borkumensis, which makes up a large part of the biomass in oil-polluted marine environments (14), and alkane-degrading strains such as Sphingomonas sp. HXN-200, Oleomonas sagaranensis HXN-1400, and several Mycobacterium sp., isolated from a trickling-bed bioreactor set up to remove hexane from an air stream (39). They contain one or more alkane-hydroxylating P450 enzymes that belong to the CYP153 family (21, 37), instead of—or in addition to—the well-studied terminal alkane-hydroxylating enzyme systems, such as AlkB.
Eleven CYP153 genes, responsible for aliphatic alkane degradation, have now been cloned. Since CYP153 enzymes are class I P450 proteins requiring the presence of an electron-delivering protein system (ferredoxin and ferredoxin reductase protein), eight CYP153 genes together with a CYP153 family ferredoxin and ferredoxin reductase protein have been functionally expressed in Pseudomonas putida and Pseudomonas fluorescens, allowing the host to use aliphatic alkanes ranging from pentane to dodecane as the sole carbon and energy source (37).
The application of these enzymes in biotransformations was recently reported. With recombinant P. putida cells the terpene limonene was converted with a high yield into perillyl alcohol, a putative anticancer agent (4). Sphingomonas sp. HXN-200, which contains five different CYP153 enzymes, was shown to hydroxylate piperidines, pyrrolidines, and azetidines to useful pharmaceutical intermediates (19, 41). This broad substrate specificity and the ability to hydroxylate the terminal, most unreactive carbon atom of aliphatic alkanes make the CYP153 enzymes an interesting P450 family for the synthesis of pharmaceutical intermediates but also from an environmental perspective. However, the catalytic, biochemical, and biophysical properties of isolated CYP153 P450 enzymes, which could explain these features, have not been studied yet.
In this paper we describe the detailed characterization of one member of the CYP153 family, CYP153A6 from Mycobacterium sp. HXN-1500. Purification and characterization as well as kinetics, substrate binding, and modeling studies indicate why this enzyme hydroxylates C6 to C11 alkanes with ≥95% specificity for the terminal alkane position.
MATERIALS AND METHODS
Strain and growth conditions.
P. putida GPo12 (pGEc47ΔB) containing a broad-host-range expression vector with a polycistronic construct of the CYP153A6 P450 enzyme, ferredoxin, and ferredoxin reductase from Mycobacterium sp. HXN-1500 was described previously (pCom8-PFRA6) (41). The recombinant strain was grown on minimal E2 agar medium (3.5 g/liter NaNH4HPO4 · 4H2O, 7.5 g/liter K2HPO4 · 3H2O, 3.7 g/liter KH2PO4, 2 mM MgSO4, 15 g/liter Difco agar) with n-octane provided as vapor. All chemicals were from Sigma Aldrich or Acros and of the highest purity grade.
Large-scale cell material production.
To obtain sufficient cell material for enzyme purification and characterization, and for biotransformations, P. putida GPo12 (pGEc47ΔB-pCom8-PFRA6) was cultured on minimal E2 medium with 34.8 mM (NH4)2SO4, 1.87 mM NH4Cl, 0.1 mM CaCl2, 1 ml/liter trace-element solution (17) with 1 to 2% liquid octane added as the sole carbon and energy source. A two-liter fermentor (BioEngineering, Switzerland) was inoculated with a 100-ml preculture, and cells were grown for 24 h to a turbidity at 450 nm of 53 with continuous feeding of octane. These cells were used to inoculate a 30-liter fermentor (MBR Bioreactor, Switzerland). After reaching a turbidity at 450 nm of 45 after 15 h, the fermentation was stopped and cells were harvested by centrifugation. The cell paste was pelleted (15.000 × g, 15 min) and stored at −80°C until use.
Purification of CYP153A6.
Cell paste of P. putida GPo12 (pGEc47ΔB-pCom8-PFRA6) was resuspended in buffer A (50 mM potassium-phosphate buffer [pH = 7.4], 5% glycerol) with 1 mM dithiotreitol (DTT) (Gerbu, Germany), 200 μM phenylmethylsulfonyl fluoride (PMSF) at a concentration of 20 g cell dry weight/liter. Five millimolar EDTA was added, and the suspension was shaken at 200 rpm for 15 min at 30°C in an orbital platform. To separate the cells from the suspension, the solution was ultracentrifuged at 150,000 × g for 20 min. The supernatant that contained the P450 protein was further used for purification.
All solutions used in the purification of CYP153A6 contained 1 mM DTT, 200 μM PMSF, and 1 to 5 mM EDTA. The supernatant was brought to 40% (NH4)2SO4 saturation with a saturated (NH4)2SO4 solution (60 min at 0°C) and centrifuged (150,000 × g, 20 min). (NH4)2SO4 was added to the supernatant to 60% saturation, and after slow stirring for 60 min at 0°C the solution was centrifuged (150,000 × g, 20 min). The resulting pellet containing CYP153A6 was resuspended in buffer A with 1 M (NH4)2SO4 and centrifuged (15,000 × g, 5 min), and the supernatant was loaded on a phenyl-Sepharose (Pharmacia Biotech) hydrophobic interaction column (10 by 1.25 cm) preequilibrated with buffer A-1 M (NH4)2SO4. A 60-min linear gradient to buffer A-0 M (NH4)2SO4 was applied without the elution of CYP153A6. Subsequently, CYP153A6 was eluted from the column with demineralized water. Immediately after elution, a 1/10 volume of a 10-times-concentrated stock solution of buffer A was added to the pooled protein sample to stabilize the protein solution.
