

Abstract
Background
Allopregnanolone (ALLO) and structurally related endogenous neurosteroids are potent modulators of GABAA receptor function at physiologically relevant concentrations. Accumulating evidence implicates a modulatory role for ALLO in behavioral processes underlying ethanol self-administration, discrimination and reinstatement. The purpose of this study was to evaluate the impact of exogenous neurosteroid challenges with the agonist ALLO and the partial agonist/antagonist epipregnanolone (EPI) on the microarchitecture of ethanol drinking patterns.
Methods
Male C57BL/6J mice were initiated to consume an unsweetened 10% v/v ethanol solution (10E) by a saccharin fading procedure during daily 2-hour limited access sessions beginning 1 hour after dark phase onset. Cumulative lick responses were recorded for 10E and water using lickometer circuits. After establishing 10E intake baselines, mice were habituated to vehicle injection (VEH; 20% w/v β-cyclodextrin; i.p.), and then were treated with either VEH or neurosteroid immediately prior to the drinking session. Each mouse received a series of ALLO doses (3.2, 10, 17 and 24 mg/kg) alone and EPI doses (0.15, 1, 3 and 10 mg/kg) alone in a counterbalanced within-group design.
Results
The GABAA receptor positive modulator, ALLO, dose-dependently modulated overall ethanol intake throughout the 2-hr session with the 3.2 mg/kg dose eliciting a significant increase whereas the 24 mg/kg dose produced a significant suppression of ethanol intake versus vehicle pretreatment. ALLO-evoked alterations in intake corresponded with a significant, dose-dependent alterations in bout frequency and inter-bout interval. ALLO also elicited robust, dose-dependent elevations in 10E licks during the initial 5-minutes of access, but subsequently resulted in a dose-dependent suppression of 10E licks during session minutes 20–80. In contrast, the partial agonist/antagonist neurosteroid, EPI, exhibited no influence on any consumption parameter evaluated.
Conclusions
The present findings suggest that GABAA receptor-active neurosteroids may modulate the regulatory processes that govern the onset, maintenance, and termination of drinking episodes. The differential influence of ALLO and EPI on ethanol intake patterns may reflect an alteration in GABAergic inhibitory tone that is likely due to each neurosteroid’s pharmacological profile at GABAA receptors. Manipulation of endogenous ALLO may prove a useful strategy for diminishing excessive intake and protecting against the loss of regulatory control over drinking.
Keywords: Allopregnanolone, Epipregnanolone, Drinking Patterns, Lickometer, Bout Microarchitecture
INTRODUCTION
The endogenous neurosteroid allopregnanolone (ALLO, 3α-hydroxy-5α-pregnan-20-one) is a potent positive modulator of GABA-induced chloride influx through the GABAA receptor ionophore (e.g., Lambert et al., 1995). Consistent with its positive modulation of GABAA receptors, ALLO possesses anesthetic, anticonvulsant, sedative-hypnotic, anxiolytic, locomotor stimulant, and muscle relaxant properties (e.g., Finn et al., 1997; Gasior et al., 1999). ALLO and related neurosteroids are known to selectively interact with GABAA receptors in the nanomolar to low micromolar range of concentrations (e.g., Lambert et al., 2003), and their concentrations episodically vary in concert with reproductive cycle-related hormonal fluctuations (Concas et al., 1998; Corpechot et al., 1997; Genazzani et al., 2002; Herbison, 2001; Palumbo et al., 1995) and in response to environmental and physiological stressors. For example, acute foot shock stress was found to elevate plasma and brain ALLO levels in rats by as much as 3- to 4-fold (Barbaccia et al., 2001). Ethanol, known to activate the stress axis, also augmented endogenous brain ALLO levels when injected in rats (Barbaccia et al., 1999; Morrow et al., 2001) or self-administered by male mice (Finn et al., 2004). Collectively, these findings indicate that in vivo fluctuations in endogenous ALLO levels (1–100 nM) are physiologically relevant, since this range of concentrations can potentiate the action of GABA at GABAA receptors in vitro (Biggio et al., 2001; Gee et al., 1988; Morrow et al., 1987; Paul and Purdy, 1992).
Ethanol influences the function of multiple neurotransmitter receptor ion channels and voltage-gated channels (Crews et al., 1996). Its propensity to modulate GABAA receptor complex function has been extensively documented (Grobin et al., 1998; Mihic et al., 1997). Some behavioral and pharmacological manifestations of ethanol (presumably via an action at GABAA receptors) are similarly exhibited by ALLO (Grobin et al., 1998; Morrow et al., 2001). Thus, the interaction of ALLO with ethanol’s behavioral and neurochemical effects has recently received considerable attention.
Several studies have demonstrated a modulatory role for ALLO in ethanol self-administration and reinstatement. In 1998, Janak and colleagues demonstrated ALLO’s propensity to enhance ethanol-reinforced instrumental responding in male rats (Janak et al., 1998). Specifically, ALLO increased the number of active lever presses and resultant ethanol dipper presentations primarily by enhancing responses during the initial run of a 30-minute operant session, and in the absence of a concomitant change in response rate. A more recent study compared the efficacy of ALLO, the GABAA receptor agonist muscimol, and the GABAB receptor agonist baclofen as modulators of operant responding for ethanol (Janak and Gill, 2003). ALLO dose-dependently augmented ethanol-reinforced lever pressing behavior, whereas muscimol and baclofen decreased this measure at the doses examined. ALLO also selectively enhanced ethanol-reinforced responding when a sucrose solution was concurrently available, suggesting specificity for ALLO in modulating ethanol self-administration in male rats. Consistent with these findings, it was demonstrated in our laboratory that ALLO dose-dependently increased consumption of unsweetened 5% and 10% ethanol solutions during the first hour of 2-hour limited access sessions in male mice, without concomitantly altering water intake (Sinnott et al., 2002). ALLO was also recently shown to promote ethanol reinstatement (Nie and Janak, 2003); ALLO priming injections were found to dose-dependently reinstate extinguished operant responding of male rats on an active lever previously associated with 10% ethanol, an effect that failed to occur for a lever previously paired with a 5% sucrose solution.
