

Abstract
Replication of human immunodeficiency virus type 1 (HIV-1) in primary blood lymphocytes, certain T-cell lines (nonpermissive cells), and most likely in vivo is highly dependent on the virally encoded Vif protein. Evidence suggests that Vif acts late in the viral life cycle during assembly, budding, and/or maturation to counteract the antiviral activity of the CEM15 protein and possibly other antiviral factors. Because HIV-1 virions produced in the absence of Vif are severely restricted at a postentry, preintegration step of infection, it is presumed that such virions differ from wild-type virions in some way. In the present study, we established a protocol for producing large quantities of vif-deficient HIV-1 (HIV-1/Δvif) from an acute infection of nonpermissive T cells and performed a thorough examination of the defect in these virions. Aside from the expected lack of Vif, we observed no apparent abnormalities in the packaging, modification, processing, or function of proteins in Δvif virions. In addition, we found no consistent defect in the ability of Δvif virions to perform intravirion reverse transcription under a variety of assay conditions, suggesting that the reverse transcription complexes in these particles can behave normally under cell-free conditions. Consistent with this finding, neither the placement of the primer tRNA3Lys nor its ability to promote reverse transcription in an in vitro assay was affected by a lack of Vif. Based on the inability of this comprehensive analysis to uncover molecular defects in Δvif virions, we speculate that such defects are likely to be subtle and/or rare.
Human immunodeficiency virus type 1 (HIV-1), like other lentiviruses, has a number of regulatory genes in addition to the gag, pol, and env genes that are common to all replication-competent retroviruses. The vif (for viral infectivity factor) regulatory gene of HIV-1, which is conserved in all known lentiviruses except equine infectious anemia virus, encodes a protein that acts as a positive modulator of viral infectivity in certain cellular environments. In particular, Vif is required for efficient replication of HIV-1 in primary T cells and in some T-cell lines (nonpermissive cells), such as CEM, H9, and HUT78, where it stimulates single-round infectivity on the order of 10- to 100-fold (6, 9, 19, 20, 22, 24, 53, 55, 60, 63, 65, 67). However, Vif is dispensable for replication in other T-cell lines (permissive cells), such as CEM-SS (6, 19, 20, 24, 53, 55, 60, 63, 65, 67). In vivo, Vif is essential for pathogenic infections of simian immunodeficiency virus (SIV) in rhesus macaques, caprine arthritis encephalitis virus in goats, and feline immunodeficiency virus in cats (16, 28, 29, 34, 35, 43). Taken together, these observations suggest that vif plays a vital role in HIV-1 replication and pathogenesis in vivo.
The exact nature of Vif function is not well established. Vif is expressed late in the viral life cycle, and in situ immunofluorescence studies have indicated that it largely colocalizes with Gag in HIV-1-infected cells (58). In addition, subcellular fractionation experiments have demonstrated that Vif and Gag are targeted to similar membrane-free, cytoplasmic complexes, hypothesized to represent HIV-1 assembly intermediates (57). Whether Vif interacts directly with Gag in these complexes is controversial (8, 31, 33, 54, 57, 68, 69). In contrast, there is compelling evidence that interactions with the host cell are essential for Vif function. First, Vif activity is influenced by the species derivation of the virus-producing cell, as evidenced by the greater potency of HIV Vif proteins in human cells relative to many SIV Vif proteins (62; N. C. Gaddis et al., unpublished results) and the noted replicative enhancement of murine leukemia virus (a simple retrovirus that does not encode a Vif protein) in the presence of Vif (54, 62). Second, heterokaryons between permissive and nonpermissive cells display the nonpermissive phenotype, suggesting that nonpermissive cells naturally express an antiviral activity that is overcome by Vif (45, 59). Recently, we have used gene transfer experiments to show that the CEM15 protein (also termed APOBEC3G [37]) is at least partially responsible for this antiviral activity (55); whether Vif interacts directly with this protein is unknown. Based on this evidence, an appealing model is that Vif acts at the site of virus assembly, budding, and/or maturation to counteract a host antiviral mechanism and thereby impart infectivity upon progeny virions.
Virions produced in nonpermissive cells in the absence of the vif gene are impaired at a postentry step of infection and therefore fail to establish proviruses. Studies have suggested that either reverse transcription fails to proceed to completion or that reverse transcripts are unstable, possibly depending on the identity of the target cell (14, 15, 18, 46, 60, 67). Correspondingly, several groups have found Δvif virions to be partially defective in endogenous reverse transcriptase (RT) assays (17, 18, 27, 46, 48). Whether these findings are indicative of a specific problem with the reverse transcription complex, structural abnormalities in the viral core (6, 9, 32, 48), or some other molecular defect in virions is unknown. To date, no reproducible quantitative or qualitative difference in the protein content of wild-type and Δvif virions has been reported, aside from a difference in Vif incorporation (6, 11, 22, 38, 39, 42, 61) and a controversial difference in Gag processing (6, 56). Recent work has suggested, however, that primer 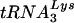 -genomic RNA complexes from Δvif virions may be unable to prime efficient reverse transcription (17).
-genomic RNA complexes from Δvif virions may be unable to prime efficient reverse transcription (17).
Because HIV-1 does not replicate in nonpermissive cells in the absence of Vif, a major obstacle for the detailed analysis of Δvif virions has been obtaining sufficient quantities of virus. Here, we developed a method for producing significant quantities of HIV-1/Δvif from acute infection of nonpermissive T cells. With the exception of the expected lack of Vif, analysis of these Δvif virions revealed no noticeable difference in the quantity, processing, or posttranslational modification of any viral proteins. Additionally, the performance of Δvif virions in the endogenous RT reaction was not consistently diminished compared to wild-type virions. Finally, the 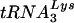 primer appeared to be correctly placed on purified genomes of Δvif virions and was able to promote efficient initiation of reverse transcription in an in vitro assay. These results suggest either that the lack of Vif in Δvif virions is responsible for their reduced infectivity or, in our view more likely, that the relevant defect is not easily detectable in the absence of additional insights into the function(s) of Vif in virus-producing cells.
primer appeared to be correctly placed on purified genomes of Δvif virions and was able to promote efficient initiation of reverse transcription in an in vitro assay. These results suggest either that the lack of Vif in Δvif virions is responsible for their reduced infectivity or, in our view more likely, that the relevant defect is not easily detectable in the absence of additional insights into the function(s) of Vif in virus-producing cells.
MATERIALS AND METHODS
Molecular clones.
The HIV-1 proviral expression vectors pIIIB, pIIIB/Δvif, pIIIB/ΔvifΔenv, pYU-2, and pYU-2/Δvif, as well as the vesicular stomatitis virus (VSV) G glycoprotein expression vector pHIT/G have been described previously (21, 26, 55, 59, 63). The CCR5-expressing retroviral vector pMIGR1-P/CCR5 is a derivative of the MSCV-based pMIGR1 vector (51) in which the gfp gene of pMIGR1 was replaced by the gene for puromycin resistance from LP-M and the ccr5 gene was inserted upstream of the internal ribosome entry site.
Cells and cell lines.
The human cell lines HUT78, CEM-SS-CCR5, C8166-CCR5/HIV-CAT, and 293T were maintained as previously described (21, 63). Standard retrovirus-mediated transduction of HUT78 cells with pMIGR1-P/CCR5 was used to establish the HUT78-CCR5 cell line, which was then maintained in complete medium supplemented with 40 ng of puromycin per ml. Peripheral blood mononuclear cells (PBMCs) from healthy adult donors were purified, activated, and maintained by standard procedures (60).
Production, purification, and quantification of viruses.
Viral stocks for the analysis of HIV-1YU-2 and HIV-1YU-2/Δvif replication were generated by transient transfection of 293T cells, as described previously (21). The virus was quantified by an enzyme-linked immunosorbent assay (ELISA) for soluble p24Gag concentration (NEN).
In order to generate high-titer virus from acutely infected T cells for investigation of the defect in Δvif virions, 293T cells were first transiently cotransfected with pHIT/G and pYU-2, pYU-2/Δvif, pIIIB, pIIIB/Δvif, or pIIIB/ΔvifΔenv to generate pseudotyped viral stocks, as described previously (57). In experiments involving kinetic PCR, the pseudotyped stocks were then treated with 1 mg of DNase (Roche Molecular Biochemicals) per ml in the presence of 10 mM MgCl2 for 30 min at 37°C to minimize plasmid contamination in the PCRs. HUT78 cells were challenged with the pseudotyped viruses by spin infection at 1,200 × g and 25°C for 2.5 h. After infection, the input virus was removed by extensive washing with phosphate-buffered saline (PBS), and the T cells were incubated in fresh media for 24 to 36 h to allow viral production to reach a high level. The cells were then washed again with PBS and placed in the appropriate medium for viral production. After production, the virus-containing supernatants were centrifuged for 5 min at 500 × g and filtered through 0.45-μm-pore-size filters. The infectivity of the virions was determined by infection of C8166-CCR5/HIV-CAT reporter cells as described previously (21, 60).