Fractions containing CYP153A6 were collected and applied to a Source 15Q (Pharmacia Biotech) column (7 by 1.25 cm) equilibrated with buffer A. After washing, a 60-ml gradient to buffer A-0.5 M NaCl was applied. P450 eluted at approximately 0.15 M NaCl. Fractions containing the protein were concentrated using an ultrafiltration unit (Millipore) (cutoff, 5 kDa), and a 500-μl sample was applied to a BioSec gel filtration column (Fractogel EMD BioSEC650; Merck), which was eluted with buffer A. After pooling, P450 was concentrated and fractions were stored at −20°C until use.
Alkane hydroxylation activity assay.
Cell extract was prepared by disruption of a 20-g-cell-dry-weight/liter cell suspension in buffer A using two rounds of French press. The concentration of CYP153A6 in the cell extract was determined from CO difference spectra. After ultracentrifugation (150,000 × g, 20 min), 1-ml supernatant samples were pipetted into 10-ml screw-caped glass tubes, and 1 mM DTT, 2.5 mM NADH (Gerbu, Germany), and substrate were added. The concentration of added substrate was approximately 6 mM, well above the solubility of the different alkanes, ranging from 0.02 to 100 μM (Table 1). The glass tubes were shaken in an orbital shaker at 200 rpm and 30°C. To extract the substrate and products, 1 ml of ethylacetate or hexane with 0.01% tetradecane as an internal standard was added to the tubes. After vigorous mixing, the samples were centrifuged (15,000 × g, 5 min); the ethylacetate or hexane layer was harvested, dried with Na2SO4, stored in a capped GC vial, and analyzed by GC (Fisons Instruments MFC800, Italy) (carrier gas, H2; flow rate, 2 ml/min). Samples were analyzed on an Optima-5 column (Macherey-Nagel, Switzerland), using the following temperature program: 5 min isotherm at 40°C, ramp of 10°C per min to 140°C, ramp of 20°C per min to 280°C, 5 min isotherm at 280°C. The reaction product concentrations were calculated from the peak integrated area using the integrated area of 0.01% tetradecane as the standard. Specific activities were calculated based on product and CYP153A6 concentrations.
TABLE 1.
Activity of CYP153A6 in cell extractsa and substrate binding parameters towards various substrates of purified protein
| Substrate | Activity (min−1)b | Solubilityc (μM) | Apparent Kd value (μM)b | High spind (%) |
|---|---|---|---|---|
| Hexane | 0.7 (0.3) | 93.4 | 3.7 (0.4) | 40 |
| Heptane | 21.2 (4.2) | 24.4 | 0.48 (0.13) | 78 |
| Octane | 60.8 (10.6) | 4.0 | 0.17 (0.03) | 87 |
| Nonane | 49.7 (11.1) | 1.22 | 0.022 (0.016) | 100 |
| Decane | 13.8 (3.0) | 0.27 | 0.010 (0.009) | 90 |
| Undecane | 0.4 (0.1) | <0.01 | 0.020 (0.003) | 38 |
| 2-Methyl octane | 35 (5) | ∼1e | 0.020 (0.008) | 95 |
| 2,5-Dimethyl hexane | ND | ∼1-2 | 1.5 (0.15) | 41 |
| Limonene | 31.2 (1.4) | 0.84 | 4.7 (0.7) | 23 |
| p-Cymene | 38.9 (7.5) | 1.49 | 5.8 (1.1) | 37 |
| Ethyl cyclohexane | ND | ∼7.8f | 4.2 (0.5) | 17 |
The CYP153A6 concentration in the cell extract was ∼2 μM; ND, not determined.
Numbers in parentheses are errors.
Solubility of the substrates in water at 20°C (29).
Percentage of high spin is given relative to nonane.
Estimate for 4-methyl octane from http://srdata.nist.gov/solubility/index.asp.
At 77°C from http://srdata.nist.gov/solubility/index.asp.
Because 2-octanol and 1-octanal could not be separated on the Optima-5 column, a Lipodex A column (Macherey-Nagel, Switzerland) was used to distinguish between the two products with the following temperature program: 5 min isotherm at 40°C, ramp of 10°C per min to 140°C, ramp of 20°C per min to 180°C, 5 min isotherm at 180°C. The conversion of limonene to perillyl alcohol and the hydroxylation of p-cymene were measured as described earlier on a Optima-5 column (41). However, a different GC temperature program was used to analyze the samples: 1 min isotherm at 80°C, ramp of 10°C per min to 140°C, ramp of 20°C per min to 280°C, 5 min isotherm at 280°C.
A gas chromatography-mass spectrometry (GC-MS) method was used to confirm the assignment of the peak that migrated at the retention time of 2-octanol. Samples were separated on a Lipodex column, as described above, using a Fisons 8000 series GC equipped with a Fisons MD800 MS. The GC-MS spectra were analyzed with MassLab software. To identify 2-octanol, the fragmentation pattern of the presumed 2-octanol peak was compared to the fragmentation pattern of pure 2-octanol and a reference fragmentation pattern (http://webbook.nist.gov/chemistry/name-ser.html).
UV-Vis characterization.
UV-visible (UV-Vis) spectra of purified CYP153A6 were recorded on a Cary 1E spectrophotometer (Varian, Germany) using a 0.5-ml quartz cuvette with a 1-cm path length. Measurements were performed in buffer A at 20°C. CYP153A6 was reduced by the addition of 10 mM dithionite from a freshly prepared 1 M stock solution, and the carbon monoxide complex was formed by slow bubbling with CO gas for approximately 30 to 60 s. P450 concentrations were determined by the difference in absorption at A450 to A490 between the reduced and the reduced CO-bound spectrum with an extinction coefficient of 91 M−1 cm−1 (27).
