

Abstract
The colony formation assay is the most commonly used titration method for defining the concentration of replication-incompetent murine leukemia virus-derived retroviral vectors. However, titer varies with target cell type and number, transduction time, and concentration of polycation (e.g., Polybrene). Moreover, because most of the viruses cannot encounter target cells due to Brownian motion, their short half-lives, and the requirement for target cell division for activity, the actual infectious retrovirus concentration in the collected supernatant is higher than the viral titer. Here we correlate the physical viral particle concentration with the infectious virus concentration and colony formation titer with the help of a mathematical model. Ecotropic murine leukemia retrovirus supernatant, collected from the GP+E86/LNCX retroviral vector producer cell line, was concentrated by centrifugation and further purified by a sucrose density gradient. The physical concentration of purified viral vectors was determined by direct particle counting with an electron microscope. The concentrations of fresh and concentrated supernatant were determined by a quantitative reverse transcriptase activity assay. Titration of all supernatants by neomycin-resistant colony formation assay was also performed. There were 767 ± 517 physical viral particles per infectious CFU in the crude viral supernatant. However, the infectious viral concentration determined by mathematical simulation was 143 viral particles per infectious unit, which is more consistent with the concentration determined by particle counting in purified viral solution. Our results suggest that the mathematical model can be used to extract a more accurate and reliable concentration of infectious retrovirus.
The murine leukemia virus (MuLV)-based vector is the most widely used retroviral vector in gene therapy due to its ability to stably integrate its transgene into host chromosomal DNA with low immunogenicity (23). Determining the number of infectious retroviral entities in the supernatant is important. Currently, the titration method, which is performed by overlaying viral supernatant onto target cells (e.g., NIH 3T3 cells) after serial dilution, is widely used to represent infectious viral concentration as number of colonies per volume (i.e., CFU per milliliter). However, this method has several limitations as a quantitative indicator of infectious retrovirus concentration for a given viral supernatant.
In static retroviral transduction systems, retroviral particles move in random motion with short half-lives. Only a small fraction of infectious retrovirus particles can transduce target cells successfully (7, 26). Even if retroviruses can be homogeneously mixed with suspended target cells, the virus will bind to the receptor on the cell surface and reach equilibrium according to its equilibrium dissociation constant (4, 35). At equilibrium, some viral particles bind onto the cells but others remain suspended in solution as free particles. As a result, the titer assay reflects only successfully transduced cells and underestimates the real infectious viral particle concentration in the supernatant. In addition, the titers determined by different research groups can vary due to inconsistent conditions used for the same titration method such as target cell type, target cell number, polycation (e.g., Polybrene) concentration, incubation temperature, and exposure time for transduction. Therefore, development of a universally applicable method to determine the concentration of infectious retroviral particles in the supernatant would be highly useful.
Unlike the titration method mentioned above, counting viral particles via electron microscopy (EM) and measurement of a viral component (e.g., reverse transcriptase [RT]) can both provide the number of virus particles in the supernatant (5). However, the total number of viral particles derived from these methods cannot distinguish infectious from noninfectious viral particles. Since it is a daunting task to enumerate and differentiate infectious and noninfectious colloidal virus particles directly, developing a mathematical model to describe the quantity of infectivity (e.g., number of colonies) can be an alternative method for estimating the concentration of infectious retrovirus in a supernatant (2, 20, 22, 28, 29). The concentration of infectious retrovirus measured by Tavoloni using repeated titration of the supernatant showed that the value obtained was 5- to 20-fold higher than the viral titer (28). Andreadis et al. also estimated that only 1 to 8% of bulk infectious retrovirus was counted by determining the viral titer (2). Recently, we have shown that more accurate infectious retrovirus concentrations can be determined with an extended mathematical model and experimentally determined infectivity (e.g., colony count) (15, 16). With this new method, a consistent value of concentration was obtained under different transduction conditions, e.g., different concentrations of Polybrene (15). We have also shown that the efficiencies of retroviral transduction with different tropisms can be compared quantitatively (16).
In this study, viral concentrations measured by titration and particle counting (e.g., EM and RT assay) and those derived from the new mathematical model method were compared. Our data indicate that the infectious retrovirus concentration is 170-fold underestimated by our colony formation assay for measuring virus titer. The results confirm the validity of this new method and further suggest that the method used in this study should be applicable to the measurement of the infectious retrovirus concentration of any retrovirus system.
MATERIALS AND METHODS
Retroviral production and titer assay.
Ecotropic Moloney MuLV-derived retrovirus (MoMuLV) was produced from GP+E86/LNCX, as reported previously (35). To determine infectious titer, NIH 3T3 cells were plated onto six-well plates (2.5 × 104 cells/well) 1 day before transduction in D10 medium, which is Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Irvine Scientific, Santa Ana, Calif.). Diluted viral supernatant (10−1 to 10−6; 0.5 ml) with 8 μg of Polybrene (Sigma, St. Louis, Mo.)/ml was added to each well and incubated for 2 h unless specified otherwise, followed by replacement with 2 ml of fresh D10 medium. After overnight culture, the medium was replaced with fresh medium containing 0.6 mg of G418 (Gibco BRL, Rockville, Md.)/ml and cultured for an additional 7 days. The resistant colonies were counted after staining with 1% (wt/vol) methylene blue in methanol (Sigma). The retroviral titer, defined as CFU per milliliter, was calculated by multiplying the number of colonies by the dilution factor and 1/V, where V is the volume of virus-containing medium added to the plate. The deactivation rate constant (kd) and the half-life of virus were obtained by plotting ln (T/T0) versus incubation time (t) at 37°C, where T and T0 are the titers with incubation for arbitrary time and without incubation, respectively.
