

Abstract
Proteins internalized into the endocytic pathway are usually degraded. Efficient proteolysis requires denaturation, induced by acidic conditions within lysosomes, and reduction of inter- and intrachain disulfide bonds. Cytosolic reduction is mediated enzymatically by thioredoxin, but the mechanism of lysosomal reduction is unknown. We describe here a lysosomal thiol reductase optimally active at low pH and capable of catalyzing disulfide bond reduction both in vivo and in vitro. The active site, determined by mutagenesis, consists of a pair of cysteine residues separated by two amino acids, similar to other enzymes of the thioredoxin family. The enzyme is a soluble glycoprotein that is synthesized as a precursor. After delivery into the endosomal/lysosomal system by the mannose 6-phosphate receptor, N- and C-terminal prosequences are removed. The enzyme is expressed constitutively in antigen-presenting cells and induced by IFN-γ in other cell types, suggesting a potentially important role in antigen processing.
Reduction, oxidation, and isomerization of protein disulfide bonds in the cytosol and endoplasmic reticulum (ER) of eukaryotic cells are carried out by enzymes of the thioredoxin family (1). Protein disulfide isomerase and related molecules catalyze the formation and isomerization of protein disulfide bonds in the ER (2–4). Thioredoxin and glutaredoxin catalyze reduction of disulfide bonds in the cytosol and nucleus (1, 5). These enzymes use oxidized cofactors (e.g., oxidized glutathione) as electron sinks or reduced cofactors (e.g., glutathione) as electron donors for oxidation or reduction of protein disulfide bonds, respectively (5, 6). Members of the thioredoxin family often share little sequence similarity but do possess a common active site (WCGH/PCK) and folding pattern (7–10). The cysteine residues in the active site are believed to act by transferring electrons between themselves and either the substrate protein or cofactors (9).
Disulfide bond reduction also occurs in the endocytic pathway. Most proteins that enter the endocytic pathway are degraded in lysosomes to small peptides and free amino acids. Denaturation of proteins is a prerequisite for lysosomal proteolysis (reviewed in ref. 11) and is facilitated by the acidic pH of the lysosome (12). A reducing environment within the endocytic pathway also facilitates denaturation by cleaving disulfide bonds in substrate proteins (reviewed in ref. 13). Various groups have demonstrated reducing activity in lysosomes (14–17) and endosomes (18). Although the presence of excess cysteine favors the reduction of disulfide bonds (16), the process is not favored at low pH (19), and no enzyme(s) that catalyzes reduction in these compartments has been described. We have now defined such an enzyme and have named it Gamma interferon-inducible lysosomal thiol-reductase (GILT). GILT was originally described by Luster et al. (20) and called IP30. It is synthesized as a 224-aa precursor that is transported to endocytic compartments by mannose-6-phosphate receptors (M6PR) and has thiol reductase activity with an acidic pH optimum. Induction by IFN-γ suggests a possible immunological function, and the potential role of GILT in MHC class II-restricted antigen processing is discussed.
Materials and Methods
Cells and Antibodies.
The human B-lymphoblastoid cell lines (B-LCL) used were previously described (21). The J3 melanoma cell line was a gift from Janet Lee, Memorial Sloan–Kettering Cancer Center, New York. COS-7 cells were from the American Type Culture Collection. The antibodies used were all previously described (21, 22), except for the rabbit anti-GILT serum, which was raised to affinity-purified mature GILT.
GILT Sequencing and Production of GILT-Expressing Cell Lines.
Full-length human GILT cDNA was amplified from a Raji cDNA library (gift from J. Douhan and L. Glimcher, Harvard University) or T2 cDNAs by PCR by using pfu DNA polymerase (Stratagene) and standard PCR conditions. Primers GILT-5 (5′-GTCCCGGGATCCGCCACCATGGATAGTCGCCACACC-3′; plus strand, 5′-end) and GILT-3 (5′-TCGGATCCTACGTATCATGGGATGCATAAAAT-3′; minus strand, 3′-end) were designed from the published sequence (20). Cleavage sites for enzymes SmaI and BamHI to the GILT-5 primer and SnaBI and BamHI to GILT-3 primer were added, and a consensus Kozak sequence was placed in the GILT-5 primer. After sequencing, GILT cDNA was cloned into the expression vector RSV.Neo. The melanoma cell line J3 was transfected with the construct (yielding J3/GILT) or vector alone (yielding J3/Neo) by electroporation at 960 μF and 200 mV and selected in 1 mg/ml neomycin. GILT-positive clones were identified by indirect immunofluorescence and Western blotting (21).
Metabolic Radiolabeling and Immunoprecipitations.
Pala cells (106–107) were labeled in Brefeldin A with l-[35S]methionine and l-[35S]cysteine (Amersham) (0.5–1.0 mCi), chased, and immunoprecipitations performed as previously described (21).