Spectral determinations of Kd for the binding of various substrates were performed by the consecutive addition of 0.25- to 1-μl aliquots from stock solutions at 20°C (maximum, 5 μl) to the CYP153A6 solution. Stock solutions (0.25 to 5 mM) were made in ethanol, and protein concentrations were typically around 0.1 mg/ml. To determine Kd, the difference between A392 and A418 was taken, corrected with the A392-to-A418 value at 0 μM substrate, and plotted against the substrate concentration. The data were fitted with the following equations (26) using the program Igor Pro (WaveMetrics).
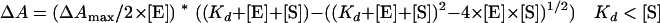 |
(1) |
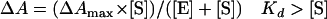 |
(2) |
where E and S are enzyme and substrate, respectively. The temperature dependence of UV-visible spectra of enzyme and enzyme-substrate complexes at pH 7.4 was measured by raising the temperature of the cuvette from 10 to 40°C in steps of 5°C. After the temperature was raised, the cuvette was left for 5 min to accommodate to the temperature before a spectrum was recorded. To measure a possible pH-dependent change in the UV-visible spectrum, spectra of the native enzyme were recorded at the appropriate pH in 50 mM Na acetate, 50 mM K phosphate buffer, and 5% glycerol before and after 5 μM octane was added.
EPR spectroscopy.
Oxidized samples of CYP153A6 were concentrated to approximately 200 μM in 50 mM potassium phosphate buffer (pH 7.4). Twenty percent glycerol was added in order to obtain a good glass upon freezing in liquid nitrogen. Sufficient amounts of octane were added to the enzyme sample from an ethanol stock solution. To obtain an enzyme-heptane complex, a sample of CYP153A6 was diluted with heptane-saturated buffer A, concentrated to 150 μM, and 20% glycerol was added. Measurements were performed at 10 K and several different microwave powers. The spectrometer used for electron paramagnetic resonance (EPR) measurements was a Bruker E500 working at X-Band (microwave frequency of 9.5 GHz). The spectrometer was equipped with a Bruker ER 4122 super-high-quality factor cavity and an Oxford continuous-helium-flow cryostat.
Homology modeling and docking.
To identify suitable templates for modeling, a BLAST search was performed with the sequence of CYP153A6 and X-ray structures present in the RCSB protein data bank at the SwissModel website (http://swissmodel.expasy.org//SWISS-MODEL.html) (7). Among the most similar proteins of which X-ray structures were determined is P450terp (CYP108, 1CPT; 29% identity). P450cam (CYP101, 1T87; 23% identity) was included in the alignments because it was engineered to accept aliphatic alkanes as the substrate. The ClustalW method was used in the MegAlign program of the DNAstar program package to align the protein sequences of CYP108, CYP101, and 11 CYP153 genes (37). The alignments were manually checked, and if necessary adjusted, with respect to conserved structural elements such as the proximal cysteinate and the catalytic threonine/aspartate pair. To validate the alignment further, the secondary structure of 1CPT was compared with the secondary structure predicted for CYP153A6 using PSIPRED (16). Once an acceptable alignment was produced, a protein model of CYP153A6 was generated using Modeler 8 (30). To include the cofactor, a heme molecule was docked in the generated protein model as described below using AutoDock 2.4. Fifty simulations were run, and the heme coordination with the lowest energy was implemented in the protein structure.
Substrate docking experiments were performed using AutoDock 2.4. Three-dimensional structures of alkanes were created and optimized in ChemDraw 3D Ultra. Preparation of substrate and enzyme was performed using the AutoDockTool program. All bonds of the substrate were defined to be rotational, and the enzyme was assumed to be rigid. Polar hydrogen atoms and Kollmann united atom charges were added to the enzyme, while nonpolar hydrogen atoms were removed from the substrate. Atomic solvation parameters were assigned using the AddSol program within AutoDockTools. The docking simulation was carried out using the Monte Carlo simulated annealing method, which is a grid-based method in which the substrate performs a random walk in protein space. Each grid point was precalculated with 0.375 Å between the grid points. The grid was centered in the protein center. Fifty simulations were run.
RESULTS AND DISCUSSION
Enzyme production and purification.
CYP153A6 was expressed in P. putida GPo12 (pGEc47ΔB-pCom8-PFRA6) cells by cultivating the recombinant with octane as the sole carbon source. Growth rates up to 0.33 h−1 were reached in a 30-liter bioreactor on minimal medium with a maximum of 2% octane as the second phase. Analysis of cell extracts of isolated P. putida GPo12 (pGEc47ΔB-pCom8-PFRA6) cells showed that five percent of the total protein was P450 protein. This corresponds to a production level of 300 mg CYP153A6 per liter culture volume.
Cell extracts for purification were obtained by incubating cells in 50 mM phosphate buffer, 5% glycerol, 5 mM EDTA, 1 mM DTT, 0.2 mM PMSF, followed by centrifugation. The CYP proteins were released from the cytoplasm by treatment with EDTA to chelate membrane-stabilizing metal ions. Subsequent incubation in 50 mM phosphate buffer resulted in an osmotic shock that released the CYP proteins, presumably by lysis or permeabilization of the cells (10, 34). CO difference spectra showed that the supernatant contained approximately 90% of the CYP protein content in comparison to the amount of CYP protein in cell extracts obtained by use of a French press. In addition, the resulting activity found after EDTA treatment is around 80 to 90% of that found after use of a French press, showing that most of the CYP present in the cell is released into the supernatant. However, the first method affords a better separation of supernatant and cell debris. CYP153A6 was purified as previously described with several adjustments (41): a 40 to 60% (NH4)2SO4 cut was further purified by hydrophobic interaction chromatography, anion exchange chromatography, and a gel filtration chromatography step at 4°C. Protein samples were kept on ice. In a typical purification run, approximately 10 mg enzyme was purified to homogeneity (>98%) based on overloaded sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. A purification yield of 34.5% and a purification factor of 20 were obtained. The hydrophobic interaction chromatography purification step resulted in a significant loss of P450 enzyme, but this step was necessary to separate active P450 enzyme from a protein that absorbs at 420 nm but does not bind CO.