For some experiments, virions produced by the HUT78 cells were purified by layering the viral supernatants onto a 20% (wt/vol) sucrose (in PBS) cushion and centrifuging them at 100,000 × g for 1.5 h at 4°C in an SW28 rotor (Beckman Instruments, Inc.). In some cases, virions were further purified by using 20 to 60% (wt/vol) continuous sucrose gradients (in PBS) (22).
Analysis of replication.
PBMC, HUT78-CCR5 cells, and CEM-SS-CCR5 cells were infected with wild-type or Δvif HIV-1YU-2 stocks containing ca. 40, 5, and 10 ng of p24Gag per ml, respectively. Cells were maintained at ca. 2 × 106 cells per ml for the PBMC infections or 1 × 106 cells per ml for HUT78-CCR5 and CEM-SS-CCR5 infections. At 6-day intervals, freshly activated cells were added to the PBMC infections. Viral replication was monitored by measuring the soluble p24Gag concentration in the supernatants of the challenged cells.
Antibodies and Western analysis.
Viral proteins from sucrose gradient-purified virions were resolved by standard reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrophoretically to nitrocellulose. Individual proteins were detected by initial incubation with the murine monoclonal antibodies 319 (Vif), 24-2 (capsid [CA]), 17-3 (matrix [MA]), 4D1 (nucleocapsid [NC]), IN-2 (integrase [IN]), and MAb21 (reverse transcriptase [RT]; obtained from the National Institutes of Health [NIH] AIDS Research and Reference Reagent Program); the rabbit polyclonal antibodies R1169 (gp120 [a generous gift of Robert Doms]) and α-cyclophilin A (α-CypA); and the rat polyclonal antibody UP39 (Vpr). The membranes were then hybridized with appropriate horseradish peroxidase-conjugated mouse, rabbit, and rat secondary antibodies. Proteins were visualized by enhanced chemiluminescence and autoradiography.
Two-dimensional gel analysis.
For experiments involving radiolabeling of virions, HUT78 cells infected with wild type or Δvif HIV-1YU-2 were washed several times with PBS 30 to 36 h after infection and resuspended at ca. 106 cells per ml in labeling medium (RPMI 1640 medium lacking cysteine and methionine but containing 10% dialyzed fetal bovine serum, 1% l-glutamine, 1% penicillin-streptomycin, and 1 mCi of 35S-Express [NEN] per sample). Silver stain samples were treated similarly except that they were resuspended in regular complete medium instead of labeling medium. After 12 h of virus production, virions were purified by sucrose gradient centrifugation and lysed in isofocusing sample buffer (8 M urea, 1.7% [vol/vol] NP-40, 1.7% [vol/vol] 3/10 ampholytes [Bio-Rad], 1 mM sodium orthovanadate [Sigma], 1 μM microcystin LR [Sigma], and Complete protease inhibitor cocktail [Roche Molecular Biochemicals]). Prior to electrophoresis, 10% (vol/vol) β-mercaptoethanol was added to each sample. Viral proteins were separated in two dimensions, first by nonequilibrium pH gradient electrophoresis (NEPHGE; pH 3.0 to 10) and then by standard SDS-PAGE under reducing conditions. The resolved proteins were transferred to a nitrocellulose membrane by electroelution and visualized either by autoradiography using a Biomax Transcreen LE intensifying screen (Kodak) for radiolabeled samples or by silver staining for unlabeled samples.
Reversed-phase (RP)-HPLC.
Virions generated in HUT78 cells were pelleted through sucrose cushions, disrupted in 8 M guanidine-HCl (Pierce) with or without 50 mM dithiothreitol (DTT; Calbiochem) and fractionated by high-performance liquid chromatography (HPLC) to isolate viral proteins. HPLC was performed at a flow rate of 300 μl per min on a 2.1-by-100-mm Poros R2/H narrow-bore column (Boehringer Mannheim GmbH) with aqueous acetonitrile-trifluoroacetic acid solvents and a Shimadzu HPLC system equipped with LC-10AD pumps, SCL-10A system controller, CTO-10AC oven, FRC-10A fraction collector, and SPD-M10AV diode array detector. The gradient of buffer B (0.1% trifluoracetic acid in acetonitrile) was as follows: 10 to 36.5% for 12 min, 36.5 to 37% for 4 min, 37 to 41% for 7 min, 41 to 70% for 12 min, and 70% for 5 min. A temperature of 55°C was maintained during HPLC separation. Peaks were detected by UV absorption at 206 and 280 nm and then analyzed by sequencing with an automated Applied Biosystems, Inc., 477 Protein Sequencer, by SDS-PAGE with Coomassie or silver staining, and by immunoblot analysis with enhanced chemiluminescence.
In vitro kinase assay.
Virions produced in HUT78 cells were purified by sucrose gradient centrifugation and resuspended in kinase buffer (50 mM HEPES [pH 7.5], 0.5% NP-40, 5 mM MnCl2, 0.2 mM sodium orthovanadate [Sigma], 1 μM microcystin LR [Sigma], and Complete protease inhibitor cocktail without EDTA [Roche Molecular Biochemicals]). Kinase reactions were performed at 20°C for 30 min in 50 μl of kinase buffer containing virus corresponding to 50 ng of p24Gag and 50 μCi of [γ-32P]ATP. The reactions were halted by adding gel loading buffer, followed by heating for 5 min at 95°C. Phosphorylated proteins were resolved by reducing SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography with a Biomax Transcreen HE intensifying screen (Kodak).
Pulse-chase analysis.
Approximately 30 h after infection of HUT78 cells with HIV-1YU-2 or HIV-1YU-2/Δvif, the cells were washed several times with PBS and incubated in medium without cysteine and methionine for 30 min at 37°C. The cells were then pelleted, resuspended in labeling medium (depletion medium supplemented with 0.4 mCi of 35S-Express [NEN])/ml, and placed at 37°C for 15 min. After labeling, the samples were washed once in chase medium (complete RPMI 1640 medium containing 20 mM methionine and 20 mM cysteine), pelleted, resuspended in complete medium, and placed at 37°C. At 0, 1.5, 3, 6, 12, and 24 h after the addition of chase medium, one-sixth of each sample was removed, the cells were pelleted, and the viral supernatants were filtered through 0.45-μm-pore-size filters. Both the cells and the virions were then lysed in radioimmunoprecipitation assay (RIPA) buffer (final concentration: 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 10 mM Tris-HCl [pH 7.5], 1 mM EDTA), and p24Gag (CA) and its precursors were immunoprecipitated with murine monoclonal antibody 24-4. The immunoprecipitated proteins were separated under reducing conditions by SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography using a Biomax Transcreen LE intensifying screen (Kodak).
Kinetic PCR.
Strong-stop and second-strand-transfer reverse transcription products were detected by kinetic (fluorescence-monitored) PCR on an ABI 7700 kinetic PCR instrument (Applied Biosystems) by using molecular beacon technology (66). The forward primer for both the strong-stop and second-strand-transfer reactions was 5′-GCTAGCTAGGAAACCCACTGCTTA-3′. The reverse primers were 5′-GCTAGAGATTTTCCACACTGACT-3′ for the strong-stop reaction and 5′-CTGCGTCGAGAGAGCTCCTCTGGTT-3′ for the second-strand-transfer reaction. The molecular beacon for both the strong-stop and second-strand-transfer reactions was 5′-GCGAGTCACACAACAGACGGGCACACACTACTCGC-3′, labeled at the 5′ end with the reporter fluorochrome 6-carboxyfluorescein and at the 3′ end with the quencher DABCYL (4-[4′-dimethylamino-phenylazo]-benzene) (Midland Certified Reagent Company). The kinetic PCRs were performed in a 50-μl volume containing 5 μl of 10× Buffer II (Applied Biosytems); 5 mM MgCl2; 250 μM (each) dATP, dCTP, dGTP, and dTTP (Promega); 500 nM concentrations of each primer; a 200 nM concentration of molecular beacon; and 1.25 U AmpliTaq Gold (Applied Biosystems). The cycling conditions were 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C.
Exogenous and endogenous RT assays.
Exogenous RT assays were performed as previously described (62). For the endogenous RT reaction, virions produced in HUT78 cells were pelleted in triplicate at 100,000 × g for 2 h at 4°C in a Beckman SW28 rotor. The viral pellets were resuspended on ice at 200 ng of p24Gag per ml in endogenous RT buffer lacking salt (50 mM Tris-HCl [pH 8]; 5 μg of melittin [Sigma]/ml; 2 mM DTT; 2 mM magnesium acetate; 0.1 mM [each] dATP, dCTP, dGTP, and dTTP [Promega]); these pH and melittin concentration optima were each determined empirically (data not shown). Aliquots of the resuspended pellets were then mixed with equal volumes of endogenous RT buffer containing various concentrations of NaCl in duplicate. The RT reactions were incubated for 6 to 8 h at 37°C and stopped by the addition of an equal volume of 2× PCR lysis buffer (20 mM Tris-HCl [pH 8], 2 mM EDTA, 0.002% SDS, 0.002% Triton X-100). The RT products were quantified by means of kinetic PCR against strong-stop and second-strand-transfer cDNAs. The majority of virus particles assayed by this method were inferred as being intact since reactions carried out in the absence of melittin resulted in levels of cDNA synthesis that were ∼10-fold lower than those seen with 5 μg of melittin/ml (at 150 mM NaCl [data not shown]).