GILT Purification.
B-LCL were extracted in 0.15 M NaCl/0.01 M Tris (TS), pH 6.9, containing 2% C12E9 for 1 hr on ice at 4 × 107 cells per milliliter. The extract was centrifuged at 1,200 × g for 10 min and further clarified at 100,000 × g for 1 hr at 4°C. The extract was passed through a mouse IgG-Biogel A15 m followed by a MAP.IP30-Biogel A15 m column equilibrated in TS, pH 6.9/0.1% C12E9. After washing with 0.3 M NaCl, TS, pH 6.9/0.6% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), followed by TS, pH 6.9, 0.6% CHAPS, the column was eluted with 0.1 M sodium acetate, pH 3.5/0.6% CHAPS, and the eluate dialyzed against 0.15M NaCl.
Deglycosylation and Matrix-Assisted Laser Desorption Ionization (MALDI)-MS Analysis of Tryptic Peptides.
Affinity-purified GILT (20 μg) was heated at 100°C for 5 min in 0.02 M sodium phosphate buffer/0.1%SDS/5 mM DTT, pH 7.5 and alkylated by using iodoacetamide (25 mM). The sample was adjusted to 1% CHAPS/10 mM EDTA/0.2% 2-mercaptoethanol and incubated overnight at 37°C with N-glycosidase F (3 units). After SDS/PAGE, the material was subjected to tryptic digestion and peptides analyzed by MALDI-MS.
Electrophoresis and Western Blotting.
One- and two-dimensional SDS/PAGE and fluorography were performed as previously described (21). Bands were quantitated with a Bio-Rad GS-250 Molecular Imager. Western blotting was performed as previously described (21). For M6P detection, blots were incubated with biotinylated M6PR (a gift from Peter Lobel, Rutgers University, NJ) followed by streptavidin-horseradish peroxidase (HRP; Molecular Probes). Bands were detected with SuperSignal CL-HRP (Pierce) and Kodak Biomax MR film.
In Vitro Assay for Thiol Reductase Activity.
Affinity-purified rabbit anti-mouse F(ab′)2 (Jackson ImmunoResearch) was iodinated by the chloramine T method (23, 24) and quantitated by absorbance at 280 nm. F(ab′)2 was denatured by boiling in 0.2% SDS and diluted in 0.1% Triton X-100. Assays were carried out at 37°C. Each reaction (20 μl) contained 10,000 cpm SDS-denatured F(ab′)2. For Km determinations, F(ab′)2 was used at concentrations between 125 pM and 116 nM containing 80,000–640,000 cpm. GILT or thioredoxin (Spirulina sp., Sigma) was preactivated with 50 μM DTT for 10 min at 37°C (final concentration, 25 μM). The reaction was terminated by the addition of iodoacetamide to 5 mM, the samples analyzed by nonreducing SDS/PAGE, and individual bands quantitated as above.
Immunoelectron Microscopy.
Raji cells were treated with IFN-γ overnight and processed for electron microscopy, as previously described (25). Ultrathin cryosections were indirectly single or double immunolabeled with 10 nm protein-A/gold particles or 10 and 15 nm protein-A/gold particles, as previously detailed (26). Rabbit anti-human invariant chain C terminus, rabbit anti-HLA-DR (27), MAP.IP30, and R.IP30N were the antibodies used.
Assay for Cellular GILT Activity.
J3/GILT and J3/Neo cells were incubated at 37°C for 1 hr in dialyzed FBS, washed twice with cold Hanks' balanced salt solution (HBSS) containing 10% FBS and 400 μM 5,5′-dithio-bis (2-nitrobenzoic acid) (DTNB, Sigma), to inhibit cell-surface reduction (19), and suspended in cold HBSS/FBS containing DTNB. The cells were incubated at 0°C for 30 min with 125I-H5C6 (CD63 mAb), washed twice at 0°C, and resuspended in HBSS/FBS at 37°C. At different time points, N-ethylmaleimide (20 mM final concentration) was added and the cells extracted in 0.1% Triton X-100 containing protease inhibitors. The extracts were analyzed by nonreducing SDS/PAGE and the individual bands quantitated.
PCR-Based Mutagenesis.