Spectral characterization.
The UV-visible light spectra of oxidized CYP153A6 showed typical P450 absorption bands (Fig. 1). The absorption maximum of the Soret band was located at 418 nm. The α- and β-absorption bands, which are lower in intensity, were found at 567 and 538 nm, respectively. Addition of sodium dithionite had no effect on the position of the Soret band but reduced the absorption intensity. Upon reduction and CO binding, the enzyme showed the expected absorption maximum of 448 nm, Moreover, the α- and β-absorption bands changed to a single spectral feature at 545 nm. Small amounts of the P420 form were also observed.
FIG. 1.

UV-Vis spectra of purified oxidized CYP153A6 (– – –), dithionite-reduced CYP153A6 (· · · ·), and carbon monoxide complex (—). The insert shows the CO difference spectrum.
Catalytic properties.
The CYP153 enzymes are the first soluble P450 enzymes that specifically display hydroxylating activity towards the terminal position of alkanes. Hydroxylation activity was measured using cell extracts prepared by French press treatment of P. putida GPo12 (pGEc47ΔB-pCom8-PFRA6). Although the preparation of cell extracts with the described EDTA method is easier, the French press method was used because this yielded higher hydroxylation activities. Possibly, the EDTA treatment resulted in lower ferredoxin and ferredoxin reductase concentrations due to incomplete release in the supernatant. In the assays, a CYP concentration of at least ∼0.05 mg/ml was necessary to obtain significant and reproducible hydroxylation activities. Below this concentration, a sharp decrease in activity was observed, which could result from reduced interaction of the three components of the P450 enzyme system.
The rate of product formation from aliphatic alkanes was determined by GC analysis. Hexane was hydroxylated with a turnover number of 0.74 min−1, while octane, the preferred substrate, was hydroxylated with a turnover number of 60 min−1 (Table 1). All measurements were performed at pH 7.4, although subsequent measurements of activity as a function of pH showed an optimum around pH 6.8 to 7.0, with apparent pH values at which a residue in the enzyme-substrate complex involved in catalysis is (de)protonated of 6.1 and 7.5. Below pH 5 and above pH 9.5 the protein precipitated. The maximum turnover was 70 min−1, and fitting of the data indicated a maximal theoretical turnover of 90 min−1. With alkanes over 10 carbons in length, markedly lower kcat values were measured (e.g., 0.4 min−1 for undecane). Other substrates, such as limonene or p-cymene, were hydroxylated at the terminal methyl group giving perillyl alcohol and p-cumic alcohol, respectively, with turnover numbers near 35 min−1 (Table 1). Other terminal alkane-hydroxylating P450 enzymes, belonging to the CYP52 family, are membrane bound and catalyze terminal alkane hydroxylation with a reported turnover rate in the order of 27 min−1 (31, 32). P450cam from Pseudomonas putida and P450BM-3 from Bacillus megaterium were engineered to improve the enzyme-substrate fit for alkanes such as octane, butane, propane and even ethane, resulting in turnovers up to 750 min−1 (5, 6, 12, 28, 42). However, these enzymes have low regiospecificity with at most 10% hydroxylation at terminal positions, corresponding to an effective terminal hydroxylation activity of 50 to 100 min−1, in the same range as that of the CYP153s.
The regiospecificity of CYP153A6 is an interesting property of the enzyme. GC-MS analysis of in vitro octane hydroxylation experiments showed a major 1-octanol peak and a small peak (≤5%) identified as 2-octanol based on retention time and MS fragmentation spectrum (Fig. 2). The formation of minor amounts of 2-hydroxylated products in in vitro studies was not observed in growing cultures (37). The observation that P. putida GPo12 (pGEc47ΔB) can only metabolize 1-alkanols, not 2- or 3-alkanols, suggests that in vivo only 1-hydroxylation occurs (8, 38).
FIG. 2.

GC analysis of octane hydroxylation assays. The upper GC trace shows the standards, while the lower panel shows the analysis of the extracted products.
Substrate binding studies.
Binding of substrates to the P450 enzymes can induce a shift of the heme iron spin state towards the high-spin form, leading to a change in the Soret region of the P450 absorption spectrum. To determine if alkanes and other substrates are bound in the active site, spectral binding titrations were performed (Fig. 3). Almost all tested aliphatic alkanes induced a shift of the Soret band from 418 nm to 392 nm with an isosbestic point at 406 nm, which is typical for type I binding. The extent to which the alkanes induce a spin state perturbation varied between the different alkanes.
FIG. 3.

Titration curves of various alkanes: C6, first row left; C7, first row middle; C8, first row right; C9, second row left; C10, second row middle; C11, second row right; 2-methyl octane, third row left; 2,5-dimethyl hexane, third row middle; p-cymene, third row right; limonene, fourth row left; ethyl-cyclohexane, fourth row right. The inserts show the differences in absorption between A392 and A418 upon addition of micromolar concentrations (μM) of substrate and the fitted trace from which a Kd value was determined.