Concentration and purification of viral supernatant.
Virus purification was by a method previously described (33). Fresh viral supernatant (FV) from producer cell line GP+E86/LNCX was centrifuged at 9,500 rpm in Beckman rotor JA-14 at 4°C for 12 h. The pellet was resuspended in TNE buffer (0.01 M Tris-HCl [pH 7.2], 0.1 M NaCl, 0.001 M EDTA), and the suspension was named centrifuged virus (CV). The CV was further purified through a 10 to 60% linear sucrose gradient in a Beckman SW-40 rotor at 30,000 rpm for 2 h at 20°C. The virus band was recovered with a needle punch, suspended in TNE buffer, and pelleted by centrifugation at 17,000 rpm in Beckman rotor JA-17 for 2 h at 4°C. The pellet was then resuspended in TNE buffer at 1/1,000 of the original supernatant volume, and the suspension was named purified virus (PV).
Viral particle counting under EM.
The PV stock was mixed with an equal volume of a polystyrene particle solution (diameter, 102 ± 3 nm; Duke Scientific Corp., Palo Alto, Calif.) of a known concentration. The mixture was added to a copper mesh with 300 grid (Ted Pella, Redding, Calif.) precoated with Formvar (Fluka, Milwaukee, Wis.) and air dried. The grid was stained with 1% uracyl acetate (Fluka). The viral particles were examined under EM, and both polystyrene beads and viral particles were counted. The viral particle concentration of this viral stock was then determined from the ratio of the viral particle number to the polystyrene bead number.
RT assay.
The RT assay was performed as previously described (32). Briefly, 10 μl of virus sample was mixed with 20 μl of an RT cocktail which contained 15 μg of poly(A)/ml, 7.5 μg of oligo(dT)/ml, 15 μM dTTP, 75 mM Tris (pH 8.3), 30 mM dithiothreitol, 0.9 mM MnCl2, 94 mM NaCl, 0.075% Nonidet P-40, and 15 μCi of α-S35-dTTP (1 Ci/μmol; ICN, Costa Mesa, Calif.). Following a 1-h incubation at 37°C and a 5-min heat inactivation at 95°C, an aliquot (10 μl) of the reaction solution was transferred to Whatman DE81 filter paper. The filter paper was dried, washed with 2× SSC (0.3 M NaCl plus 0.03 M sodium citrate), rinsed with ethanol, and dried again. The radioactivity on the filter paper was counted in 3 ml of scintillation fluid with a Beckman LS 8000 scintillation counter.
Transduction system.
Two sets of NIH 3T3 cells (2.5 × 104 per well) were prepared on six-well plates for titration and cell count. Viral vector suspensions, FV, CV, and PV, were diluted by factors of 104, 2 × 106, and 5 × 105, respectively, with D10 containing 8 μg of Polybrene/ml to obtain the desired range for colony number. The suspensions were overlaid onto sets of wells containing NIH 3T3 cells. At different times during incubation (up to 48 h), transduction was terminated by replacing the retrovirus-containing supernatant with 2 ml of D10. After 24 h of culture, medium was replaced with D10 supplemented with 0.6 mg of G418/ml. After a 7-day G418 selection, colonies were stained with methylene blue, washed with deionized water, and counted. For the other set of culture wells, the cells were trypsinized and counted at various times during the transduction period by using a hemocytometer with trypan blue exclusion. The growth kinetics of NIH 3T3 cells can therefore be determined. The area of one stained cell was determined to be 376.1 ± 148.0 μm2 by averaging the areas of 100 randomly selected cells with the National Institutes of Health (Bethesda, Md.) Image software (version 1.62). The pixels occupied by the cells and a known standard area of a hemocytometer were determined by the software. The area occupied by a cell was calculated with the aid of a precalibrated microscopic area.
Freeze-thaw of viral supernatant.
Since it is not possible to simultaneously titer the supernatants from the same batch before and after freezing, titers measured at different times were compared to that of a reference supernatant, which was previously prepared and stored at −80°C. The freshly harvested supernatant was aliquoted into two sets (2 ml each). One of them (without freeze-thaw), along with the reference supernatant, was titered immediately after collection. The other set of supernatants was stored at −80°C after different repeats of freezing at −80°C for 1.5 h and thawing by shaking in a 37°C water bath only until the last piece of ice disappeared so that the thermal decay of virus could be kept minimal. This process was repeated up to eight times. Stored supernatants along with the reference supernatant were titered, and results were normalized with the titer of the reference supernatant.
Antibodies.
The rat monoclonal antibody 83A25, specific for the C-terminal region of MuLV gp70, was obtained as a stock of hybridoma supernatant from L. Evans (9). Anti-rat and anti-mouse immunoglobulin G conjugated with fluorescein isothiocyanate (FITC) was purchased from Kirkegaard & Perry Laboratories, Inc. (Gaithersburg, Md.).
Virus-cell binding and immunofluorescence assays.
The virus bound on the cell surface was quantified by indirect immunofluorescence using a fluorescence-activated cell sorting (FACS) analysis described previously (34). Briefly, 1 ml of virus was mixed with NIH 3T3 cells (2 × 105 cells) at the specified temperature for 2 h or as indicated. The cells were then centrifuged at 16,000 × g for 15 s, the supernatant was discarded, and the cells were washed once with 1 ml of ice-cold wash buffer (phosphate-buffered saline [PBS] containing 10% goat serum). The cells were resuspended in 250 μl of monoclonal antibody 83A25 and incubated at 4°C for 1 h. They were then washed again with 1 ml of cold wash buffer and resuspended in 100 μl of wash buffer containing 1 μl (0.5 μg) of fluorescein-labeled goat anti-rat immunoglobulin G for 30 min at 4°C. After a final wash, they were fixed in 300 μl of 4% paraformaldehyde in PBS. The fluorescence intensity of the cells was analyzed on a FACStar Plus flow cytometer (Becton Dickinson, Franklin Lakes, N.J.). The measured mean channel number of the samples was converted to fluorescence intensity (f) according to the formula log f = a · MN + b, in which MN is the mean channel number as measured by FACS and a and b are constants derived from a linear regression of a standard curve generated with fluorescein-labeled RCP-70-5 microbeads with known fluorescence intensities (Spherotech, Inc., Libertyville, Ill.). Background fluorescence was measured from cells mixed with D10 and the antibodies and was subtracted from the experimental values after the fluorescence conversion.