GILT was subcloned from pBluescript KS+/GILT into the vector pcDNA 3.1(−) puro (a gift from Jaana Karttunen), and the construct was used as the template for PCR-based mutagenesis. The mutagenic primers used were C46S sense (5′-CTATGAAGCACTGAGCGGTGGCTGCCGAGCCTTCCTG-3′), C46S antisense (5′-CAGGAAGGCTCGGCAGCCACCGCTCAGTGCTTCATAG-3′), C49S sense (5′-CTATGAAGCACTGTGCGGTGGCAGCCGAGCCTTCCTG-3′), C49S antisense (5′-CAGGAAGGCTCGGCTGCCACCGCACAGTGC- TTCATAG-3′), C46S/C49S sense (5′-CTATGAAGCACTGAGCGGTGGCAGCCGAGCCTTCCTG-3′), and C46S/C49S antisense (5′-CAGGAAGGCTCGGCTGCCACCGCTCAGTGCTTCATAG-3′). For each construct, two fragments encompassing the mutation were PCR amplified in reactions containing either GILT-5 and the mutagenic antisense primers or GILT-3 and the mutagenic sense primers. In a second reaction, the fragments were annealed, extended, and amplified with GILT-5 and GILT-3, and the PCR products were cloned again into pcDNA 3.1 (−) puro.
Transient Transfections in COS-7 Cells.
COS-7 cells were seeded overnight in T75 flasks, grown to 80% confluency, and transfected by using CellFECTIN (Life Technologies, Grand Island, NY). After 48 h, the cells were harvested and lysed in 1% Triton X-100 in TS, pH 6.9. GILT was immunoprecipitated with MAP.IP30-A15 m beads, eluted with 120 μl 0.1% Triton X-100/100 mM NaCl/50 mM acetate, pH 3.5, and the pH was adjusted to pH 4.5 with 1 N NaOH before assay.
Results
Nucleotide and Predicted Amino Acid Sequence of GILT.
We sequenced GILT cDNAs amplified from mRNA isolated from the T − B hybrid cell line T2 and a cDNA library derived from the B cell line Raji. Comparison with the published sequence (20) revealed a number of differences (Fig. 1A). These resulted in a shift in the reading frame from nucleotide position 470 and a different predicted amino acid sequence in the C-terminal half of the molecule, as well as a number of additional amino acid changes. On the basis of our sequence, GILT is a 261-aa protein comprised of a 37-aa signal peptide and 224-aa proform. The proform has 11 cysteine residues and 3 potential N-linked glycosylation sites at residues 37, 69, and 82. Rabbit antisera against peptides derived from the predicted N-terminal (R.IP30N) and C-terminal (R.IP30C) amino acid sequences (21) were able to immunoprecipitate a 35-kDa protein from a detergent extract of Pala B-LCL labeled with [35S]methionine [Fig. 1B; (21)], confirming the sequence. A similar protein was precipitated from human monocytic cells (U937, MonoMac 6) and several B-LCL (data not shown). Immunoprecipitation of the 35-kDa protein was inhibited only by the specific peptides against which the sera were raised [(21); data not shown].
Figure 1.

(A) GILT cDNA and predicted amino acid sequence. The predicted amino acid sequence of GILT is shown in bold above its nucleotide sequence (GenBank accession no: AF097362). The signal peptide sequence is italicized, propeptide sequences are underlined, potential glycosylation sites are boxed, and cysteines are circled. The active site (CXXC) is enclosed by an oval. (B) Secretion and intracellular processing of GILT. Pala cells were labeled with [35S]methionine for 1 hr and chased for different periods of time. Secreted GILT was precipitated from supernatants with R.IP30N or R.IP30C sera. Intracellular GILT was precipitated from cell extracts with R.IP30N, R.IP30C, or MAP.IP30 antibodies. Samples were analyzed by SDS/PAGE (12%) under reducing conditions and visualized by autoradiography. (Left). The intensity of specific bands is plotted as relative intensity (PD, pixel density) vs. time (hr) (Right). In the lower panel the filled squares represent mature GILT.
Secretion and Intracellular Processing.
Pala cells were labeled for 1 hr in the presence of Brefeldin A (21), washed to remove the Brefeldin A, and chased at 37°C in the presence of unlabeled methionine and cysteine. Cells and supernatants were collected and analyzed by immunoprecipitation. Fig. 1B shows that a small fraction (<20%) of GILT was secreted as the proform, whereas the majority remained intracellular and was proteolytically processed. GILT precipitated with R.IP30N or R.IP30C was detectable in the supernatant (Fig. 1B Upper) as a 35-kDa protein as early as 2 hr, reached the maximum level at 6 hr, and remained the same throughout the chase (21). At early time points, these sera also precipitated a 35-kDa protein from detergent extracts of the cells (Fig. 1B Lower). At later chase points, the mAb raised to mature GILT [MAP.IP30; (21)] precipitated an additional species from the extracts with an Mr of 30 kDa. The appearance of the 30-kDa protein paralleled the disappearance of the 35-kDa protein, reaching a maximum at 6 hr and then remaining steady throughout the chase. The 35-kDa species is the GILT proform, and the 30-kDa species is the mature form.