Octane and nonane induced the largest change, while hexane and undecane hardly changed the position of the Soret band. Plots of absorption change versus alkane concentration were hyperbolic. Fitting of these data with equations (equation 1, Kd, <1 μM; equation 2, Kd, >3 μM) generated apparent Kd values (Table 1). These should be interpreted with care, because at higher alkane chain lengths very sharp turning points were observed, which could be due to the very low solubility of these alkanes (near the fitted Kd) affecting the actual binding affinity. Especially for undecane, for which the solubility is really lower than the measured Kd value, substrate limitation might play a role. The observed change in low-spin/high-spin distribution is also indicative for this. For all tested alkanes, the fitted Kd values were in the range of ∼20 nM to 3.7 μM. The maximal difference in absorption upon alkane addition, and thus the amount of high-spin formation, was dependent on chain length. Nonane yielded, for example, the largest difference, which was set to 100% for comparison with other substrates, while hexane induced 40% high-spin features. And although enzyme activity and high-spin formation were linked, no linear correlation was found; a turnover of 0.4 min−1 generated a 40% high-spin conversion, while a turnover of 60 min−1 corresponded to 87% high-spin.
For the native enzyme, the spin state was not temperature dependent between 10 and 40°C, and only a low-spin spectrum was obtained. In the presence of shorter alkane substrates, i.e., heptane, at temperatures around 10°C, a high-spin signal was found, which partially converted into a low-spin spectrum at 40°C. This is in contrast to reported low-spin-to-high-spin conversion with increasing temperature (33, 35). With increasing alkane chain length, this effect was significantly reduced: with nonane and undecane, high-spin spectra were observed at all temperatures.
The effect of methyl side groups on substrate binding was assessed by titrations with 2-methyl-octane and 2,5-dimethyl-hexane (Fig. 3). 2-Methyl-octane, like octane, yielded almost 100% high-spin formation, and the Kd value decreased 10-fold to ∼20 nM, which equals the Kd value for nonane. An additional methyl group on the other end in 2,5-dimethyl-hexane did not change the high-spin formation compared to hexane; 41% high spin was observed. The Kd value of ∼1.5 μM was between the Kd values of hexane and octane. To further determine if the presence of an ionic group could play a role in the binding and orientation of substrate, the effect of pH on the UV-Vis spectra was examined. Although a minor effect on the formation of the high-spin species with hexane and heptane was observed at pH values between 6 and 7, no changes were measured when the UV-Vis spectra of CYP153A6 in the absence and presence of octane were recorded between pHs 5 and 9 (not shown).
Other substrates for CYP153A6, such as the terpenes limonene or p-cymene, did not induce strong spectral shifts either. Only 37% high spin was observed with p-cymene (Kd, 5.8 μM), while limonene induced 23% high spin (Kd, 4.7 μM).
Thus, alkanes appear to bind tightly, as shown by the temperature dependence of enzyme-alkane complexes and their extremely low dissociation constants. Furthermore, steric effects in the active site also appear to play a role, as shown by the increase in the binding constants of cyclic and aromatic substrates, such as limonene and p-cymene. These substrates do not exhibit clear type I binding (33). However, this could also result from their low solubility (29).
EPR spectroscopy.
To provide more evidence for the coordination of aliphatic alkanes in the substrate binding pocket, EPR studies with the absence or presence of substrate were performed. The EPR spectrum at 10 K of purified CYP153A6 at pH 7.4 showed a rhombic signal with one major species with g values at 1.90, 2.25, and 2.46 and another less intense feature with g values at 1.92, 2.25, and 2.41 (Fig. 4), reflecting a typical low-spin P450 heme. The observed heterogeneity is also shared by other CYPs (11, 18, 20). No inactive P450 was observed which could be associated with these low-spin signals. Addition of octane from a stock solution in ethanol did not yield the signals at lower field that are indicative of a high-spin species (data not shown). However, the spectrum changed in favor of the minor species in the native sample, forming almost one single low-spin signal with g values of 1.92, 2.25, and 2.41. The addition of pure ethanol, however, yielded the same signal (data not shown). To judge if an enzyme-octane complex was formed, a UV-Vis spectrum of a 100-fold-diluted sample was recorded at room temperature. This showed the Soret band at 418 nm, indicative of a low-spin species. Apparently, not enough octane can be supplied to a 200 μM protein sample, probably due to the low solubility of this alkane in buffer. None of the other compounds tested had a better solubility and induced a 100% conversion to a high-spin state. However, heptane also binds to the enzyme, as judged from the 78% high-spin features in the UV-Vis spectrum, and has almost sixfold higher solubility than octane (29). A sample of CYP153A6 was diluted in heptane-saturated buffer and then concentrated. Its EPR spectrum revealed several low-spin signals at 10 K and 0.063 mW, as well as a small high-spin signal (not shown). Apparently the high-spin/low-spin ratio at 10 K was much lower than the ratio at room temperature observed with UV-Vis measurements, where 78% high spin is observed. This temperature effect has also been observed for other CYPs and could result from a thermal spin equilibrium or a change in heme coordination by freezing effects (1, 11, 20). The different low-spin features could be assigned to three species with g values of 1.92, 2.25, and 2.48, g values of 1.90, 2.25, and 2.46, and g values of 1.97, 2.23, and 2.41 based on their relative intensities and saturation behavior. These were quite similar or identical to the spectra of other bacterial cytochromes P450 (11, 18, 20, 22, 25). The low-spin signals at g values of 1.90, 2.25, and 2.46 and g values of 1.92, 2.25, and 2.48 saturated at a lower microwave power than the g = 1.97, 2.23, 2.41 feature (not shown). A high-spin signal with g values at 1.68, 3.63, and 7.97 was observed, indicative of substrate binding and subsequent removal of the water molecule from the heme iron. Its intensity increased upon increasing the power to 10 mW due to its short relaxation times (not shown). To rule out the possibility that the low solubility of heptane prevented the formation of the enzyme-substrate complex (as was the case with octane), the sample was diluted in heptane-saturated buffer. In a fivefold-diluted sample, the high-spin feature remained unchanged. The relative presence of the low-spin feature with g values of 1.97, 2.23, and 2.41 was significantly enhanced while maintaining its saturation behavior (not shown). A 10-fold dilution showed the complete loss of the g = 1.90, 2.25, 2.46 and g = 1.92, 2.25, 2.48 features, leaving only the high-spin signal and the g = 1.97, 2.23, 2.41 low-spin species (Fig. 4). The remaining low-spin signal was also observed with P450BioI. Here, the signal was suggested to be a lipid-complexed enzyme form. On the basis of extensive EPR studies on P450cam, Lipscomb proposed that the g = 1.97 signal is unique for a substrate-complexed enzyme in which the substrate displaces the bound water molecule and coordinates to the heme iron (20). A second possibility is that residues, such as N- or S-containing amino acids, which are located near the heme are forced by the substrate into a position such that they interact directly with the heme iron. This bond would be rather weak, and an equilibrium with an off form is reasonable, which could give rise to the high-spin spectrum in UV-visible spectroscopy (20). A spectrum at room temperature with a maximum at 392 nm was observed without a significant shoulder at 418 nm, which is indicative of a high-spin species. Thus, a high-spin enzyme-substrate complex is observed as well as a low-spin spectrum with g values of 1.97, 2.23, and 2.41, which appears to be a low-spin enzyme-heptane complex.