Statistical analysis.
All of the experimental data were obtained in triplicate and are presented as means ± standard deviations. The number of batches used to determine the ratio of viral particles to titer (Cp/titer) is also presented. Statistical comparison by analysis of variance was done at a significance level (P) of <0.05 based on Student's t test.
RESULTS
Virus concentration by physical particle counting.
MoMuLV vector supernatant was purified by centrifugation twice, first by direct spin and second by linear sucrose gradient ultracentrifugation. The purified viral vectors were examined under EM (Fig. 1). The viral particle is spherically shaped with a shell and a condensed core, and its diameter was measured to be approximately 100 nm. Some viral particles show tails, which might be result from the high-speed spin, as reported previously (27). The viral particles in the purified viral suspension, as well as the premixed polystyrene beads, were counted under EM. The viral particle concentration was determined by comparing the viral particle number to polystyrene bead number with a known concentration before mixing. There were 1.40 × 1010 viral particles per ml in one batch of the purified viral suspension with an infectious titer of 1.12 × 108 CFU/ml, as determined by neomycin-resistant-colony formation (Table 1). The average number of viral particles per infectious CFU was 122 ± 27 (n = 4).
FIG. 1.


Ecotropic MuLV under EM. Ecotropic retrovirus produced from GP+E86/LNCX producer cells was mixed with polystyrene standard-size beads (102 ± 3 nm) and photographed under transmission EM. Arrowheads and arrow, viral particles, some of which have a tailed structure.
TABLE 1.
Comparison of infectious retrovirus concentration by titration, EM, RT activity assay, and mathematical model
| Samplea | Vol (ml) | Titer (CFU/ml) | Cpb (particles/ml) | Cic (CFU/ml) | k (10−2 cm/h) | Cp/titerd | Ci/titer |
|---|---|---|---|---|---|---|---|
| FV | 1,000 | 5.05 × 106 | 3.70 × 109 | 7.23 × 108 | 2.77 | 767 ± 517 (6) | 143 |
| CV | 1 | 5.70 × 108 | 1.57 × 1011 | 9.90 × 1010 | 1.50 | 206 ± 37 (4) | 174 |
| PV | 1 | 1.12 × 108 | 1.40 × 1010 | 2.00 × 1010 | 1.27 | 122 ± 27 (4) | 179 |
FV, CV, and PV are viral supernatants collected directly from producer cell culture, concentrated by centrifugation at 12,000 × g for 12 h, and purified by centrifugation through a 10 to 60% sucrose gradient, respectively.
Cp for PV was determined by counting viral particles under EM and comparing the number with the number of polystyrene beads of known particle concentration that were mixed with the viral supernatant. Cp for FV and CV was determined by RT activity assay and normalized with a purified viral suspension with known viral particle number.
Ci is the concentration of infectious retrovirus before it was diluted to form separated colonies (i.e., Ci = C0 × FD, where C0 is the infectious retrovirus concentration, which is determined by the mathematical model with experimental data after dilution, and FD is the dilution factor, equal to 104, 2 × 106, and 5 × 105 for FV, CV, and PV, respectively).
The Cp/titer is the mean ± standard deviation of repeated batches of viral supernatants (number of batch is in parentheses). The other data were obtained from a single batch of supernatant.
Virus concentration by RT activity assay.
Both purified and nonpurified viral supernatants were analyzed for their RT activity and their infectious titer by G418 selection. The RT activities in both purified and nonpurified viral suspensions are linearly correlated to the viral particle concentrations in the suspensions at a range of 105 to 109 CFU/ml (i.e., log T = 1.0333 × log cpm + 3.767 [coefficient of determination {R2} = 0.9987], where T and cpm represent titer and RT activity of a supernatant, respectively). Since the RT activity per milliliter (6 ± 4 × 10−6 cpm/particle) for the purified supernatant was known, the viral particle concentration in the nonpurified viral supernatant was determined through a quantitative RT activity assay. The viral particle concentrations of one batch of fresh and one-step-centrifuged viral supernatants were 3.70 × 109 and 1.57 × 1011 particles per ml, respectively (Table 1). The ratio of viral particles to infectious CFU (Cp/titer) for the fresh viral supernatant was 767 ± 517 (n = 6). After one-step concentration by centrifugation, the viral particles per titer were significantly lowered to 206 ± 37 (n = 4); this value was further lowered to 122 ± 27 (n = 4) after sucrose gradient purification.
Growth kinetics of target NIH 3T3 cells.
Target cells should be at exponential growth phase to get maximum retroviral transduction efficiency, as retroviruses can only transduce actively dividing cells (11, 21); the specific growth rate (μ) of target cells can be calculated from the equation X = X0exp (μt), where X is the cell number at an arbitrary time t and X0 is the cell number at the inception of the transduction. To achieve exponential growth, cells were inoculated 1 day before overlaying supernatant (i.e., after lag growth phase) at low inoculation density (i.e., negligible level of contact inhibition). The growth of target NIH 3T3 cells fits well to the exponential growth curve, and μ was calculated to be 0.0445 h−1 (Fig. 2).