The N terminus of purified mature GILT was sequenced by Edman degradation. The N-terminal residues were a mixture of Asn-32 and Ala-33 of the proform. To determine the C terminus, tryptic peptides of deglycosylated GILT (see Materials and Methods) were analyzed by matrix-assisted laser desorption ionization-MS. A peptide corresponding to residues 169 to Lys-206 was identified. Thus, 31–32 residues from the N terminus and, at the most, 18 residues from the C terminus of the proform are cleaved to generate the mature 30-kDa form of GILT. These cleavage sites are close to dilysine motifs, at residues 29, 30 and 206, 207, respectively. A large number of lysosomal enzymes are converted from precursor zymogen forms to mature active enzymes by proteolysis of N- or C-terminal propeptides (28), and many are cleaved at similar dibasic sequences (29, 30). Treatment of B-LCL with chloroquine, which neutralizes the acidic pH of endosomes and lysosomes, or the sulfhydryl protease inhibitor leupeptin causes the accumulation of the proform of GILT at the expense of the mature form (21), consistent with the maturation step occurring in the endocytic pathway.
Intracellular Localization.
The Burkitt's lymphoma cell line Raji was examined by electron microscopy. In MHC class II-expressing cells, late endosomes (LEs) and lysosomes have collectively been termed MIICs (31). Recently, an additional type of endosome was identified and characterized in B-lymphocytes, the so-called early MIIC, which is positioned at the interface between early endosomes (EEs) and LEs, and which represents the main entrance site to the endocytic pathway for MHC class II-invariant chain complexes (32). Early MIICs differ from typical EEs in morphology and contain less transferrin receptors (27). All these endocytic subcompartments were abundantly present in Raji cells, and their morphology is illustrated in Fig. 2. The labeling patterns of the proform of GILT (detected by using R.IP30N) and mature GILT (detected by MAP.IP30) showed little overlap. The proform was found in the ER, Golgi complex (not shown), and EEs/early MIICs together with invariant chain (Fig. 2A). There is a steep decrease of invariant chain labeling downstream of EEs in the endocytic pathway (27), probably because of proteolysis. Mature GILT was found only in multivesicular LEs and multilaminar lysosomes, i.e., MIICs, together with MHC class II molecules (Fig. 2B). As previously described, the MIICs in Raji cells are also positive for HLA-DM (33). These morphological data strongly suggest that the proform of GILT enters the endocytic system in EEs and is converted to the mature form immediately thereafter in multivesicular LEs where the majority of invariant chain degradation also occurs.
Figure 2.

Intracellular localization of GILT. Ultrathin cryosections of Raji cells double immunolabeled for the C terminus of invariant chain [rabbit anti-human invariant chain C terminus (IiC)] and the proform of GILT (Gi-N) (A), and MHC class II and mature GILT (Gi) (B). Gold sizes in nanometers are indicated on the plates. In A, IiC and proGILT colocalize in early endosomes ★, whereas MIICs ⋆ are negative. In B, MHC class II is present both in an early endosome ★ and in MIICs ⋆. Mature GILT can be seen only in MIICs. Bars (A and B) = 200 nm. (C) GILT is derivatized with M6P. Affinity-purified mature GILT was separated on isoelectrofocusing followed by SDS/PAGE under reducing conditions. Separated proteins were visualized by silver staining (Left) or transferred to Immobilon P membrane and probed with biotinylated M6PR followed by avidin coupled to horseradish peroxidase (Right).
GILT Is Mannose-6 Phosphorylated.
GILT is a soluble protein localized in lysosomes. Frequently, N-linked glycans of soluble lysosomal hydrolases bear M6P and are targeted to the endocytic pathway by the M6PR (34). We assayed for the presence of M6P on purified GILT by using biotinylated M6PR as a probe. Affinity-purified GILT was separated by two-dimensional PAGE, and its purity was analyzed by silver staining. A single spot with a pI of 4.8 corresponding to GILT was observed (Fig. 2C Left). Western blotting and probing with biotinylated M6PR clearly revealed the presence of M6P on GILT (Fig. 2C Right).
Mature GILT Has Thiol Reductase Activity Optimal at Low pH.
The amino acid sequence of GILT does not have significant homology with any known protein. There is a CXXC motif at positions 46–49 which, although not precisely the functional motif of the thioredoxin family, suggested possible thiol reductase activity. In addition, alignments of the GILT sequence with existing folding patterns in the database [ucla-doe structure-prediction server; (35)] revealed that the protein has a weak similarity to thioredoxin. Conventionally, a turbidimetry assay measuring insulin precipitation on reduction is used to measure thiol reductase activity (36), but this could not be adapted for analysis at low pH because of spontaneous insulin precipitation. We therefore developed a new assay by using 125I-F(ab′)2 as a substrate. Reduction of F(ab′)2 into Fab and heavy and light chains was analyzed by nonreducing SDS/PAGE followed by autoradiography. Initial experiments indicated that F(ab′)2 reduced more readily when denatured. We therefore used SDS-denatured F(ab′)2 as a substrate in all the experiments described except for some determinations of the Michaelis constant, Km (see below).