FIG. 4.

EPR spectra of CYP153A6 at 10 K. Upper panel, 150 μM native enzyme; lower panel, 15 μM CYP153A6 in heptane-saturated buffer. The insert shows a blow-up of the low-spin signals between 260 and 380 mT. Conditions: microwave frequency, 9.47 GHz; power, 0.06 mW (native) or 10 mW (heptane saturated); modulation, 10 G at 100 kHz; temperature, 10 K.
Structural insight by homology modeling and docking.
Attempts to prepare diffracting crystals of CYP153A6 have not been successful yet. However, to gain information on the structure of CYP153A6 and to understand the terminal hydroxylation of aliphatic alkanes, we built a three-dimensional homology model of CYP153A6 based on the structures of other bacterial P450 enzymes (Fig. 5). The validity of the sequence alignment was checked by verifying the correct positioning of catalytically relevant residues and by comparing secondary structure elements. All P450 proteins have a very specific fold. The sequence identity between CYP153A6 and its template was 29%, and the identical residues were observed over the full gene sequence. The model suggested that the active site of CYP153A6 is predominantly hydrophobic in nature, as is the case for other CYPs. Residues lining the active site include Met101, Ile103, and Met306, which are located near the heme propionyl groups; Leu252, Val255, Gly256, Gly257, and Thr260 in the central I-helix; and Leu303, Phe407, and Val408.
FIG. 5.

Homology model of CYP153A6. Left panel: cartoon representation of the structural model of CYP153A6 generated with Modeler. The heme cofactor is presented space filled. Alkane substrates (pentane to undecane) were docked in the protein and shown to coordinate exclusively in the active site in a terminal position. Right panel: the coordination of undecane, in red, towards the heme cofactor (green) in CYP153A6. The central I-helix is shown, while residues coordinating the active site and substrates are depicted as stacks. The figure was generated using PyMol.
In silico docking of C5 to C11 alkanes showed that all of these molecules coordinate in exactly the same way in >90% of the docking runs, with the terminal carbon near the heme iron and the carbon chain in a bent conformation (forced by Phe407) alongside the I-helix towards the G-helix (Fig. 5). The aforementioned residues are all located within a 4.5-Å distance of the docked alkane molecule. With carbon chain lengths from 10 and higher or 5 and lower, many of the substrate molecules were also docked on the surface of the modeled CYP153A6 (up to 50%).
Comparison of the CYP153A6 active site residues with the structure of P450cam shows that more than half of the identified CYP153A6 active site residues are completely conserved in P450cam. In particular, the residues that are located in the central I-helix, to which the alkanes coordinate, are identical. An interesting observation is that in mutant P450cam, engineered for alkane hydroxylation, two of these conserved I-helixresidues were mutated to more hydrophobic residues. Val247Leu allowed propane hydroxylation, while Gly248Ala closed a cavity in the active site that was proposed to bind small molecules distant from the heme (42). Further examination of the homology model showed that several active site residues in CYP153A6 are not homologous to their counterparts in P450cam, which could indicate sites that determine terminal hydroxylation. These include the hydrophobic Ile103, located near the propionyl group in CYP153A6, which is aligned to the polar Thr101 in P450cam. This residue was mutated in P450cam (Thr101Leu) to allow ethane hydroxylation (42). Finally, at the positions corresponding to Met101 and Met306 in CYP153A6, Ile99 and Gly298 are present in P450cam. Thus, our experimental and in silico results suggest that the model is valid and might be useful for future structure-function studies.
Conclusion.
We demonstrate that CYP153A6 is the first soluble cytochrome P450 enzyme that preferentially hydroxylates unreactive aliphatic alkanes with high regioselectivity on terminal positions. Although short-chain aliphatic and cyclic alkanes bind in the active site, as shown by substrate binding, EPR, and molecular modeling studies, and are subsequently hydroxylated, medium-to-long-chain alkanes are preferred. Molecular insight into the binding of substrates provides a basis for understanding the preference of CYP153s to activate terminal carbon atoms, which allows their hosts to metabolize and grow on alkane molecules.
Acknowledgments
We thank Annik Perrenoud for help with GC-MS analysis. Johannes Michel is acknowledged for helpful discussions during purification of CYP153A6.
This work was supported by a grant from the Swiss National Funds (no. 205321-101674).