Isolation and quantification of viral genomic RNA.
Sucrose gradient-purified virions from HUT78 cells were lysed in buffer containing 10 mM Tris-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, and 0.5% SDS. Genomic RNA was then isolated by performing three phenol-chloroform extractions, followed by ethanol precipitation in the presence of 20 μg of glycogen (Roche Molecular Biochemicals) per ml as a carrier. The integrity of the genomic RNA was confirmed by Northern blotting, as described previously (55), and the level of RNA was normalized between samples by means of a primer extension assay. The primer 5′-GCTAGAGATTTTCCACACTGACT-3′, which binds HIV-1 genomic RNA within the U5 region of the long terminal repeat, was added to the RNA samples at 1 ng per μl and extended for 30 min at 37°C in RT reaction buffer (50 mM Tris-HCl [pH 8]; 60 mM KCl; 3 mM MgCl2; 10 mM DTT; 100 μM [each] dATP, dCTP, dGTP, and dTTP [Promega]; 1 U of RNasin [Promega]/μl; 2.5 ng of purified HIV-1 RT [obtained from Stephen Hughes]/μl). The reactions were then heated for 20 min at 95°C to halt extension, and the primer extension products were quantified by means of kinetic PCR by using the strong-stop primer set.
tRNA placement assay.
Analysis of the placement of the 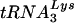 primer on the HIV-1 genomic RNA was done in a similar fashion to that described previously (3). Briefly, the primer 5′-CCCCGCACTTAATACTGACGC-3′, which binds “upstream” of the
primer on the HIV-1 genomic RNA was done in a similar fashion to that described previously (3). Briefly, the primer 5′-CCCCGCACTTAATACTGACGC-3′, which binds “upstream” of the 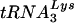 primer-binding site on the HIV-1 genome, was 5′ end labeled with 32P. The labeled primer was then mixed with the RNA samples at 1 ng per μl in RT buffer and extended for 30 min at 37°C. The cDNA products of the primer extension were ethanol precipitated, resolved on a denaturing 6% polyacrylamide-urea gel, and visualized by autoradiography.
primer-binding site on the HIV-1 genome, was 5′ end labeled with 32P. The labeled primer was then mixed with the RNA samples at 1 ng per μl in RT buffer and extended for 30 min at 37°C. The cDNA products of the primer extension were ethanol precipitated, resolved on a denaturing 6% polyacrylamide-urea gel, and visualized by autoradiography.
tRNA extension assay.
To examine the ability of the native 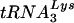 primer to prime reverse transcription, purified HIV-1 genomic RNA-primer
primer to prime reverse transcription, purified HIV-1 genomic RNA-primer 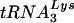 complexes were incubated for 30 min at 37°C in RT buffer containing various concentrations of deoxynucleoside triphosphates (dNTPs). Reactions were stopped by heating them for 20 min at 95°C, and the quantity of cDNA product formed was measured by kinetic PCR by using the strong-stop primers.
complexes were incubated for 30 min at 37°C in RT buffer containing various concentrations of deoxynucleoside triphosphates (dNTPs). Reactions were stopped by heating them for 20 min at 95°C, and the quantity of cDNA product formed was measured by kinetic PCR by using the strong-stop primers.
RESULTS
Strategy for production of Δvif virions.
It has previously been shown that the infectivity of Δvif HIV-1 produced in nonpermissive cells is severely reduced compared to wild type, whereas for virions produced in permissive cells, the infectivity of wild type and Δvif HIV-1 is similar (6, 9, 22, 24, 45, 53, 60). Therefore, when trying to determine the molecular basis for the infectivity phenotype observed in nonpermissive cells, differences between wild-type and Δvif virions that are seen in both nonpermissive and permissive cells are unlikely to dictate infectivity. In this investigation, we focused on uncovering defects particular to Δvif virions produced in nonpermissive cells.
Because HIV-1/Δvif does not replicate in nonpermissive cells, we optimized conditions to achieve a maximal output of virions from an acute infection. The strategy we developed is illustrated in Fig. 1A. Briefly, wild type or Δvif HIV-1YU-2 (an R5-tropic isolate) pseudotyped with the G protein of VSV was generated by transient cotransfection of 293T (permissive) cells. These pseudotyped viruses then were used to spin infect HUT78 (nonpermissive) cells. After challenge, the input virus was removed by vigorous washing of the infected T cells to prevent residual viral particles from contaminating later analyses. A second round of washing was also done closer to the time of analysis to ensure that the majority of virions being studied were recently assembled. Viral supernatants from the T cells were harvested for characterization ca. 36 to 48 h after infection, at which time production of Δvif viruses by nonpermissive cells was optimal.
FIG. 1.

Optimization of viral production from acute infection of T cells. (A) Strategy for high-level production of HIV-1/Δvif from acute infection of nonpermissive cells. See text for details. (B) Analysis of the benefits of VSV-G pseudotyping, spin infection, and use of HIV-1YU-2 for optimal viral output after acute infection. Pseudotyped or nonpseudotyped preparations of the indicated viruses were generated by transient transfection of 293T cells and used to infect nonpermissive HUT78 cells, either by spin infection or passive diffusion. The infected cells were then washed extensively to remove residual input particles and incubated for ∼30 h in growth medium to allow viral production to reach a high level. The cells were then washed again and placed in fresh growth medium for ∼10 h. The quantity of soluble p24Gag produced during this period was measured by ELISA.
As depicted in Fig. 1B, use of HIV-1YU-2 instead of the X4-tropic isolate HIV-1IIIB in this procedure resulted in a 15- to 35-fold enhancement in viral production. We suspect that much of this substantial improvement in yield is due to the lack of functional CCR5 expression in HUT78 cells (41), which averts virus-cell and cell-cell fusion, virus spread, and virus-induced (Env-associated) cytopathicity for the R5-tropic HIV-1YU-2. This hypothesis is supported by the finding that deletion of the env gene of HIV-1IIIB also results in a significant (although not quite as great) increase in viral output (Fig. 1B). Consistent with the dependence of HIV-1YU-2 replication on Vif expression in nonpermissive CCR5-expressing cells (Fig. 2), challenges of the T-cell-indicator cell line C8166-CCR5/HIV-CAT showed that wild-type HIV-1YU-2 generated by using our protocol and HUT78 cells was generally ∼40-fold more infectious than Δvif HIV-1YU-2 (data not shown), a finding in line with what has been observed for other HIV-1 isolates (6, 9, 22, 24, 45, 53, 60). Because the initial pseudotyped Δvif viruses from 293T cells are as infectious as Vif-expressing viruses, the dramatic differential in infectivity of the final virus stocks illustrates that little, if any, of the initial inocula is carried forward into the final virus preparations. Other features of our procedure that were important for achieving optimal yield were pseudotyping with the G protein of VSV and inoculation by spin infection rather than by passive diffusion (Fig. 1B). The ability to produce large quantities of infection-deficient Δvif virions enabled us to perform a number of studies that had been previously problematic.
FIG. 2.

Vif is required for replication of HIV-1YU-2 in PBMC and HUT78-CCR5 T cells. PBMC (A), HUT78-CCR5 cells (B), or CEM-SS-CCR5 cells (C) were infected with 293T-derived stocks of HIV-1YU-2 or HIV-1YU-2/Δvif at 40, 5, or 10 ng of p24Gag per ml, respectively. The PBMC infections were maintained at ∼2 × 106 cells/ml, and freshly stimulated PBMC were added every 6 days. The HUT78-CCR5 and CEM-SS-CCR5 infections were maintained at ∼106 cells/ml. Replication was monitored by measuring the concentration of soluble p24Gag in the supernatants of the infected cells by ELISA.
Protein composition of virions.