FIG. 2.

Growth kinetics of NIH 3T3 cells. After inoculation (2.5 × 104 cells/well with area of 9.8 cm2) for 1 day, the number of cells (X) was determined at 0, 12, 24, 36, and 48 h. μ was determined to be 0.0445 h−1 from the slope for a least-square linear fit of ln (X/X0) to t.
Deactivation rate of retroviral infectivity.
Since retrovirus decay follows the Arrhenius equation with first-order decay [i.e., T = T0 exp(kdt)] (18), kd can be obtained as the negative slope by plotting ln (T/T0) versus t. After the freshly collected viral supernatants were incubated at 37°C for different times, their infectious titers on NIH 3T3 cells were measured. The data were plotted, and the value of kd for MoMuLV at 37°C was determined to be 0.1186 h−1 and the corresponding half-life was 5.84 h (Fig. 3). A consistent value of kd was obtained with FV, CV, and PV (data not shown).
FIG. 3.

Decay kinetics of ecotropic retrovirus. The titers of MoMuLV supernatants incubated at 37°C for different times were measured and plotted as ln (T/T0). The kd of ecotropic MoMuLV was the slope determined by a linear least-square fit of ln (T/T0) versus t. kd was 0.1186 h−1, and the corresponding half-life was 5.84 h.
Determination of infectious retrovirus concentration.
The mathematical model used to quantitate the processes of retroviral diffusion, deactivation, and uptake was recently developed (see Appendix A) (15). The concentration profiles of infectious retrovirus in the supernatant (C) can be depicted mathematically as
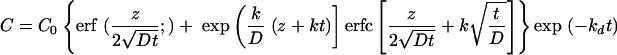 |
where C0 represents the initial concentration of infectious retrovirus, z is the depth of supernatant overlaid on target cells, D is the diffusion coefficient of the retrovirus (6.5 × 10−8 cm2/s) (7), k is the transduction rate constant, and t is the exposure time of target cells to supernatant.
The number of colonies (NC), a quantity reflecting the number of infectious retroviruses taken up by the target cells, can be expressed as
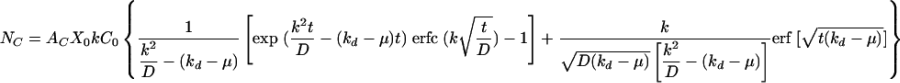 |
(1) |
where AC is the area occupied by one target cell. As for the other parameters, X0 (2.57 × 104 cells/well at t = 0) and AC (376.1 ± 148.0 μm2) were measured directly from the plates being transduced and kd and μ were determined experimentally; then k and C0 could be obtained by nonlinear regression of equation 1 with a time course profile of NC measured experimentally. Based on equation 1, the number of colonies does not increase significantly after 12 h of incubation (i.e., 90% of maximum colony number) and the titer based on the colony formation assay is not linearly proportional to the incubation time.
By using experimentally determined parameters X0, AC, μ, and kd shown above, NC values at different times were curve fitted with equation 1 to obtain unknown constants C0 and k for the fresh, concentrated, and purified ecotropic MoMuLV suspensions (Fig. 4 and Table 1). Numbers of colonies at different periods of time from the inception of transduction (i.e., up to 48 h) were determined by counting stained NIH 3T3 cell colonies, which were assumed to have originated from one transduced cell. Since each of the viral supernatants was diluted in order to obtain separated colonies, the concentration of infectious retrovirus (Ci) was obtained by multiplying C0 by the dilution factor (FD) (Table 1). Results show that the titer represents only a small fraction of the infectious retroviral particles existing in the supernatant. The higher k (2.77 × 10−2 cm/h) of fresh supernatant compared with those of concentrated (k = 1.50 × 10−2 cm/h) and purified (k = 1.27 × 10−2 cm/h) virus indicates that freshly collected ecotropic retrovirus transduces target NIH 3T3 cells more efficiently. The values of Ci per titer are consistent for all viral solutions in a range of 143 to 179 and are close to the ratio of particle concentration to infectious CFU of PV (Cp/titer = 122 for PV) (Table 1).
FIG. 4.

Colony formation for different viral supernatants. After NIH 3T3 cells on six-well plates were transduced by overlaying FV (•), CV (▴), or PV (▪) for different times, they were subjected to G418 selection for 1 week to form colonies (NC). The time course profiles of formed colonies (NC) are curve fitted with equation 1. Simulated values of C0 and k are summarized in Table 1. Triplicate data were taken, with standard deviations shown by the bars.
Effect of freeze-thaw on retroviral infectivity.
The ecotropic MoMuLV suspension produced from GP+E86/LNCX cells lost approximately 80% its viral titer after two or three freeze-thaw processes (Fig. 5), while more cycles did not significantly further reduce the infectivity titer; titer stabilized at around 15% of the original infectivity titer.
FIG. 5.

Effect of freezing and thawing on viral activity. The retroviral supernatant was stored at −80°C and thawed (see Materials and Methods). This freeze-thaw process was repeated seven more times. Each time after the thaw, the viral supernatant was titered and the titer was compared to the titer of fresh collected supernatant to generate a relative titer.
Repeated binding and titration.