REFERENCES
- 1.Adachi, S., S. Nagano, K. Ishimori, Y. Watanabe, I. Morishima, T. Egawa, T. Kitagawa, and R. Makino. 1993. Roles of proximal ligand in heme proteins: replacement of proximal histidine of human myoglobin with cysteine and tyrosine by site-directed mutagenesis as models for P-450, chloroperoxidase, and catalase. Biochemistry 32:241-252. [DOI] [PubMed] [Google Scholar]
- 2.Asperger, O., R. Muller, and H. P. Kleber. 1983. Isolation of cytochrome-P450 and the corresponding reductase system from Acinetobacter. Acta Biotechnol. 3:319-326. [Google Scholar]
- 3.Ayala, M., and E. Torres. 2004. Enzymatic activation of alkanes: constraints and prospective. Appl. Catal. A Gen. 272:1-13. [Google Scholar]
- 4.Belanger, J. T. 1998. Perillyl alcohol: applications in oncology. Altern. Med. Rev. 3:448-457. [PubMed] [Google Scholar]
- 5.Bell, S. G., E. Orton, H. Boyd, J.-A. Stevenson, A. Riddle, S. Campbell, and L.-L. Wong. 2003. Engineering cytochrome P450cam into an alkane hydroxylase. Dalton Trans. 2003:2133-2140. [Google Scholar]
- 6.Bell, S. G., J. A. Stevenson, H. D. Boyd, S. Campbell, A. D. Riddle, E. L. Orton, and L. L. Wong. 2002. Butane and propane oxidation by engineered cytochrome P450(cam). Chem. Comm. 2002:490-491. [DOI] [PubMed] [Google Scholar]
- 7.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosetti, A., J. B. van Beilen, H. Preusting, R. G. Lageveen, and B. Witholt. 1992. Production of primary aliphatic alcohols with a recombinant Pseudomonas strain, encoding the alkane hydroxylase enzyme system. Enzyme Microb. Technol. 14:702-708. [Google Scholar]
- 9.Craft, D. L., K. M. Madduri, M. Eshoo, and C. R. Wilson. 2003. Identification and characterization of the CYP52 family of Candida tropicalis ATCC 20336, important for the conversion of fatty acids and alkanes to α,ω-dicarboxylic acids. Appl. Environ. Microbiol. 69:5983-5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Smet, M. J., J. Kingma, and B. Witholt. 1978. The effect of toluene on the structure and permeability of the outer and cytoplasmic membranes of E. coli. Biochim. Biophys. Acta 506:64-80. [DOI] [PubMed] [Google Scholar]
- 11.Girvan, H. M., K. R. Marshall, R. J. Lawson, D. Leys, M. G. Joyce, J. Clarkson, W. E. Smith, M. R. Cheesman, and A. W. Munro. 2004. Flavocytochrome P450BM3 mutant A264E undergoes substrate-dependent formation of a novel heme iron ligand set. J. Biol. Chem. 279:23274-23286. [DOI] [PubMed] [Google Scholar]
- 12.Glieder, A., E. T. Farinas, and F. H. Arnold. 2002. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 20:1135-1139. [DOI] [PubMed] [Google Scholar]
- 13.Green, A., S. Rivers, M. Cheeseman, G. Reid, L. Quaroni, I. Macdonald, S. Chapman, and A. Munro. 2001. Expression, purification and characterization of cytochrome P450 Biol: a novel P450 involved in biotin synthesis in Bacillus subtilis. J. Biol. Inorg. Chem. 6:523-533. [DOI] [PubMed] [Google Scholar]
- 14.Harayama, S., Y. Kasai, and A. Hara. 2004. Microbial communities in oil-contaminated seawater. Curr. Opin. Biotechnol. 15:205-214. [DOI] [PubMed] [Google Scholar]
- 15.Hawkes, D. B., G. W. Adams, A. L. Burlingame, P. R. Ortiz de Montellano, and J. J. De Voss. 2002. Cytochrome P450cin (CYP176A), isolation, expression, and characterization. J. Biol. Chem. 277:27725-27732. [DOI] [PubMed] [Google Scholar]
- 16.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195-202. [DOI] [PubMed] [Google Scholar]
- 17.Lageveen, R. G., G. W. Huisman, H. Preusting, P. Ketelaar, G. Eggink, and B. Witholt. 1988. Formation of polyesters by Pseudomonas oleovorans: effect of substrates on formation and composition of poly-(R)-3-hydroxyalkanoates and poly-(R)-3-hydroxyalkenoates. Appl. Environ. Microbiol. 54:2924-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawson, R. J., D. Leys, M. J. Sutcliffe, C. A. Kemp, M. R. Cheesman, S. J. Smith, J. Clarkson, W. E. Smith, I. Haq, J. B. Perkins, and A. W. Munro. 2004. Thermodynamic and biophysical characterization of cytochrome P450 BioI from Bacillus subtilis. Biochemistry 43:12410-12426. [DOI] [PubMed] [Google Scholar]
- 19.Li, Z., and D. L. Chang. 2004. Recent advances in regio- and stereoselective biohydroxylation of non-activated carbon atoms. Curr. Org. Chem. 8:1647-1658. [Google Scholar]
- 20.Lipscomb, J. D. 1980. Electron paramagnetic resonance detectable states of cytochrome P450cam. Biochemistry 19:3590-3599. [DOI] [PubMed] [Google Scholar]
- 21.Maier, T., H.-H. Foerster, O. Asperger, and U. Hahn. 2001. Molecular characterization of the 56-kDa CYP153 from Acinetobacter sp. EB104. Biochem. Biophys. Res. Commun. 286:652-658. [DOI] [PubMed] [Google Scholar]