Fig. 3A shows assays of thioredoxin and the purified GILT preparation shown in Fig. 2C over a pH range of 4.0 to 7.0. With thioredoxin, maximum reduction was observed at pH 7.0 and 6.0, and no reduction was seen at pH 4.0. With GILT, reduction of F(ab′)2 was maximal between pH 4.0 and pH 5.0. In reactions that included identical concentrations of lysozyme as a control protein, detectable F(ab′)2 reduction, caused by the low level of DTT (25 μM) used in the assay was seen only at pH 7.0. The activity of GILT was also assayed kinetically at pH 4.0, stopping the reaction at different times by using excess iodoacetamide. With a control protein (lysozyme), no detectable reduction of F(ab′)2 was seen (Fig. 3B). In the presence of GILT, reduction of F(ab′)2 was detectable as early as 5 min and was almost complete by 20 min.
Figure 3.

GILT is an acid optimal thiol reductase. (A) Lysozyme (negative control), GILT (550 nM), or thioredoxin (1.04 μM) was incubated with 125I-F(ab′)2 (55 nM) under different pH conditions for 45 min at 37°C. (B) Lysozyme or GILT (550 nM) was incubated with 125I-F(ab′)2 (55 nM) at pH 4.0 and 37°C, and the reaction was stopped with excess iodoacetamide at different periods of time. (C) To quantitate the effect of substrate denaturation on GILT activity, 125 pM to 1 nM SDS-denatured 125I-F(ab′)2 (Left) and 14.5 nM to 116 nM native 125I-F(ab′)2 (Right) were incubated for 10 min at 37°C with 69 nM or 1.1 μM GILT, respectively, stopping the reaction by the addition of iodoacetamide. Samples were separated by nonreducing SDS/PAGE (12%) and visualized by autoradiography. In A and B, the gels are shown, and the positions of F(ab′)2, Fab, H′ and light chains are indicated (Left). The intensity of specific bands (pixel density) was quantitated and plotted in A as percent reduced heavy and light chains after background subtraction and in B as percent of maximum. In C, the bands were converted to molarity of substrate. The results are presented as Lineweaver–Burke plots.
The use of SDS-denatured F(ab′)2 as a substrate should theoretically decrease the energy requirement for reduction by eliminating the need to break noncovalent protein–protein interactions. To quantitate this effect, we performed experiments using both denatured and native 125I-F(ab′)2 as a substrate. The reaction was carried out at pH 4.0 for 10 min to determine initial rates, and the samples were analyzed by nonreducing SDS/PAGE. Fig. 3C shows the results in the form of Lineweaver–Burke plots. The Km value determined was 1.2 × 10−9 M when denatured F(ab′)2 was used as a substrate and 1.0 × 10−7M when native F(ab′)2 was used. The 100-fold higher Km for the native substrate presumably reflects the additional energy required to break noncovalent interaction between the light-chain and heavy-chain fragments. Similar Km values were obtained for thioredoxin (data not shown).
Active Site Determination by Mutagenesis.
To determine whether the CXXC motif at 46–49 constitutes the active site, the two cysteine residues were mutated individually and together to serine. Wild-type GILT and the mutants were expressed in COS-7 cells, isolated by immunoprecipitation from detergent extracts by using MAP.IP30, and assayed for activity (Fig. 4). Mutation of either or both cysteine residues abolished reducing activity (Fig. 4 B and C). Mutagenesis did not significantly affect intracellular processing as determined by Western blot analysis (Fig. 4A and data not shown) or subcellular localization as determined by immunofluorescence (data not shown).
Figure 4.

Identification of the GILT active site by mutagenesis. Wild-type GILT and mutants with Cys-46, Cys-49, or both converted to serine residues were expressed in duplicate in COS-7 cells, immunoisolated, and assayed for thiol reductase activity. GILT and all variants were equally well expressed as determined by Western blotting with rabbit anti-GILT serum (A). B shows the level of reduction of F(ab′)2 by three different concentrations of wild-type and mutant GILT eluates from duplicate transfections, by eluates of various dilutions from a mock transfection, and by purified GILT (0.5 μg). C quantitates the level of reduction by the different eluate concentrations.
GILT Catalyzes Disulfide Reduction in Vivo.
To determine whether in vitro reduction by GILT reflects a true lysosomal enzymatic activity, a cDNA expression construct encoding GILT was transfected into the GILT-negative melanoma cell line, J3. Fig. 5A shows that a mAb specific for the lysosomal marker CD63 was endocytosed from the cell surface specifically into GILT-positive intracellular vesicles in GILT-expressing J3 cells. J3 cells expressing the vector only (J3/Neo) internalized the mAb into similar vesicles that were GILT negative. In both cases, these vesicles were positive for the lysosomal marker Lamp-1 (data not shown).