A repeated mixing of viral suspension with serial sets of fresh cells to deplete viral particles was performed. The viral binding on the cell surface and remaining RT activity were analyzed. After 1 ml of viral supernatant (6 × 109 particles/ml) was mixed with either 105 or 107 NIH 3T3 cells for 30 min at 15°C, the virus-containing supernatant was recovered and an aliquot of the recovered viral supernatant was taken to measure its RT activity and its infectious titer. The rest of the recovered viral supernatant was mixed with another set of fresh cells. The viruses on the cells were analyzed by an indirect immunofluorescence assay for quantification of viral particles bound on the cell surface. After viruses were mixed with 105 cells five times, the amounts of viral vectors bound to the cell surface remained similar (Fig. 6a). In contrast, the bound viral particles on the cell surface decreased to nearly background level when the viral supernatant was mixed with 107 cells five times. Similarly, RT activity, representing viral particle concentration in the viral supernatant, did not change significantly (at a level of 6 × 108 particles/ml) after the viral supernatant was mixed with 105 cells five times, while the RT activity decreased about 40% (from 6 × 108 to 3 × 108 particles/ml) after viral supernatant was mixed with 107 cells five times (Fig. 6b).
FIG. 6.

Repeated mixing of viral supernatant with fresh cells. A virus supernatant (6 × 109 particles/ml) was mixed with either 105 (▪) or 107 (▴) NIH 3T3 cells for 30 min at 15°C. The cells and virus supernatant were separately recovered by centrifugation. The recovered viral supernatant was mixed with another set of fresh cells, and this procedure was repeated five times. (a) The recovered cells were analyzed by an indirect immunofluorescence assay, and the fluorescence on the cell surface was measured. f, fluorescence intensity determined by flow cytometry. (b) Recovered viral supernatants were analyzed for their RT activity. The RT activity was converted into RT titer (number of particles per milliliter) by comparing the RT activity to the purified viral suspension with known particle concentration by the EM method.
DISCUSSION
The methods for measuring infectious retroviral particles in the supernatant harvested from cell culture medium can be categorized as direct and indirect. Direct methods involve quantifying a physical property: counting physical particles under EM or measuring a viral indispensable component such as reverse transcriptase to quantify retroviral particles. The most conventional method is indirect and uses titration based on the number of target cells which are successfully transduced by encountering a retrovirus. However, virus titration is a limited assay. When a viral supernatant (in six serial dilutions, 10−1 to 10−6) was titered five consecutive times by incubation with cells on a series of plates for 20 min at 37°C, the viral titers measured for these series of incubation were not significantly different with each repeat (data not shown), indicating that only a small fraction of virus was actually measured. The data reported here show that there is a difference of 2 orders of magnitude (Cp/titer ranged from 122 to 767) between titer, as determined by colony formation assay, and number of physical particles, as determined by direct count under EM or indirectly by RT activity assay. This indicates that the real concentration of infectious retrovirus can be underestimated by the titration method.
The discrepancy results from several factors. Since retroviruses have a relatively short half-lives of 5 to 8 h (Fig. 3) (7), the presence of a retroviral particle does not necessarily translate into biological activity (i.e., infectivity). Virus counting under EM cannot distinguish infectious from noninfectious viruses and, therefore, overestimates the active viral concentration. On the other hand, diffusion limits retroviral adsorption, and multiple virus entries per cell can cause an underestimation of the infectious retrovirus number (7, 29). It has been reported that only one-third (33%) of retroviruses remain infectious in the supernatant harvested after 24 h due to spontaneous decay at 37°C (18). After being frozen at −80°C and thawed, approximately 60% of activity is further lost (Fig. 5). Therefore, only 13% of the initially produced retroviruses preserve infectious capability after being thawed from −80°C storage. More than 80% of retroviruses in the supernatant are inactive particles. In consideration of factors of decay (33%) and freezing and thawing (40%), the infectious particle concentration in FV can be estimated as 4.9 × 108 CFU/ml (3.7 × 109 [Cp] × 0.33 × 0.4), which is close to the Ci (7.2 × 108 CFU/ml; Table 1) calculated by our mathematical model. After purification, the total viral particle number was reduced by 264-fold, which is greater than the loss of total infectious viruses by titer (45-fold). It is likely that there are a number of retrovirus transduction inhibitors, which could be noninfectious retroviral particles, soluble envelope proteins released from the producer cell surface or from viral particles, or proteoglycans in the freshly collected viral supernatant (3, 19, 30). The decrease in the ratio Cp/titer in Table 1 implies that significant amounts of inhibitors were removed by centrifugation and purification. Note also that Cp is close to Ci in the PV suspension, which indicates that most of the particles in the PV suspension are infectious.
Virus particles in freshly collected supernatant cannot be directly counted by EM for the following reasons. (i) The concentration of retrovirus is too low to be observed on a limited area of grid. The fresh virus is approximately 20 to 100 times less concentrated than the purified virus. (ii) The impurity of the FV prevents observation under EM. Sugar and proteins in the solution are burned under the high-energy beam, resulting in carbon black. Therefore, the virus particle count in the fresh virus preparation was measured by an RT activity assay. The possibility of overestimating retroviral particles by RT assay was not accounted for because of the following facts. After ultracentrifugation (35,000 rpm with a Beckman SW-40 rotor, which is equivalent to 218,000 × g), the majority (>90%) of the RT activity was recovered in the pellet; total RT activity in supernatant was about 100-fold lower than that in the pellet (data not shown). However, after lower-speed centrifugation (9,500 rpm with Beckman rotor JA-14, which is equivalent to 14,000 × g), less than 10% of RT activity was recovered in the pellet: total retroviral particles in FV (determined by RT activity assay) was 3.70 × 109 × 1,000 particles, compared to the total in CV, 1.57 × 1011 × 1 particles (Table 1). These data indicate that most RT is confined in particles with different sizes. Ultracentrifugation can spin down all particles, leaving not much free RT in solution. There are lots of less-condensed or “defect” particles, e.g., bare particles, which contain RT but which cannot be centrifuged down at lower speed.