- 22.McLean, K. J., M. R. Cheesman, S. L. Rivers, A. Richmond, D. Leys, S. K. Chapman, G. A. Reid, N. C. Price, S. M. Kelly, J. Clarkson, W. E. Smith, and A. W. Munro. 2002. Expression, purification and spectroscopic characterization of the cytochrome P450 CYP121 from Mycobacterium tuberculosis. J. Inorg. Biochem. 91:527-541. [DOI] [PubMed] [Google Scholar]
- 23.McLean, M. A., S. A. Maves, K. E. Weiss, S. Krepich, and S. G. Sligar. 1998. Characterization of a cytochrome P450 from the acidothermophilic archaea Sulfolobus solfataricus. Biochem. Biophys. Res. Commun. 252:166-172. [DOI] [PubMed] [Google Scholar]
- 24.Merkx, M., D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Muller, and S. J. Lippard. 2001. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: a tale of two irons and three proteins. Angew. Chem. 40:2782-2807. [DOI] [PubMed] [Google Scholar]
- 25.Miles, J., A. Munro, B. Rospendowski, W. Smith, J. McKnight, and A. Thomson. 1992. Domains of the catalytically self-sufficient cytochrome P-450 BM-3. Genetic construction, overexpression, purification and spectroscopic characterization. Biochem. J. 288:503-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murataliev, M. B., and R. Feyereisen. 2000. Functional Interactions in cytochrome P450BM3. Evidence that NADP(H) binding controls redox potentials of the flavin cofactors. Biochemistry 39:12699-12707. [DOI] [PubMed] [Google Scholar]
- 27.Omura, T., and R. Sato. 1964. The carbon monoxide-binding pigment of liver microsomes. J. Biol. Chem. 239:2370-2378. [PubMed] [Google Scholar]
- 28.Peters, M. W., P. Meinhold, A. Glieder, and F. H. Arnold. 2003. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc. 125:13442-13450. [DOI] [PubMed] [Google Scholar]
- 29.Riddick, J. A., W. B. Bunger, and T. K. Sakano. 1986. Organic solvents: physical properties and methods of purification, vol. 2. Wiley, New York, N.Y.
- 30.Sali, A., and T. L. Blundell. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779-815. [DOI] [PubMed] [Google Scholar]
- 31.Scheller, U., T. Zimmer, D. Becher, F. Schauer, and W. H. Schunck. 1998. Oxygenation cascade in conversion of n-alkanes to α,ω-dioic acids catalyzed by cytochrome P450 52A3. J. Biol. Chem. 273:32528-32534. [DOI] [PubMed] [Google Scholar]
- 32.Scheller, U., T. Zimmer, E. Kaergel, and W.-H. Schunck. 1996. Characterization of the n-alkane and fatty acid hydroxylating cytochrome P450 forms 52A3 and 52A4. Arch. Biochem. Biophys. 328:245-254. [DOI] [PubMed] [Google Scholar]
- 33.Schenkman, J. B., S. G. Sligar, and D. L. Cinti. 1981. Substrate interaction with cytochrome P-450. Pharm. Ther. 12:43-71. [DOI] [PubMed] [Google Scholar]
- 34.Schwaneberg, U., C. Otey, P. C. Cirino, E. Farinas, and F. H. Arnold. 2001. Cost-effective whole-cell assay for laboratory evolution of hydroxylases in Escherichia coli. J. Biomol. Screening 6:111-117. [DOI] [PubMed] [Google Scholar]
- 35.Sligar, S. G. 1976. Coupling of spin, substrate, and redox equilibria in cytochrome P450. Biochemistry 15:5399-5406. [DOI] [PubMed] [Google Scholar]
- 36.van Beilen, J. B., and E. G. Funhoff. 2005. Expanding the alkane oxygenase toolbox: new enzymes and applications. Curr. Opin. Biotechnol. 16:308-314. [DOI] [PubMed] [Google Scholar]
- 37.van Beilen, J. B., E. G. Funhoff, A. van Loon, A. Just, M. Bouza, R. Holtackers, M. Röthlisberger, and B. Witholt. 2006. Cytochrome P450 alkane hydroxylases of the CYP153 family are common in alkane-degrading eubacteria lacking integral-membrane alkane hydroxylases. Appl. Environ. Microbiol. 72:59-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Beilen, J. B., J. Kingma, and B. Witholt. 1994. Substrate specificity of the alkane hydroxylase system of Pseudomonas oleovorans GPo1. Enzyme Microb. Technol. 16:904-911. [Google Scholar]
- 39.van Beilen, J. B., Z. Li, W. A. Duetz, T. H. M. Smits, T. Plaggemeier, B. Witholt, and K.-H. Engesser. Hexane-degrading bacteria isolated from a trickling-bed bioreactor contain integral-membrane non-heme iron as well as soluble cytochrome P450 alkane hydroxylases. Submitted for publication.
- 40.van Beilen, J. B., Z. Li, W. A. Duetz, T. H. M. Smits, and B. Witholt. 2003. Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci. Technol. 58:427-440. [Google Scholar]
- 41.van Beilen, J. B., D. Lüscher, R. Holtacker, B. Witholt, and W. A. Duetz. 2005. Biocatalytic production of perillyl alcohol from limonene using a novel Mycobacterium sp. cytochrome P450 alkane hydroxylase expressed in P. putida. Appl. Environ. Microbiol. 71:1737-1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu, F., S. Bell, J. Lednik, A. Insley, Z. Rao, and L. Wong. 2005. The heme monooxygenase cytochrome P450(cam) can be engineered to oxidize ethane to ethanol. Angew. Chem. 44:4029-4032. [DOI] [PubMed] [Google Scholar]