Figure 5.
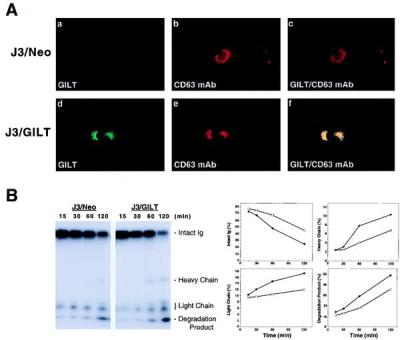
Lysosomal reduction in GILT-expressing cells. (A) Immunofluorescence detection of internalized mAb to CD63 (H5C6) in J3/Neo or J3/GILT cells was performed as described (22). After internalization, cells were fixed, permeabilized, and stained for internalized mAb and for GILT (rabbit anti-GILT serum) by using appropriate conjugated secondary antibodies. The internalized mAb colocalizes with GILT. (B) The reduction status of internalized 125I-H5C6 (CD63 mAb) in J3/Neo or J3/GILT transfectants at different periods of time was analyzed by nonreducing SDS/PAGE. (C) Individual bands were quantitated as described and plotted as percent of total in each lane vs. time (min). Open squares represent J3/Neo and closed circles, J3/GILT.
As a substrate to examine reduction after endocytosis, we used 125I-labeled CD63-specific mAb. The mAb was allowed to bind at 0°C, after which the cells were washed and warmed to 37°C. Aliquots of cells were detergent extracted at intervals in the presence of N-ethylmaleimide to prevent further reduction, and the reduction status of the IgG was examined by nonreducing SDS/PAGE followed by autoradiography. The results show that the apparent rate of IgG reduction to heavy and light chains was increased approximately 2-fold in J3/GILT vs. J3/Neo, the vector control (Fig. 5B). Enhanced proteolysis was also evident in the presence of GILT, measured by the appearance of 125I-labeled degradation products at the dye front (see Fig. 5B Lower Right). Thus, the data support the hypothesis that GILT serves to enhance the reduction of internalized proteins and consequently their rate of lysosomal degradation.
Discussion
GILT bears no significant homology to any other human protein currently in the database. In initial attempts to determine its function, we were unable to demonstrate either proteolytic or protease inhibitory activity. The presence of a CXXC motif at positions 46–49, which bears little similarity to the active motif of thioredoxin family members (WCGH/PCK) beyond the spacing of the cysteine residues, suggested the possibility that GILT might be a thiol reductase. Although thioredoxin family members share minimal sequence identity, they do have a common fold, and GILT showed reasonable matching with the thioredoxin fold. Indeed, GILT proved to be capable of catalyzing the reduction of the interchain disulfide bonds of F(ab)′2 and intact IgG, and this activity was abolished by mutagenesis of either of the two putative active-site cysteines (Fig. 4).
In contrast to thioredoxin, which is more efficient at neutral pH, GILT is optimally active between pH 4 and 5, consistent with its function being mediated in late endocytic compartments and lysosomes (Fig. 3A). Reduction, oxidation, and isomerization of disulfide bonds are chemically favored at neutral pH (19); see also Fig. 3A, where spontaneous reduction of F(ab)′2 by DTT in the absence of GILT is seen only at pH 7.0. Presumably, as for other lysosomal enzymes, key structural features of GILT evolved to facilitate both stability and activity under acidic conditions.
Thioredoxin depends on two accessory molecules for reduction, namely the enzyme thioredoxin reductase and the cofactor NADPH (37). Protein disulfide isomerase and the related proteins that catalyze disulfide bond isomerization in the ER appear to require only reduced and oxidized glutathione (6). GILT requires the addition of a reducing agent for activity; we have used DTT. Glutathione does not activate GILT activity in vitro, but l-cysteine does (data not shown), and the latter may be the active reducing agent in vivo. Conceivably other cofactors may exist that further amplify GILT activity.
We propose that the primary function of GILT is to facilitate complete unfolding of proteins destined for lysosomal degradation by releasing structural constraints imposed by intra- and interchain disulfide bonds. This is supported by the data shown in Fig. 5, which shows that the presence of GILT in a melanoma cell line results in a 2-fold increase in the rate of reduction of internalized IgG. Whether reduction in the absence of GILT is caused by a constitutive thiol reductase is unknown. Several groups have observed that thiols activate lysosomal degradation of proteins (38–42). The mechanisms by which they do this presumably include facilitation of denaturation as well as direct activation of thiol proteases. Evidence suggesting that GILT provides such a “housekeeping” function is that a GILT homologue appears to be present in Caenorhabditis elegans. Although the level of amino acid identity is low, all the cysteine residues are conserved. The similar protein in the mouse is much more conserved, exhibiting approximately 70% homology at the amino acid level.