Having optimized conditions for production of Δvif virions, we first wished to analyze whether the absence of vif results in a quantitative or qualitative change in the virion protein profile. Sucrose gradient-purified virions produced in nonpermissive HUT78 cells were lysed, separated by standard reducing SDS-PAGE, transferred to nitrocellulose, and probed with antibodies to various viral and nonviral proteins (Fig. 3). As seen before, there were no obvious differences between wild-type and Δvif virions in p55Gag, CA, MA, RT, IN, or gp120 (1, 9, 17, 22, 39, 44, 47, 67). In addition, the HIV-1-encoded NC and Vpr proteins and the cellular protein CypA were found to be present in appropriate quantities in Δvif virions. Furthermore, hybridization of the membranes with antibodies against phosphorylated serine, threonine, and tyrosine revealed no vif-related abnormality in phosphorylation of viral proteins (data not shown). As observed previously, the only protein difference detectable in our Western analysis was the Vif protein, which was present in wild-type but not Δvif virions (Fig. 3). Importantly, treatment of virions with subtilisin, a protease that nonspecifically degrades external viral proteins and protein contaminants (50), did not eliminate the Vif signal in wild-type virus preparations, suggesting that Vif is in fact associated with virion cores (data not shown) (11, 39, 42). Of note, contrary to some reports (6, 56) but consistent with others (9, 17, 22, 39, 44, 47), there was no apparent vif-related defect in the processing of the p55Gag polyprotein. In particular, the sizes and quantities of the p55Gag-derived proteins MA, CA, and NC were normal in Δvif virions.
FIG. 3.

Western blot analysis of wild-type and Δvif HIV-1 virions. Virions produced by HUT78 cells infected with HIV-1YU-2 or HIV-1YU-2/Δvif were purified through continuous sucrose gradients, lysed, separated by SDS-PAGE, and transferred to nitrocellulose. The filters were then incubated with antibodies against the indicated proteins, followed by appropriate horseradish peroxidase-conjugated secondary antibodies. The proteins were visualized by enhanced chemiluminescence and autoradiography. The quantities of virus loaded in each lane corresponded to the indicated proteins as follows: 10 ng of p24Gag for CA; 50 ng of p24Gag for MA, NC, and IN; 100 ng of p24Gag for RT, gp120, and Vpr; and 200 ng of p24Gag for Vif and CypA.
To facilitate visualization of a broader spectrum of proteins, we next labeled virions metabolically. In addition, the samples were resolved according to both isoelectric point (by NEPHGE) and molecular mass (by SDS-PAGE) in hopes of achieving better resolution and revealing differences in protein modifications. Comparison of the protein profiles of sucrose gradient-purified wild-type and Δvif virions prepared in this manner revealed them to be strikingly similar (Fig. 4A). No reproducible differences in quantities or modifications of proteins were apparent, even with longer exposure times, with different pH ranges during NEPHGE or with different concentrations of acrylamide during SDS-PAGE (data not shown). Because metabolic labeling of proteins does not allow efficient detection of proteins with long half-lives, unlabeled virions were also subjected to two-dimensional electrophoresis and silver staining (Fig. 4B). Again, no differences between the proteins present in wild-type and Δvif virions were exposed.
FIG. 4.

Two-dimensional gel analysis of metabolically labeled (A) or silver-stained (B) wild-type and Δvif HIV-1 virions. Approximately 30 to 36 h following infection of HUT78 cells with HIV-1YU-2 or HIV-1YU-2/Δvif, the cells were metabolically labeled with [35S]methionine and [35S]cysteine (A) or incubated in fresh medium (B) for 12 h. Virions produced during this period were sucrose-gradient purified, lysed, and separated in two dimensions, first by NEPHGE and then by SDS-PAGE. The viral proteins were then transferred to nitrocellulose and visualized by autoradiography (A) or silver staining (B). The identity of the some spots was determined by Western blotting as described for Fig. 3.
As a further examination of the viral protein profile of Δvif virions, wild-type and Δvif virions produced in HUT78 cells were pelleted through a sucrose cushion, disrupted, and subjected to RP-HPLC analysis. As with the two-dimensional gel electrophoresis analysis, the HPLC chromatograms of wild-type and Δvif virions were nearly identical (Fig. 5). Minor differences did exist, for example in the CA-containing peaks. However, treatment of virions with subtilisin prior to RP-HPLC effectively eliminated these disparities (data not shown), suggesting that they were mainly attributable to contaminating serum proteins (from the growth medium) or cellular proteins on the exterior of virions. Further analysis of the RP-HPLC fractions by SDS-PAGE, followed by Coomassie blue staining, also revealed no major defects in the Δvif virions (Fig. 5). Importantly, this RP-HPLC analysis provided further evidence that the processing of p55Gag is normal in Δvif virions, since the ratios of the p55Gag derivatives MA, CA, NC, p6, and p1 were similar in wild-type and Δvif virions. In addition, side-by-side comparison of the fractions containing p6, MA, and CA on SDS-PAGE gels demonstrated that these proteins are appropriately sized in Δvif virions.
FIG. 5.

RP-HPLC examination of wild-type and Δvif HIV-1 virions. HIV-1YU-2 and HIV-1YU-2/Δvif produced in HUT8 cells were pelleted through sucrose cushions, disrupted, and separated by RP-HPLC. The identity of the peaks was determined by peptide sequencing and immunoblot analysis. The protein content of each fraction was visualized by SDS-PAGE and Coomassie blue staining. Adjacent lanes on the gels show matched fractions for wild-type (green) and Δvif (red) viruses.
Processing of viral proteins.
To address the issue of p55Gag processing more precisely, we used pulse-chase labeling of infected cells, followed by immunoprecipitation with a CA-specific monoclonal antibody to analyze the ability of PR to cleave the Gag polyprotein (Fig. 6). Comparison of the fate of the labeled p55Gag molecules clearly showed that the rate of processing of p55Gag to CA in both the producer cells (upper panels) and virions (lower panels) is unaffected by the presence or absence of Vif. In addition, the kinetics of p55Gag release from infected cells was very similar for wild-type and Δvif viruses, suggesting that budding is normal for the Δvif mutant.
FIG. 6.

Pulse-chase analysis of Gag processing. Approximately 30 h after infection of HUT78 with HIV-1YU-2 or HIV-1YU-2/Δvif, the infected cells were washed several times with PBS and incubated in growth medium lacking cysteine and methionine for 30 min at 37°C. The cells were then labeled in growth medium containing [35S]cysteine and [35S]methionine for 15 min at 37°C, washed once with chase medium containing excess cold cysteine and methionine, and placed in regular growth medium. At 0, 1.5, 3, 6, 12, and 24 h after the chase, one-sixth of each culture was removed, the cells were pelleted, and both the cells and the viral supernatants were lysed in RIPA buffer. The lysates were incubated overnight in the presence of a CA-specific monoclonal antibody, and the antibody-protein complexes were captured with agarose beads conjugated to protein G. The beads were washed with RIPA buffer, and the bound proteins were solubilized in gel loading buffer, resolved by SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography. The ∼41-kDa band represents a Gag processing intermediate.
Virion-associated kinases.
A number of kinases, including ERK2 mitogen-activated protein kinase, a 53-kDa serine/threonine kinase, and Nm23-H1, are present in HIV-1 virions and can phosphorylate various viral proteins (10, 12, 25, 36, 49). As is the case with mutations in vif, some of these phosphorylation events may have an effect on the fate of infection (10, 13, 25, 36). To characterize the activity of kinases in Δvif virions, we performed in vitro kinase assays on lysates of sucrose gradient-purified virions. The resulting 32P-labeled viral proteins were then resolved by standard SDS-PAGE and visualized by autoradiography. For both wild-type and Δvif virions, three major targets of phosphorylation by the virion-associated kinases were observed (Fig. 7). They were identified by means of immunoprecipitation to be the HIV-1-encoded CA, MA, and NC proteins (data not shown). The pattern and extent of phosphorylation seen for the Δvif virions suggests that the kinases are both present in normal quantities and active.
FIG. 7.

Examination of virion-associated kinase activity. Wild-type and Δvif HIV-1YU-2 virions produced in HUT78 cells were sucrose gradient purified and lysed in kinase buffer. Viral lysates corresponding to 50 ng of p24Gag were then incubated for 30 min at 20°C in the presence of MnCl2 and [γ-32P]ATP. Proteins that had been phosphorylated with radioactive phosphate groups during the reaction were resolved by SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography.
Activity of the reverse transcription complex.
A number of labs have observed that HIV-1/Δvif performs inefficiently in the endogenous RT assay, in which virions are “stimulated” via addition of a permeabilization agent, dNTPs, and a divalent cation to reverse transcribe their genomes with the packaged 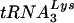 primers. To confirm this finding, we developed an assay in which strong-stop and second-strand-transfer products of the endogenous RT reaction were quantified by kinetic (fluorescence-monitored) PCR. A notable aspect of this technology is that it facilitates the parallel analysis of multiple repeats of the same and similar samples and therefore conveys an appreciation of the inherent variability of a given assay or method of sample preparation. Initially, a series of experiments was performed to identify the optimal permeabilization (5 μg/ml of melittin) and pH (pH 8) conditions for the reaction (data not shown). In addition, the time course of the reaction was examined, and an incubation time of 6 to 8 h, which is within the linear range of the assay, was chosen for subsequent experiments (data not shown).
primers. To confirm this finding, we developed an assay in which strong-stop and second-strand-transfer products of the endogenous RT reaction were quantified by kinetic (fluorescence-monitored) PCR. A notable aspect of this technology is that it facilitates the parallel analysis of multiple repeats of the same and similar samples and therefore conveys an appreciation of the inherent variability of a given assay or method of sample preparation. Initially, a series of experiments was performed to identify the optimal permeabilization (5 μg/ml of melittin) and pH (pH 8) conditions for the reaction (data not shown). In addition, the time course of the reaction was examined, and an incubation time of 6 to 8 h, which is within the linear range of the assay, was chosen for subsequent experiments (data not shown).