In the quantitative method introduced in this study, μ, AC, and the kd of a retrovirus were measured experimentally. Equation 2 was then fitted to the experimental data for NC obtained at different transduction times, representing infectious retroviral activity, in order to determine infectious retrovirus concentration at the beginning of transduction (C0) and the transduction rate constant (k). The results show consistent values of Ci/titer (143 to 179) for FV, CV, and PV, which closely agree with the ratio of physical particle number determined by EM counting to the titer for the purified viral supernatant (Cp/titer = 122). This value is 5 to 20 times higher than the result determined by the repeated-titration method (28) and 1.5 to 10 times higher than that obtained by the the mathematical approach of Andreadis et al. (2). Tavoloni empirically calculated the infectious retrovirus concentration based on the fact that only a small fraction of retrovirus is adsorbed during repeated titration (28). Since retroviral particles decay during the repeated titration, a change of titer reflects not only adsorbed retroviral particles but also loss of infectivity of retroviruses, which could result in the lower estimation of the viral concentration by Tavoloni. Andreadis et al. (2) adopted the viral diffusion model initially developed by Valentine and Allison (29), with the modification of introducing a viral decay term, and calculated the viral concentration in the power series expressions with the assumption that the retrovirus binds and internalizes infinitely fast (i.e., k→∞). As shown in our results, retroviral uptake is indeed not very fast (k ≈ 1.27 × 10−2 to 2.77 × 10−2 cm/h; Table 1) as assumed by previous researchers. As a matter of fact, for the same data for NC (i.e., NC is constant), C0 should decrease as k increases (equation 1), which could be the reason that Andreadis et al. obtained a value for Ci/titer 1.5 to 10 times lower than our result. In other words, real retroviral transduction is 1.5 to 10 times less efficient than that which would occur with a k corresponding to infinitely fast binding and internalization (e.g., physical adsorption). The model we introduced in this study is more extended than the model by Andreadis et al. and more systematically set up than the one by Tavoloni. Our method is universally applicable to various transduction systems in that it introduces a k which depends on the viral vector, target cell, and transduction conditions. In addition, C0 and k were obtained easily by curve fitting the experimental data with the closed-form mathematical expression, which can be handled more easily than the power series expression used by Andreadis et al. (2).
In our study we found that purification can change the value of k of the viral supernatant. Since k and C0 were determined by nonlinear regression of equation 1 with experimental data (i.e., number of colonies at different times of transduction), k is the lumped parameter of all steps included in retroviral transduction from initial binding of the viral particle to final expression of the transgene. For example, if there is an unsuccessful step during transduction, there is no colony formed and k becomes zero. Therefore, k is measure of the overall efficiency of all transduction steps. In our previous report (16), a 3.7-fold-higher k value was obtained by using vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped MoMuLV rather than the natural ecotropic envelope. This could be explained by the observation that VSV-G has stronger fusogenic activity than the MoMuLV envelope. VSV-G can even convert noninfectious viral particles into infectious viral particles and increase transfection of plasmid DNA (1, 24). We have also shown that the concentration of Polybrene increases k and titer (15). NIH 3T3 cells were transduced by VSV-G-pseudotyped MoMuLV with a higher k value and titer than human embryonic kidney epithelial 293 cells (data not shown). This is due to different densities of receptors on the cellular surface (14) and different rates of mitotic activity, which affects retroviral transduction (11, 21). However, it should be emphasized that the value of C0 was conserved for the same batch of retroviral supernatant with different target cells and Polybrene concentration, while k was dependent on the target cell type and Polybrene concentration (15). Therefore, k is the parameter that represents the diversity of the experimental systems consisting of various retroviruses (e.g., ecotropic and VSV-G-pseudotyped MoMuLVs) and target cells (e.g., NIH 3T3 and 293 cells) at different conditions (e.g., Polybrene concentration).
In this study, the k value for FV was higher than those for CV and PV (Table 1), which suggests that freshly collected retrovirus transduces cells more efficiently than the purified retrovirus. This could be explained in two ways. First, the infectious activity of retroviral particles could be damaged by the centrifugation and purification processes due to the fragile disulfide bond of envelope proteins (6, 10, 25). Second, the removal of soluble envelope protein in the supernatant could result in less-efficient fusion between viral and cellular membranes. Previous studies have shown that soluble ecotropic envelope protein binds to the cellular receptor, stimulates membrane fusion, and helps overcome defective retroviral transduction (12, 17, 31), although free envelope proteins normally inhibit infection by virions having the same ones on their surfaces. A defective retrovirus, which has less envelope protein than an infectious one, probably can be converted into an infectious retrovirus if it adsorbs more envelope protein onto its membrane. Thus, centrifugation and purification of retroviral particles result in less-efficient transduction by damaging linkages and removing the fusogenic activity of envelope proteins, but at the same time badly damaged noninfectious particles are also removed (i.e., significantly lower Cp/titer for CV and PV than for FV and similar Ci/titer values [Table 1]).
The infectious retrovirus concentration measured in this report closely reflects the real viral infectivity, which is underestimated by the titration method and which is overestimated by EM counting and the RT assay. The results of this study suggest that, while viral infectivity remains the same, the transduction efficiency can be increased if the retroviral particles can be forced to encounter target cells more efficiently, such as through spinoculation or flowthrough transduction (8, 13). Moreover, the method introduced here can be applied to any retrovirus- or lentivirus-mediated transduction with random motion and relatively fast decay.
Acknowledgments
This work was supported in part by grants from the American Heart Association (H.Y.), the May R. Wright Foundation (H.Y.), and Genetic Therapy Inc./Novartis; by NIH grant CA 59318 (W.F.A.); and by a grant from the Charles Lee Powell Foundation (C.-A.P.).