Whether GILT has an immunological function is not yet known, but it is striking that it is expressed in both B-LCL and monocytic cell lines and primary human monocytes (this work and ref. 20). These cells are active antigen-presenting cells. In addition, mature GILT predominantly colocalizes with intracellular MHC class II molecules and HLA-DM in B-LCL when analyzed by indirect immunofluorescence (21). Immunoelectron microscopy (Fig. 2) shows that GILT is found in both the multivesicular and multilaminar lysosome-like structures in B cells believed to be the site of MHC class II peptide loading (31). Furthermore, GILT is inducibly up-regulated by IFN-γ, as are MHC class II molecules, the associated invariant chain, and HLA-DM, which catalyses peptide loading in MIICs (43). These components are all regulated at the level of transcription by the Class II transactivator [CIITA (44)]. Although GILT does not appear to be CIITA regulated (data not shown), this does not necessarily argue against a role in antigen processing, because neither is the IFN-γ-inducible lysosomal protease cathepsin S, which is important for efficient invariant chain degradation (45).
Reduction is clearly important for efficient antigen processing (13). The 2-fold enhancement in reduction rate conferred by GILT expression (Fig. 5) may not seem particularly large. However, a similar difference in the rate of reduction of an internalized substrate between class II-expressing Chinese hamster ovary cells and a hybrid of them with a normal mouse fibroblast has been suggested to account for a major difference in their ability to process and present class II-restricted antigens (46). GILT is in the right place (i.e., the MIIC) to efficiently catalyze the reduction of internalized protein antigens and may be more effective in antigen-presenting cells than in cells such as the melanoma line shown in Fig. 5. GILT seems likely to be a significant component in the generation of MHC class II-restricted epitopes.
Acknowledgments
We thank Dr. Naveen Bangia and Tobias Dick for helpful discussions, Nancy Dometios for expert preparation of this manuscript, Kathy Stone and the Keck Foundation Biotechnology Resource Laboratory for mass spectrometry and sequence analysis, and Janice M. Griffiths for expert assistance with electron microscopy. We also thank Dr. Jeffrey Ravetch, Rockefeller University, for providing c-DNA clones, Dr. Janet Lee for cell lines, and Dr. Peter Lobel for biotinylated M6PR. This work was supported by National Institutes of Health grant AI23081 and the Howard Hughes Medical Research Institute.
Abbreviations
- ER
endoplasmic reticulum
- GILT
IFN-γ-inducible lysosomal thiol-reductase
- M6PR
mannose-6-phosphate receptors
- B-LCL
human B-lymphoblastoid cell line
- TS
0.15 M NaCl/0.01 M Tris
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no: AF097362).
References
- 1.Lundstrom-Ljung J, Holmgren A. In: Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally Related Proteins. Guzman N A, editor. New York: Dekker; 1998. pp. 297–314. [Google Scholar]
- 2.Bardwell J C A, Beckwith J. Cell. 1993;74:769–771. doi: 10.1016/0092-8674(93)90455-y. [DOI] [PubMed] [Google Scholar]
- 3.Freedman R B. Curr Opin Struct Biol. 1995;5:85–91. doi: 10.1016/0959-440x(95)80013-q. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert H F. In: Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally Related Proteins. Guzman N A, editor. New York: Dekker; 1998. pp. 341–367. [Google Scholar]
- 5.Holmgren A. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 6.Holtzman J L. Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally Related Proteins. New York: Dekker; 1998. pp. 173–236. [Google Scholar]
- 7.Holmgren A, Bjornstedt M. Methods Enzymol. 1995;252:199–210. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 8.Martin J L. Structure (London) 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 9.Chivers P T, Laboissiere C A, Raines R T. in Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally Related Proteins. New York: Dekker; 1998. pp. 481–506. [Google Scholar]
- 10.Mobbs C V, Kaplitt M, Pfaff D. in Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally Related Proteins. New York: Dekker; 1998. pp. 237–272. [Google Scholar]
- 11.Bohley P, Seglen P O. Experientia. 1992;48:151–157. doi: 10.1007/BF01923508. [DOI] [PubMed] [Google Scholar]