Using these conditions, the endogenous RT activity of wild-type and Δvif virions produced in nonpermissive cells was compared at various salt concentrations (Fig. 8). The results represent the average of three experiments performed with different virus preparations. As a control, we first showed that the vif mutant virus behaves similarly to wild type in the exogenous RT reaction (Fig. 8A), confirming the previous finding that the RT enzyme itself is fully functional in the absence of Vif (17, 27, 46). For the endogenous RT reaction, the Δvif virions appeared to display a slight (<2-fold) diminution in the accumulation of both strong-stop (Fig. 8B) and second-strand transfer (Fig. 8C) cDNAs. This was also true under less-optimal experiments where other melittin concentrations or pHs were used (data not shown). However, we suspect that this tendency is unlikely to be significant based on a number of issues. First, in one of the three experiments represented, the vif mutant was not measurably defective in the endogenous RT assay, despite displaying a 25-fold reduction in virus infectivity compared to the wild type. Second, twofold differences between duplicates within the same experiment were not uncommon, highlighting the intrinsic variability of assays of this type. Third, similar variations between the wild type and the Δvif mutant were sometimes observed in virions produced in permissive cells where no differences in virus infectivity are noted (e.g., CEM-SS cells) (data not shown).
FIG. 8.

Examination of reverse transcription complexes in Δvif virions. (A) Exogenous RT activity of wild type and Δvif HIV-1YU-2. HIV-1YU-2 and HIV-1YU-2/Δvif produced in HUT78 cells were assayed for exogenous RT activity as described previously (62). The results represent the average of four experiments by using different virus preparations. (B and C) Endogenous RT activity of wild type and Δvif HIV-1YU-2. HIV-1YU-2 and HIV-1YU-2/Δvif produced in HUT78 cells were normalized by p24Gag content, pelleted in triplicate, and resuspended on ice in endogenous RT buffer lacking salt at 200 ng of p24Gag per ml. Endogenous RT buffer containing various concentrations of NaCl was then added to aliquots of the resuspended virions in duplicate, and the samples were incubated for 6 to 8 h at 37°C. The strong-stop (B) and second-strand-transfer (C) intravirion reverse transcription products were then quantified by means of kinetic PCR. The values obtained from the triplicate samples were then averaged. The results presented represent the average of three experiments with different virus preparations.
Analysis of the genomic RNA-primer 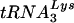 complex.
complex.
Recently, one group proposed that aberrant placement of and/or initiation from the native viral 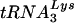 primer might be responsible for the defect they observed in endogenous reverse transcription (17). However, because the placement assay that was employed relied on initiation from the
primer might be responsible for the defect they observed in endogenous reverse transcription (17). However, because the placement assay that was employed relied on initiation from the 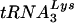 primer to measure its position, it was not possible to distinguish between the two possibilities. In order to segregate placement from initiation, we made use of an alternative primer extension-based placement assay in which the position of the annealed
primer to measure its position, it was not possible to distinguish between the two possibilities. In order to segregate placement from initiation, we made use of an alternative primer extension-based placement assay in which the position of the annealed 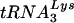 primer is mapped by virtue of its ability to impede DNA synthesis initiated from an “upstream” DNA primer (3). Accordingly, genomic RNA-primer
primer is mapped by virtue of its ability to impede DNA synthesis initiated from an “upstream” DNA primer (3). Accordingly, genomic RNA-primer 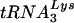 complexes were extracted from wild-type and Δvif virions (Fig. 9A), annealed to a 32P-labeled gag-specific primer, and incubated with recombinant HIV-1 RT in the presence of dNTPs. In the absence of the
complexes were extracted from wild-type and Δvif virions (Fig. 9A), annealed to a 32P-labeled gag-specific primer, and incubated with recombinant HIV-1 RT in the presence of dNTPs. In the absence of the 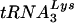 primer, the labeled primer was extended until it reached the 5′ end of the genomic RNA, resulting in a 369-nucleotide fragment. However, if the
primer, the labeled primer was extended until it reached the 5′ end of the genomic RNA, resulting in a 369-nucleotide fragment. However, if the 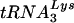 primer was correctly placed, one of a number of shorter products was generated (3). A similar ratio of full-length product to shorter products was observed for wild-type and Δvif viruses (Fig. 9B), indicating that a similar proportion of genomic RNA-primer
primer was correctly placed, one of a number of shorter products was generated (3). A similar ratio of full-length product to shorter products was observed for wild-type and Δvif viruses (Fig. 9B), indicating that a similar proportion of genomic RNA-primer 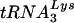 complexes exists in the two viruses. In addition, the shorter products were the same lengths for both viruses, suggesting that the
complexes exists in the two viruses. In addition, the shorter products were the same lengths for both viruses, suggesting that the 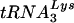 primer is correctly positioned on the genome of the vif mutant.
primer is correctly positioned on the genome of the vif mutant.
FIG. 9.

Analysis of genomic RNA-primer 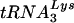 complexes. Sucrose gradient-purified HIV-1YU-2 and HIV-1YU-2/Δvif produced in HUT78 cells were lysed, and the genomic RNA-primer
complexes. Sucrose gradient-purified HIV-1YU-2 and HIV-1YU-2/Δvif produced in HUT78 cells were lysed, and the genomic RNA-primer 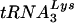 complexes were purified by phenol-chloroform extraction. (A) Northern analysis of viral RNA. The purified RNA was separated by denaturing electrophoresis and hybridized with a 32P-labeled probe against HIV-1 genomic RNA. (B) Analysis of the placement of the
complexes were purified by phenol-chloroform extraction. (A) Northern analysis of viral RNA. The purified RNA was separated by denaturing electrophoresis and hybridized with a 32P-labeled probe against HIV-1 genomic RNA. (B) Analysis of the placement of the 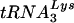 primer. A 32P-labeled primer that anneals upstream of the primer-binding site on the HIV-1 genome was added to the purified genomic RNA-primer
primer. A 32P-labeled primer that anneals upstream of the primer-binding site on the HIV-1 genome was added to the purified genomic RNA-primer 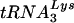 complexes and extended with recombinant HIV-1 RT in RT buffer for 30 min at 37°C. Reaction products were separated on a 6% polyacrylamide-urea gel and visualized by autoradiography. (C) Analysis of the ability of the
complexes and extended with recombinant HIV-1 RT in RT buffer for 30 min at 37°C. Reaction products were separated on a 6% polyacrylamide-urea gel and visualized by autoradiography. (C) Analysis of the ability of the  primer to promote reverse transcription. Purified genomic RNA-primer
primer to promote reverse transcription. Purified genomic RNA-primer 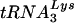 complexes were incubated for 30 min at 37°C with recombinant HIV-1 RT in RT buffer. The resulting cDNAs were quantified by kinetic PCR against strong-stop reverse transcription products.
complexes were incubated for 30 min at 37°C with recombinant HIV-1 RT in RT buffer. The resulting cDNAs were quantified by kinetic PCR against strong-stop reverse transcription products.
To examine whether 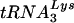 efficiently primes reverse transcription of the Δvif virus genome, wild-type and Δvif genomic RNA-primer
efficiently primes reverse transcription of the Δvif virus genome, wild-type and Δvif genomic RNA-primer 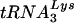 complexes were incubated with recombinant HIV-1 RT and dNTPs in the absence of added primer. The quantity of dNTPs in the reactions was varied to account for the possibility that a defect might only manifest itself at lower dNTP concentrations. By using kinetic PCR to measure the number of strong-stop cDNA copies produced in the RT reactions, the relative efficiency of priming from endogenous
complexes were incubated with recombinant HIV-1 RT and dNTPs in the absence of added primer. The quantity of dNTPs in the reactions was varied to account for the possibility that a defect might only manifest itself at lower dNTP concentrations. By using kinetic PCR to measure the number of strong-stop cDNA copies produced in the RT reactions, the relative efficiency of priming from endogenous  was determined. The data shown in Fig. 9C are one example of results obtained by using this assay. Here, the vif mutant was slightly more proficient than the wild type at all dNTP concentrations. However, depending on the RNA preparation, the activity of the Δvif genomic RNA-
was determined. The data shown in Fig. 9C are one example of results obtained by using this assay. Here, the vif mutant was slightly more proficient than the wild type at all dNTP concentrations. However, depending on the RNA preparation, the activity of the Δvif genomic RNA-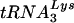 complex in this assay varied from threefold below to threefold above that of the wild type. Overall, however, it appeared that the genomic RNA-primer
complex in this assay varied from threefold below to threefold above that of the wild type. Overall, however, it appeared that the genomic RNA-primer 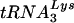 complexes from vif mutant viruses are effective templates for reverse transcription.
complexes from vif mutant viruses are effective templates for reverse transcription.