APPENDIX A
A scheme of the mathematical model is shown in Fig. A1.
Model description and its derivations.The model shown in this study is based on the diffusion of colloidal retroviral particles. Since retrovirus moves in random Brownian motion in the supernatant, the change of infectious retroviral concentration (C) at a given time t can be mathematically described by retroviral diffusion (D[∂2C/∂z2]) and the first-order viral decay (kdC) (Fig. A1, 1).
It is clear that there are infectious retroviruses mixed homogeneously (C = C0) before the transduction (t = 0). Once a retroviral particle encounters a cell at the bottom (z = 0, where z is the coordinate of the medium depth), the rate of retrovirus taken up by the target cell is proportional to the infectious retrovirus concentration in the supernatant 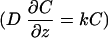 (Fig. A1, 2). The colloidal particle with a half-life (t1/2) moves only 2Dt1/2 with its activity (i.e., 400 to 600 μm with a t1/2 of 4 to 8 h). Therefore, a retrovirus more than 0.6 mm above the cell bed (z→∞) hardly has chance to encounter cells with infectivity (C = C0exp [−kdt]) (Fig. A1, 3). From the mathematical expressions of parts 1 to 3, the concentration of infectious retroviral concentration is derived as
(Fig. A1, 2). The colloidal particle with a half-life (t1/2) moves only 2Dt1/2 with its activity (i.e., 400 to 600 μm with a t1/2 of 4 to 8 h). Therefore, a retrovirus more than 0.6 mm above the cell bed (z→∞) hardly has chance to encounter cells with infectivity (C = C0exp [−kdt]) (Fig. A1, 3). From the mathematical expressions of parts 1 to 3, the concentration of infectious retroviral concentration is derived as
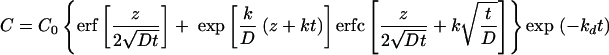 |
Because the possibility of retroviral adsorption to the target cell surface increases with the area occupied by cells (A) and the transduction time (t), the number of colonies (NC) can be expressed by
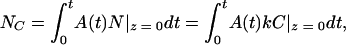 |
where N|z = 0 = kC|z = 0 = kC0exp (k2t/D)erfc (k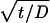 )exp (−kdt). After further derivation, the final equation used to describe the number of colonies is
)exp (−kdt). After further derivation, the final equation used to describe the number of colonies is
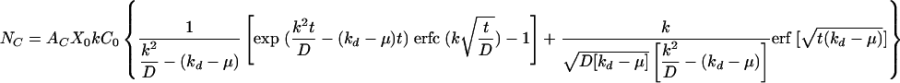 |
It should be emphasized that the rate-limiting steps were assumed to be extracellular diffusion and the reaction rate on the cell surface. However, experimentally, a colony can be formed only with successful intracellular and extracellular transduction processes (Fig. A1, 4). Therefore, k is a lumped parameter representing the apparent efficiency of all transduction processes but significantly affected by extracellular steps.
REFERENCES
- 1.Abe, A., S. T. Chen, A. Miyanohara, and T. Friedmann. 1998. In vitro cell-free conversion of noninfectious Moloney retrovirus particles to an infectious form by the addition of the vesicular stomatitis virus surrogate envelope G protein. J. Virol. 72:6356-6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreadis, S., T. Lavery, H. F. Davis, J. M. Le Doux, M. L. Yarmush, and J. R. Morgan. 2000. Toward a more accurate quantitation of the activity of recombinant retroviruses: alternatives to titer and multiplicity of infection. J. Virol. 74:3431-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batra, R. K., J. C. Olsen, D. K. Hoganson, B. Caterson, R. C. Boucher. 1997. Retroviral gene transfer is inhibited by chondroitin sulfate proteoglycans/glycosaminoglycans in malignant pleural effusions. J. Biol. Chem. 272:11736-11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Battini, J. L., P. Rodrigues, R. Muller, O. Danos, and J. M. Heard. 1996. Receptor-binding properties of a purified fragment of the 4070A amphotropic murine leukemia virus envelope glycoprotein. J. Virol. 70:4387-4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bierley, S. T., R. Raineri, J. A. Poiley, and E. M. Morgan. 1996. A comparison of methods for the estimation of retroviral burden. Dev. Biol. Stand. 88:163-165. [PubMed] [Google Scholar]
- 6.Braas, G., P. F. Searle, N. K. H. Slater, and A. Lyddiatt. 1998. Strategies for the isolation and purification of retroviral vectors for gene therapy. Bioseparation 6:211-228. [PubMed] [Google Scholar]
- 7.Chuck, A. S., M. F. Clarke, and B. O. Palsson. 1996. Retroviral infection is limited by Brownian motion. Hum. Gene Ther. 7:1527-1534. [DOI] [PubMed] [Google Scholar]
- 8.Chuck, A. S., and B. O. Palsson. 1996. Consistent and high rates of gene transfer can be obtained using flow-through transduction over a wide range of retroviral titers. Hum. Gene Ther. 7:743-750. [DOI] [PubMed] [Google Scholar]
- 9.Evans, L. H., R. P. Morrison, F. G. Malik, J. Portis, and W. J. Britt. 1990. A neutralizable epitope common to the envelope glycoproteins of ecotropic, polytropic, xenotropic, and amphotropic murine leukemia viruses. J. Virol. 64:6176-6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gliniak, B. C., S. L. Kozak, R. T. Jones, and D. Kabat. 1991. Disulfide bonding controls the processing of retroviral envelope glycoproteins. J. Biol. Chem. 266:22991-22997. [PubMed] [Google Scholar]