- 12.Okhuma S. In: Lysosomes: Their Role in Protein Breakdown. Glaumann M, Ballard J, editors. London: Academic; 1987. pp. 115–148. [Google Scholar]
- 13.Jensen P E. Semin Immunol. 1995;7:347–353. doi: 10.1006/smim.1995.0039. [DOI] [PubMed] [Google Scholar]
- 14.Grisolia S, Wallace R. Biochem Biophys Res Commun. 1976;70:22–27. doi: 10.1016/0006-291x(76)91103-7. [DOI] [PubMed] [Google Scholar]
- 15.Lloyd J B. Biochem J. 1986;237:271–272. doi: 10.1042/bj2370271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pisoni R, Acker T L, Lisowski K M, Lemons R M, Thoene J G. J Cell Biol. 1990;110:327–335. doi: 10.1083/jcb.110.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins D S, Unanue E R, Harding C V. J Immunol. 1991;147:4054–4059. [PubMed] [Google Scholar]
- 18.Shen W-C, Ryser H J-P, LaManna L. J Biol Chem. 1985;260:10905–10908. [PubMed] [Google Scholar]
- 19.Feener E P, Shen W-C, Ryser H J-P. J Biol Chem. 1990;265:18780–18785. [PubMed] [Google Scholar]
- 20.Luster A D, Weinshank R L, Feiman R, Ravetch J V. J Biol Chem. 1988;263:12036–12043. [PubMed] [Google Scholar]
- 21.Arunachalam B, Pan M, Cresswell P. J Immunol. 1998;160:5797–5806. [PubMed] [Google Scholar]
- 22.Hammond C, Denzin L K, Pan M, Griffith J M, Geuze H J, Cresswell P. J Immunol. 1998;161:3282–3291. [PubMed] [Google Scholar]
- 23.Hunter W M, Greenwood F C. Biochem J. 1964;91:43–56. doi: 10.1042/bj0910043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roche P A, Cresswell P. J Immunol. 1990;144:1849–1856. [PubMed] [Google Scholar]
- 25.Liou W, Geuze H J, Slot J W. Histochem Cell Biol. 1997;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- 26.Slot J W, Geuze H J. Eur J Cell Biol. 1985;38:87–93. [PubMed] [Google Scholar]
- 27.Kleijmeer M J, Morkowski S, Griffith J M, Rudensky A Y, Geuze H J. J Cell Biol. 1997;139:639–649. doi: 10.1083/jcb.139.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasilik A. Experientia. 1992;48:130–151. doi: 10.1007/BF01923507. [DOI] [PubMed] [Google Scholar]
- 29.Jornvall H, Persson B. Biosci Rep. 1983;3:225–232. doi: 10.1007/BF01122454. [DOI] [PubMed] [Google Scholar]
- 30.Lin H L, Day N C, Ueda Y, Martin D K, Dixon J E, Seidah N G, Akil H. Neuroendocrinology. 1993;58:94–105. doi: 10.1159/000126518. [DOI] [PubMed] [Google Scholar]
- 31.Geuze H J. Immunol Today. 1998;19:282–287. doi: 10.1016/s0167-5699(98)01269-9. [DOI] [PubMed] [Google Scholar]
- 32.Glickman J N, Morton P A, Slot J W, Kornfeld S, Geuze H J. J Cell Biol. 1996;132:769–785. doi: 10.1083/jcb.132.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanderson F, Kleijmeer M J, Kelly A, Verwoerd D, Tulp A, Neefjes J J, Geuze H J, Trowsdale J. Science. 1994;266:1566–1569. doi: 10.1126/science.7985027. [DOI] [PubMed] [Google Scholar]
- 34.Kornfeld S, Mellman I. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 35.Fisher D, Eisenberg D. Protein Science. 1996;5:947–955. doi: 10.1002/pro.5560050516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmgren A. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 37.Holmgren A. Structure (London) 1995;3:239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 38.Hanes D M, Tappel A L. Biochem Biophys Acta. 1971;245:42–53. doi: 10.1016/0005-2728(71)90006-5. [DOI] [PubMed] [Google Scholar]
- 39.Kussendrager K D, deYong Y, Bouman J M, Gruber M. Biochem Biophys Acta. 1972;279:75–86. doi: 10.1016/0304-4165(72)90242-5. [DOI] [PubMed] [Google Scholar]
- 40.Huisman W, Bouma J M W, Gruber M. Biochem Biophys Acta. 1973;297:98–109. doi: 10.1016/0304-4165(73)90053-6. [DOI] [PubMed] [Google Scholar]
- 41.Kooistra T, Millard P C, Lloyd J B. Biochem J. 1982;204:471–477. doi: 10.1042/bj2040471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mego J L. Biochem J. 1984;218:775–783. doi: 10.1042/bj2180775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mach B, Steimly V, Martinez-Soria E, Reith W. Annu Rev Immunol. 1996;14:301–331. doi: 10.1146/annurev.immunol.14.1.301. [DOI] [PubMed] [Google Scholar]
- 44.Steimle V, Otten L A, Zufferey M, Mach B. Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- 45.Riese R J, Wolf P R, Bromme D, Narkin L R, Villandangos J A, Ploegh H L, Chapman H A. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- 46.Merkel B J, Mandel R, Ryser H J-P, McCoy K L. J Immunol. 1995;154:128–136. [PubMed] [Google Scholar]