DISCUSSION
In the present study, we developed a method for producing substantial quantities of HIV-1/Δvif (typically, >100 ng of p24Gag per ml) from an acute infection of nonpermissive T cells. These particles, together with matched wild-type virions, were subjected to a detailed compositional analysis and an evaluation of their in vitro reverse transcription phenotypes. Examination of the protein profile of Δvif virions revealed no apparent abnormality in protein incorporation, with the exception of the absence of low levels of Vif itself (Fig. 3 to 5). In addition, there were no obvious defects in the posttranslational modification of viral proteins, as determined by detection with antibodies to phosphorylated residues (data not shown), analysis of the migration patterns of the proteins on two-dimensional gels (Fig. 4), and examination of the HPLC profiles of virions (Fig. 5). Also, the viral PR enzyme (Fig. 6) and the virion-associated kinases (Fig. 7) seemed to be fully active. Analysis of the activity of the reverse transcription complexes in Δvif virions by using the endogenous RT assay likewise failed to reveal a reproducible defect (Fig. 8). Individually, the RT enzyme and the genomic RNA-primer 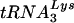 complex also functioned normally in in vitro assays (Fig. 8 and 9). Overall, aside from the presence or absence of the Vif protein itself, wild-type and vif-defective HIV-1 virions were strikingly similar.
complex also functioned normally in in vitro assays (Fig. 8 and 9). Overall, aside from the presence or absence of the Vif protein itself, wild-type and vif-defective HIV-1 virions were strikingly similar.
Our finding that cleavage of the p55Gag polyprotein in HIV-1/Δvif virions occurs with normal kinetics (Fig. 6) and generates normal mature Gag proteins (Fig. 3 to 6) is in agreement with a number of steady-state analyses (9, 17, 22, 39, 44, 47) and a previous kinetic analysis (39) but stands opposed to several other reports of aberrant p55Gag processing (6, 56). The reason for this discrepancy is not immediately obvious, but the weight of evidence now disfavors the idea that the absence of Vif results in reduced p55Gag processing. Our results also contradict recent in vitro studies that have suggested the converse notion, namely, that Vif inhibits PR action (2, 5, 23, 40, 52). Although these investigations have shown that Vif-derived peptides may inhibit PR in the context of HIV-1 infection and interfere with HIV-1 replication, demonstration of premature or excessive proteolysis of Gag during Δvif HIV-1 infection is awaited.
Our observation that Δvif virions are not consistently deficient in endogenous RT assays is somewhat surprising (Fig. 8). Although we did detect an approximate twofold decrease in the number of strong-stop and second-strand-transfer reverse transcripts generated by the vif mutant in some experiments, in other experiments the defect was negligible. Importantly, however, the infectivity of the Δvif virus preparations was measured in all cases and found to be similarly diminished irrespective of performance in the endogenous RT reaction. Analogous results were obtained with HIV-1IIIB (data not shown), indicating that our findings are not specific to HIV-1YU-2. The lack of consistent correlation between defects in infectivity and endogenous RT activity suggests that the modulation of viral infectivity by Vif does not relate directly to the biosynthetic capabilities of the reverse transcription complex. Of note, similar conclusions have previously been drawn on the basis of viral challenges of some T-cell lines (18, 60). Accordingly, we suspect that virions produced in the absence of Vif have a defect in some other area, such as virion core stability (6, 9, 32, 48), that can manifest itself as a modest reverse transcription defect under some experimental conditions.
Based on the results of the endogenous RT assays (Fig. 8), it is not surprising that we found the primer 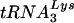 to be placed properly on the genomic RNA of Δvif virions and to be able to prime cDNA synthesis in vitro (Fig. 9). Notably, this result contradicts a previous study that found the placement of and/or initiation from the
to be placed properly on the genomic RNA of Δvif virions and to be able to prime cDNA synthesis in vitro (Fig. 9). Notably, this result contradicts a previous study that found the placement of and/or initiation from the 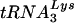 primer to be aberrant in the absence of Vif (17). In that study, the investigators were unable to distinguish between
primer to be aberrant in the absence of Vif (17). In that study, the investigators were unable to distinguish between 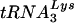 placement and the initiation of DNA synthesis. In contrast, the use of a primer extension assay allowed us to separate the two and to show that placement of the
placement and the initiation of DNA synthesis. In contrast, the use of a primer extension assay allowed us to separate the two and to show that placement of the 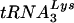 primer is normal in Δvif virions (Fig. 9B). Furthermore, we demonstrated that genomic RNA-
primer is normal in Δvif virions (Fig. 9B). Furthermore, we demonstrated that genomic RNA-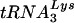 complexes from Δvif virions are able to support the synthesis of normal quantities of strong-stop cDNA in the presence of recombinant RT (Fig. 9C). Although we do not understand the reason for these discrepancies, it is worth noting that virion RNA was purified by using guanidinium isothiocyanate in the former report (17), whereas we used a more gentle method based on phenol-chloroform extraction.
complexes from Δvif virions are able to support the synthesis of normal quantities of strong-stop cDNA in the presence of recombinant RT (Fig. 9C). Although we do not understand the reason for these discrepancies, it is worth noting that virion RNA was purified by using guanidinium isothiocyanate in the former report (17), whereas we used a more gentle method based on phenol-chloroform extraction.
In the present study, we again confirmed that Vif is present in wild-type preparations of HIV-1 virions (Fig. 3) (6, 11, 22, 38, 39, 42, 61, 64), even after removal of contaminating vesicles and extravirion proteins by subtilisin treatment. Although a consensus that Vif is associated with viral cores is generally emerging (11, 39, 42), the functional relevance of Vif packaging remains unresolved. Arguing in favor of nonspecific incorporation, and possibly functional insignificance, are the observations that (i) Vif packaging levels correlate with cellular expression levels (61); (ii) Vif is preferentially present in immature, possibly aberrant, virions (64); and (iii) Vif is incorporated into murine leukemia virus particles, a virus that does not encode a Vif protein (11). Conversely, the recent finding that genome-deficient virions are unable to incorporate Vif (39) suggests that the reported association of Vif with RNA (17, 68) may facilitate Vif packaging. However, if this is true, the aforementioned murine leukemia virus result indicates that HIV-1 RNA is not unique in its ability to mediate incorporation. Clearly, a better understanding of Vif function will help us to address the significance of Vif packaging.
Regardless of whether Vif acts in virions, in the producer cells, or in both, its role seems to be the inhibition of one or more host-derived antiviral factors (30, 45, 55, 59). Accordingly, one would anticipate that Δvif virions likely have an assayable alteration (or defect) other than the lack of Vif itself. We believe that the inability of the methodologies described here to detect such a difference is most likely due either to insufficient sensitivity of the assays used or to deficiencies in our appreciation of which assays to perform. For instance, it is possible that metabolic labeling and silver staining (Fig. 4) are not adequate to discern the presence or absence of small quantities of an antiviral factor in virions or alterations to only a small proportion of any given virion protein.
We have recently reported that an epitope-tagged version of CEM15, the T-cell-expressed antiviral factor that is counteracted by Vif, is incorporated in HIV-1 particles when overexpressed in virus-producing cells (55). Once antibodies are developed to endogenous CEM15, it will be important to determine whether this protein is differentially encapsidated into wild-type and Δvif virions under physiological conditions. Since CEM15 shares significant sequence similarity with cytidine deaminases (55), it is plausible that viral (or even cellular) RNAs (messenger, genomic, or 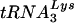 ) or DNAs are subjected to sequence modification in nonpermissive cells in the absence of Vif. One can envisage a number of potential consequences for such changes. For example, the course of reverse transcription in target cells or the fate of synthesized cDNAs could be altered. Alternatively, the coding “quality” of mRNAs in virus-producing cells could change, perhaps giving rise to mutant forms of cellular or viral proteins that exert dominant interfering effects on infectious virion production.
) or DNAs are subjected to sequence modification in nonpermissive cells in the absence of Vif. One can envisage a number of potential consequences for such changes. For example, the course of reverse transcription in target cells or the fate of synthesized cDNAs could be altered. Alternatively, the coding “quality” of mRNAs in virus-producing cells could change, perhaps giving rise to mutant forms of cellular or viral proteins that exert dominant interfering effects on infectious virion production.
Interestingly, a recent analysis of HIV-1 mRNAs in infected T cells did detect a number of guanosine-to-adenosine and cytidine-to-uridine alterations (7). Whether these changes are a result of RNA editing, and perhaps involve CEM15, or simply reflect reverse transcription errors is still a matter of debate (4). Nevertheless, in view of the potential deaminase activity of CEM15, an intriguing possibility is that Vif protects against CEM15-mediated RNA modifications via its ability to associate with RNA (17, 39, 68). In any case, it can be expected that a molecular description of CEM15 function will suggest additional aspects of Δvif virion structure, composition, and activity that should be assayed.