- 11.Hajihosseini, M., L. Iavachev, and J. Price. 1993. Evidence that retroviruses integrate into post-replication host DNA. EMBO J. 12:4969-4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones, J. S., and R. Risser. 1993. Cell fusion induced by the murine leukemia virus envelope glycoprotein. J. Virol. 67:67-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotani, H., P. B. Newton III, S. Zhang, Y. L. Chiang, E. Otto, L. Weaver, R. M. Blaese, W. F. Anderson, and G. J. McGarrity. 1994. Improved methods of retroviral vector transduction and production for gene therapy. Hum. Gene Ther. 5:19-28. [DOI] [PubMed] [Google Scholar]
- 14.Kurre, P., H. P. Kiem, J. Morris, S. Heyward, J. L. Battini, and A. D. Miller. 1999. Efficient transduction by an amphotropic retrovirus vector is dependent on high-level expression of the cell surface virus receptor. J. Virol. 73:495-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon, Y. J., and C.-A. Peng. 2002. Transduction rate constant as more reliable index quantifying efficiency of retroviral gene delivery. Biotechnol. Bioeng. 77:668-677. [DOI] [PubMed] [Google Scholar]
- 16.Kwon, Y. J., and C.-A. Peng. 2002. Engineering analysis of ex vivo retroviral transduction systems. Ann. Biomed. Eng. 30:731-742. [DOI] [PubMed] [Google Scholar]
- 17.Lavillette, D., A. Ruggieri, S. J. Russell, and F. L. Cosset. 2000. Activation of a cell entry pathway common to type C mammalian retroviruses by soluble envelope fragments. J. Virol. 74:295-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Doux, J. M., H. E. Davis, J. R. Morgan, and M. L. Yarmush. 1999. Kinetics of retrovirus production and decay. Biotechnol. Bioeng. 63:654-662. [PubMed] [Google Scholar]
- 19.MacNeill, E. C., H. Hanenberg, K. E. Pollok, J. C. M. van der Loo, M. F. Bierhuizen, G. Wagemaker, and D. A. Williams. 1999. Simultaneous infection with retroviruses pseudotyped with different envelope proteins bypasses viral receptor interference associated with colocalization of gp70 and target cells on fibronectin CH-296. J. Virol. 73:3960-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.March, K. L., J. E. Madison, and B. C. Trapnell. 1995. Pharmacokinetics of adenoviral vector-mediated gene delivery to vascular smooth muscle cells: modulation by poloxamer 407 and implications for cardiovascular gene therapy. Hum. Gene Ther. 6:41-53. [DOI] [PubMed] [Google Scholar]
- 21.Miller, D. G., M. A. Adam, and A. D. Miller. 1990. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol. 10:4239-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mittereder, N., K. L. March, and B. C. Trapnell. 1996. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J. Virol. 70:7498-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mountain, A. 2000. Gene therapy: the first decade. Trends Biotechnol. 18:119-128. [DOI] [PubMed] [Google Scholar]
- 24.Okimoto, T., T. Friedmann, and A. Miyanohara. 2001. VSV-G envelope glycoprotein forms complexes with plasmid DNA and MULV retrovirus-like particles in cell-free conditions and enhances DNA transfection. Mol. Ther. 4:232-238. [DOI] [PubMed] [Google Scholar]
- 25.Pham, L., H. Ye, F. L. Cosset, S. J. Russell, and K. W. Peng. 2001. Concentration of viral vectors by co-precipitation with calcium phosphate. J. Gene Med. 3:188-194. [DOI] [PubMed] [Google Scholar]
- 26.Shabram, P., and E. Aguilar-Cordova. 2000. Multiplicity of infection/multiplicity of confusion. Mol. Ther. 2:420-421. [DOI] [PubMed] [Google Scholar]
- 27.Stannard, L. M., F. D. van der Riet, and J. W. Moodie. 1987. The morphology of human immunodeficiency virus particles by negative staining electron microscopy. J. Gen. Virol. 68:919-923. [DOI] [PubMed] [Google Scholar]
- 28.Tavoloni, N. 1997. A simple procedure to determine the biological titer of recombinant retroviral vectors. Gene Ther. 4:150-155. [DOI] [PubMed] [Google Scholar]
- 29.Valentine, R. C., and A. C. Allison. 1959. Virus particle adsorption. 2. Theory of adsorption and experiments in the attachment of particles to non-biological surfaces. Biochim. Biophys. Acta 34:10-23. [DOI] [PubMed] [Google Scholar]
- 30.Walker, S. J., M. Pizzato, Y. Takeuchi, and S. Devereux. 2002. Heparin binds to murine leukemia virus and inhibits Env-independent attachment and infection. J. Virol. 76:6909-6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, H., R. Paul, R. E. Burgeson, D. R. Keene, and D. Kabat. 1991. Plasma membrane receptors for ecotropic murine retroviruses require a limiting accessory factor. J. Virol. 65:6468-6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willey, R. L., D. H. Smith, L. A. Lasky, T. S. Theodore, P. L. Earl, B. Moss, D. J. Capon, and M. A. Martin. 1988. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 62:139-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu, H. 2002. Preparation of pseudotyped retroviral vector, p. 149-159. In J. R. Morgan (ed.), Gene therapy protocols, 2nd ed. Humana Press, Totowa, N.J.
- 34.Yu, H., C. Empig, J. Xia, and W. F. Anderson. 1998. Quantitation of MoMuLV envelope protein on the cell surface. Virology 243:415-422. [DOI] [PubMed] [Google Scholar]
- 35.Yu, H., N. Soong, and W. F. Anderson. 1995. Binding kinetics of ecotropic (Moloney) murine leukemia retrovirus with NIH 3T3 cells. J. Virol. 69:6557-6562. [DOI] [PMC free article] [PubMed] [Google Scholar]