Acknowledgments
We thank Robert Doms for the gp120 polyclonal antibody and Stephen Hughes for the recombinant HIV-1 RT. The HIV-1 RT monoclonal antibody (MAb21) from Stephen Hughes was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH.
This work was supported by NIH research grants AI46246 (M.H.M.) and AI10460 (A.M.S.) and by funds from the NCI under contract NO1-CO-12400 (E.C. and L.E.H.). N.C.G. is a National Science Foundation predoctoral fellow. M.H.M. is an Elizabeth Glaser Scientist supported by the Elizabeth Glaser Pediatric AIDS Foundation.
REFERENCES
- 1.Akari, H., T. Uchiyama, T. Fukumori, S. Iida, H. Koyama, and A. Adachi. 1999. Pseudotyping human immunodeficiency virus type 1 by vesicular stomatitis virus G protein does not reduce the cell-dependent requirement of Vif for optimal infectivity: functional difference between Vif and Nef. J. Gen. Virol. 80:2945-2949. [DOI] [PubMed] [Google Scholar]
- 2.Baraz, L., A. Friedler, I. Blumenzweig, O. Nussinuv, N. Chen, M. Steinitz, C. Gilon, and M. Kotler. 1998. Human immunodeficiency type 1 Vif-derived peptides inhibit the viral protease and arrest viral production. FEBS Lett. 441:419-426. [DOI] [PubMed] [Google Scholar]
- 3.Beerens, N., B. Klaver, and B. Berkhout. 2000. A structured RNA motif is involved in correct placement of the tRNA(3)(Lys) primer onto the human immunodeficiency virus genome. J. Virol. 74:2227-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkhout, B., A. T. Das, and N. Beerens. 2000. HIV-1 RNA editing, hypermutation, and error-prone reverse transcription. Science 292:7. [DOI] [PubMed] [Google Scholar]
- 5.Blumenzweig, I., L. Baraz, A. Friedler, H. Danielson, C. Gilon, M. Steinitz, and M. Kotler. 2002. HIV-1 Vif-derived peptide inhibits drug-resistant HIV proteases. Biochem. Biophys. Res. Commun. 292:832-840. [DOI] [PubMed] [Google Scholar]
- 6.Borman, A. M., C. Quillent, P. Charneau, C. Dauguet, and F. Clavel. 1995. Human immunodeficiency virus type 1 Vif− mutant particles from restrictive cells: role of Vif in correct particle assembly and infectivity. J. Virol. 69:2058-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourara, K., S. Litvak, and A. Araya. 2000. Generation of G-to-A and C-to-U changes in HIV-1 transcripts by RNA editing. Science 289:1564-1566. [DOI] [PubMed] [Google Scholar]
- 8.Bouyac, M., M. Courcoul, G. Bertoia, Y. Baudat, D. Gabuzda, D. Blanc, N. Chazal, P. Boulanger, J. Sire, R. Vigne, and B. Spire. 1997. Human immunodeficiency virus type 1 Vif protein binds to the pr55Gag precursor. J. Virol. 71:9358-9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouyac, M., F. Rey, M. Nascimbeni, M. Courcoul, J. Sire, D. Blanc, F. Clavel, R. Vigne, and B. Spire. 1997. Phenotypically Vif− human immunodeficiency virus type 1 is produced by chronically infected restrictive cells. J. Virol. 71:2473-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bukrinskaya, A. G., A. Ghorpade, N. K. Heinzinger, T. E. Smithgall, R. E. Lewis, and M. Stevenson. 1996. Phosphorylation-dependent human immunodeficiency virus type 1 infection and nuclear targeting of viral DNA. Proc. Natl. Acad. Sci. USA 93:367-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camaur, D., and D. Trono. 1996. Characterization of human immunodeficiency virus type 1 Vif particle incorporation. J. Virol. 70:6106-6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartier, C., M. Deckert, C. Grangeasse, R. Trauger, F. Jensen, A. Bernard, A. Cozzone, C. Desgranges, and V. Boyer. 1997. Association of ERK2 mitogen-activated protein kinase with human immunodeficiency virus particles. J. Virol. 71:4832-4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cartier, C., P. Sivard, C. Tranchat, D. Decimo, C. Desgranges, and V. Boyer. 1999. Identification of three major phosphorylation sites within HIV-1 capsid: role of phosphorylation during the early steps of infection. J. Biol. Chem. 274:19434-19440. [DOI] [PubMed] [Google Scholar]
- 14.Chowdhury, I. H., W. Chao, M. J. Potash, P. Sova, H. E. Gendelman, and D. J. Volsky. 1996. vif-negative human immunodeficiency virus type 1 persistently replicates in primary macrophages, producing attenuated progeny virus. J. Virol. 70:5336-5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Courcoul, M., C. Patience, F. Rey, D. Blanc, A. Harmache, J. Sire, R. Vigne, and B. Spire. 1995. Peripheral blood mononuclear cells produce normal amounts of defective Vif− human immunodeficiency virus type 1 particle which are restricted for the preretrotranscription steps. J. Virol. 69:2068-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desrosiers, R. C., J. D. Lifson, J. S. Gibbs, S. C. Czajak, A. Y. Howe, L. O. Arthur, and R. P. Johnson. 1998. Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol. 72:1431-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dettenhofer, M., S. Cen, B. A. Carlson, L. Kleiman, and X.-F. Yu. 2000. Association of human immunodeficiency virus type 1 Vif with RNA and its role in reverse transcription. J. Virol. 74:8938-8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dornadula, G., S. Yang, R. J. Pomerantz, and H. Zhang. 2000. Partial rescue of the Vif-negative phenotype of mutant human immunodeficiency virus type 1 strains from nonpermissive cells by intravirion reverse transcription. J. Virol. 74:2594-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan, L., and K. Peden. 1992. Cell-free transmission of Vif mutants of HIV-1. Virology 190:19-29. [DOI] [PubMed] [Google Scholar]
- 20.Fisher, A. G., B. Ensoli, L. Ivanoff, M. Chamberlain, S. Petteway, L. Ratner, R. C. Gallo, and F. Wong-Staal. 1987. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science 237:888-893. [DOI] [PubMed] [Google Scholar]
- 21.Fouchier, R. A., B. E. Meyer, J. H. Simon, U. Fischer, and M. H. Malim. 1997. HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J. 16:4531-4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fouchier, R. A. M., J. H. M. Simon, A. B. Jaffe, and M. H. Malim. 1996. Human immunodeficiency virus type 1 Vif does not influence expression or virion incorporation of gag-, pol-, and env-encoded proteins. J. Virol. 70:8263-8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedler, A., I. Blumenzweig, L. Baraz, M. Steinitz, M. Kotler, and C. Gilon. 1999. Peptides derived from HIV-1 Vif: a non-substrate-based novel type of HIV-1 protease inhibitors. J. Mol. Biol. 287:93-101. [DOI] [PubMed] [Google Scholar]
- 24.Gabuzda, D. H., K. Lawrence, E. Langhoff, E. Terwilliger, T. Dorfman, W. A. Haseltine, and J. Sodroski. 1992. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 66:6489-6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallay, P., S. Swingler, C. Aiken, and D. Trono. 1995. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 80:379-388. [DOI] [PubMed] [Google Scholar]
- 26.Garrett, E. D., L. S. Tiley, and B. R. Cullen. 1991. Rev activates expression of the human immunodeficiency virus type 1 vif and vpr gene products. J. Virol. 65:1653-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goncalves, J., Y. Korin, J. Zack, and D. Gabuzda. 1996. Role of Vif in human immunodeficiency virus type 1 reverse transcription. J. Virol. 70:8701-8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harmache, A., P. Russo, F. Guiguen, C. Vitu, M. Vignoni, M. Bouyac, C. Hieblot, M. Pepin, R. Vigne, and M. Suzan. 1996. Requirement of caprine arthritis encephalitis virus vif gene for in vivo replication. Virology 224:246-255. [DOI] [PubMed] [Google Scholar]
- 29.Harmache, A., P. Russo, C. Vitu, F. Guiguen, J.-F. Mornex, M. Pepin, R. Vigne, and M. Suzan. 1996. Replication in goats in vivo of caprine arthritis-encephalitis virus deleted in vif or tat genes: possible use of these deletion mutants as live vaccines. AIDS Res. Hum. Retrovir. 12:409-411. [DOI] [PubMed] [Google Scholar]
- 30.Hassaine, G., M. Courcoul, G. Bessou, Y. Barthalay, C. Picard, D. Olive, Y. Collette, R. Vigne, and E. Decroly. 2001. The tyrosine kinase Hck is an inhibitor of HIV-1 replication counteracted by the viral vif protein. J. Biol. Chem. 276:16885-16893. [DOI] [PubMed] [Google Scholar]
- 31.Henzler, T., A. Harmache, H. Herrmann, H. Spring, M. Suzan, G. Audoly, T. Panek, and V. Bosch. 2001. Fully functional, naturally occurring and C-terminally truncated variant human immunodeficiency virus (HIV) Vif does not bind to HIV Gag but influences intermediate filament structure. J. Gen. Virol. 82:561-573. [DOI] [PubMed] [Google Scholar]
- 32.Höglund, S., Å. Öhagen, K. Lawrence, and D. Gabuzda. 1994. Role of vif during packing of the core of HIV-1. Virology 201:349-355. [DOI] [PubMed] [Google Scholar]
- 33.Huvent, I., S. S. Hong, C. Fournier, B. Gay, J. Tournier, C. Carrière, M. Courcoul, R. Vigne, B. Spire, and P. Boulanger. 1998. Interaction and co-encapsidation of human immunodeficiency virus type 1 Gag and Vif recombinant proteins. J. Gen. Virol. 79:1069-1081. [DOI] [PubMed] [Google Scholar]
- 34.Inoshima, Y., M. Kohmoto, Y. Ikeda, H. Yamada, Y. Kawaguchi, K. Tomonaga, T. Miyazawa, C. Kai, T. Umemura, and T. Mikami. 1996. Roles of auxiliary genes and AP-1 binding site in the long terminal repeat of feline immunodeficiency virus in the early stage of infection in cats. J. Virol. 70:8518-8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inoshima, Y., T. Miyazawa, and T. Mikami. 1998. The roles of vif and ORF-A genes and AP-1 binding site in in vivo replication of feline immunodeficiency virus. Arch. Virol. 143:789-795. [DOI] [PubMed] [Google Scholar]
- 36.Jacque, J. M., A. Mann, H. Enslen, N. Sharova, B. Brichacek, R. J. Davis, and M. Stevenson. 1998. Modulation of HIV-1 infectivity by MAPK, a virion-associated kinase. EMBO J. 17:2607-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jarmuz, A., A. Chester, J. Bayliss, J. Gisbourne, I. Dunham, J. Scott, and N. Navaratnam. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79:285-296. [DOI] [PubMed] [Google Scholar]
- 38.Karczewski, M. K., and K. Strebel. 1996. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 70:494-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan, M. A., C. Aberham, S. Kao, H. Akari, R. Gorelick, S. Bour, and K. Strebel. 2001. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J. Virol. 75:7252-7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kotler, M., M. Simm, Y. S. Zhao, P. Sova, W. Chao, S. F. Ohnona, R. Roller, C. Krachmarov, M. J. Potash, and D. J. Volsky. 1997. Human immunodeficiency virus type 1 (HIV-1) protein Vif inhibits the activity of HIV-1 protease in bacteria and in vitro. J. Virol. 71:5774-5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee, B., M. Sharron, L. J. Montaner, D. Weissman, and R. W. Doms. 1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA 96:5215-5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu, H., X. Wu, M. Newman, G. M. Shaw, B. H. Hahn, and J. C. Kappes. 1995. The Vif protein of human and simian immunodeficiency viruses is packaged into virions and associates with viral core structures. J. Virol. 69:7630-7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lockridge, K. M., M. Chien, G. A. Dean, K. S. Cole, R. C. Montelaro, P. A. Luciw, and E. E. Sparger. 2000. Protective immunity against feline immunodeficiency virus induced by inoculation with vif-deleted proviral DNA. Virology 273:67-79. [DOI] [PubMed] [Google Scholar]
- 44.Madani, N., and D. Kabat. 2000. Cellular and viral specificities of human immunodeficiency virus type 1 Vif protein. J. Virol. 74:5982-5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madani, N., and D. Kabat. 1998. An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J. Virol. 72:10251-10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nascimbeni, M., M. Bouyac, F. Rey, B. Spire, and F. Clavel. 1998. The replicative impairment of Vif− mutants of human immunodeficiency virus type 1 correlates with an overall defect in viral DNA synthesis. J. Gen. Virol. 79:1945-1950. [DOI] [PubMed] [Google Scholar]
- 47.Ochsenbauer, C., T. Wilk, and V. Bosch. 1997. Analysis of vif-defective human immunodeficiency virus type 1 (HIV-1) virions synthesized in “non-permissive” T lymphoid cells stably infected with selectable HIV-1. J. Gen. Virol. 78:627-635. [DOI] [PubMed] [Google Scholar]
- 48.Öhagen, A., and D. Gabuzda. 2000. Role of Vif in stability of the human immunodeficiency virus type 1 core. J. Virol. 74:11055-11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ott, D. E., L. V. Coren, D. G. Johnson, B. P. Kane, R. C. Sowder II, Y. D. Kim, R. J. Fisher, X. Z. Zhou, K. P. Lu, and L. E. Henderson. 2000. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 266:42-51. [DOI] [PubMed] [Google Scholar]
- 50.Ott, D. E., L. V. Coren, D. G. Johnson, R. C. Sowder II, L. O. Arthur, and L. E. Henderson. 1995. Analysis and localization of cyclophilin A found in the virions of human immunodeficiency virus type 1 MN strain. AIDS Res. Hum. Retrovir. 11:1003-1006. [DOI] [PubMed] [Google Scholar]
- 51.Pear, W. S., J. P. Miller, L. Xu, J. C. Pui, B. Soffer, R. C. Quackenbush, A. M. Pendergast, R. Bronson, J. C. Aster, M. L. Scott, and D. Baltimore. 1998. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 92:3780-3792. [PubMed] [Google Scholar]
- 52.Potash, M. H., G. Bentsman, T. Muir, C. Krachmarov, P. Sova, and D. J. Volsky. 1998. Peptide inhibitors of HIV-1 protease and viral infection of peripheral blood lymphocytes based on HIV-1 Vif. Proc. Natl. Acad. Sci. USA 95:13865-13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakai, H., R. Shibata, J.-I. Sakuragi, S. Sakuragi, M. Kawamura, and A. Adachi. 1993. Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J. Virol. 67:1663-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seroude, V., G. Audoly, P. Gluschankof, and M. Suzan. 2002. Viral and cellular specificities of caprine arthritis encephalitis virus Vif protein. Virology 292:156-161. [DOI] [PubMed] [Google Scholar]
- 55.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646-650. [DOI] [PubMed] [Google Scholar]
- 56.Simm, M., M. Shahabuddin, W. Chao, J. S. Allan, and D. J. Volsky. 1995. Aberrant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J. Virol. 69:4582-4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simon, J. H. M., E. A. Carpenter, R. A. M. Fouchier, and M. H. Malim. 1999. Vif and the p55Gag polyprotein of human immunodeficiency virus type 1 are present in colocalizing membrane-free cytoplasmic complexes. J. Virol. 73:2667-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simon, J. H. M., R. A. M. Fouchier, T. E. Southerling, C. B. Guerra, C. K. Grant, and M. H. Malim. 1997. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J. Virol. 71:5259-5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simon, J. H. M., N. C. Gaddis, R. A. M. Fouchier, and M. H. Malim. 1998. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 4:1397-1400. [DOI] [PubMed] [Google Scholar]
- 60.Simon, J. H. M., and M. H. Malim. 1996. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J. Virol. 70:5297-5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simon, J. H. M., D. L. Miller, R. A. M. Fouchier, and M. H. Malim. 1998. Virion incorporation of human immunodeficiency virus type 1 Vif is determined by intracellular expression level and may not be necessary for function. Virology 248:182-187. [DOI] [PubMed] [Google Scholar]
- 62.Simon, J. H. M., D. L. Miller, R. A. M. Fouchier, M. A. Soares, K. W. C. Peden, and M. H. Malim. 1998. The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. EMBO J. 17:1259-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simon, J. H. M., T. E. Southerling, J. C. Peterson, B. E. Meyer, and M. H. Malim. 1995. Complementation of vif-defective human immunodeficiency virus type 1 by primate, but not nonprimate, lentivirus vif genes. J. Virol. 69:4166-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sova, P., D. Volsky, L. Wang, and W. Chao. 2001. Vif is largely absent from human immunodeficiency virus type 1 mature virions and associates mainly with viral particles containing unprocessed Gag. J. Virol. 75:5504-5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sova, P., and D. J. Volsky. 1993. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type 1. J. Virol. 67:6322-6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyagi, S., and F. R. Kramer. 1996. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 14:303-308. [DOI] [PubMed] [Google Scholar]
- 67.von Schwedler, U., J. Song, C. Aiken, and D. Trono. 1993. vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 67:4945-4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang, H., R. J. Pomerantz, G. Dornadula, and Y. Sun. 2000. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J. Virol. 74:8252-8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zimmerman, C., K. C. Klein, P. K. Kiser, A. R. Singh, B. L. Firestein, S. C. Riba, and J. R. Lingappa. 2002. Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature 415:88-92. [DOI] [PubMed] [Google Scholar]