Numerous studies have been conducted to evaluate the ability of ALLO and ethanol to substitute for one another within rodent and primate drug discrimination paradigms (e.g., Bowen et al., 1999; Engel et al., 2001; Grant et al., 1996; Hodge et al., 2001). Notably, ALLO fully substituted for ethanol in these procedures, and this substitution for subjective drug effects was not generalizable to muscimol (Engel et al., 2001), consistent with the selectivity of ALLO in the enhancement of ethanol-reinforced operant responding (Janak and Gill, 2003). Collectively, these studies suggest that ALLO may be intimately involved in modulating the regulatory processes underlying ethanol consumption and reward.
In humans, quantity-frequency measures (size of each drinking episode and number of drinking episodes) of ethanol intake are utilized to estimate habitual use and can help to diagnose individuals in which a regulatory loss of control has likely occurred (Feunekes et al., 1999). Furthermore, patterns of human ethanol consumption impact projected health risks associated with ethanol abuse (Tenth Special Report to U.S. Congress, 2000), with binge drinking being associated with greater health risk when compared to consumption in moderation. Recent findings among human populations suggest that overall mean consumption alone is an insufficient indicator of alcohol-related problems and social harm, but rather identification of problematic drinking episodes (bouts) will likely provide more effective preventive strategies (Bobak et al., 2005; Gmel et al., 2001). Thus, in depth assessments of ethanol consumption patterns within animal models may provide valuable predictions in the characterization of regulatory mechanisms (e.g., physiological states or environmental cues) that underlie the loss of intake control in humans. Microanalysis procedures for ethanol bout architecture (i.e., bout size, bout frequency and lick rate), have been reliably utilized in evaluating the impact of the estrous cycle (Ford et al., 2002), strain and genetic selection (Samson, 2000), gene knockout or null mutations (Olive et al., 2000; Risinger et al., 2000), circadian rhythms (Boyle et al., 1997; Goldstein and Kakihana, 1977; Millard and Dole, 1983) and, importantly, drug treatment effects (e.g., Czachowski et al., 2001a; Czachowski et al., 2001b; Middaugh et al., 1999b; Samson and Chappell, 2003; Smith et al., 1999) on ethanol self-administration patterns in rodent models. It has similarly been observed in humans that the efficacy of therapeutic intervention (i.e., naltrexone) was contingent upon the demonstration of a drinking pattern routine that was susceptible to manipulation (Anton et al., 2004).
The purpose of the present study was to assess the microarchitecture of ethanol drinking patterns in response to exogenous neurosteroid challenges. No studies to our knowledge have investigated the impact of GABAA receptor-active neurosteroids (with the exception of ALLO) on ethanol intake drinking patterns. In addition to extending our previous findings with ALLO (Sinnott et al., 2002), the partial agonist/antagonist neurosteroid epipregnanolone (EPI; 3β-hydroxy-5β-pregnan-20-one; Belelli et al., 1990; Prince and Simmonds, 1992) was examined. These experiments were intended to provide further insight into the modulatory activity of neurosteroids on ethanol consummatory behavior. Based on previous findings in our laboratory and others, it was hypothesized that neurosteroid agonists, such as ALLO, would alter drinking patterns in a manner consistent with elevated abuse liability whereas a partial agonist/antagonist neurosteroid would exhibit a diminished or negligible influence. Identification of a neurosteroid candidate that elicits desirable alterations in ethanol self-administration patterns might result in the development of an alternative treatment strategy in the management of maladaptive ethanol intake control.
MATERIALS AND METHODS
Animals
Twenty-four male C57BL/6J (B6) mice (6 weeks of age; approximately 20 grams) were purchased from The Jackson Laboratory (Bar Harbor, ME). Each mouse was individually housed and acclimated to a reverse light/dark schedule (12hr/12hr; lights off at 0900 hrs) for a minimum of 7 days. All mice were provided ad libitum access to rodent chow and tap water in standard shoebox cages. Mice were weighed and handled daily throughout acclimation and experimental phases of the study. The local Institutional Animal Care and Use Committee approved all procedures in accordance with the guidelines of the Institutional Care and Use Committee of the National Institute on Drug Abuse, National Institutes of Health, and the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996).
Apparatus
Lickometer chambers consisted of plastic shoebox cages that were modified by the addition of a custom four-walled Plexiglas insert, resulting in an inner floor area of 7” × 4” and a height of 7”. A raised, stainless steel wire floor (VWR, Tualatin, OR) was positioned within this inner area. Two small portholes were located along the back wall of the insert to permit access to metal sipper tubes. A perforated Plexiglas lid was attached to the top of each chamber to retain the mice inside the chambers, while allowing for sufficient ventilation. Stainless steel sippers (double ball-bearing; Ancare, Bellmore, NY) were adjoined to polystyrene serological pipettes (10 ml reservoir; VWR) that were mounted within the outer area formed by the Plexiglas insert. These adapted drinking tubes permitted volume measurements to the nearest 0.05 ml. The chamber floor and two metal sippers formed open electrical circuits attached to a dual lickometer device (MED Associates, Inc., St. Albans, VT), which permitted the measurement of cumulative lick records for water and ethanol solutions. Lickometers were interfaced to an IBM compatible computer running MED-PC IV software (MED Associates, Inc.).
Ethanol Consumption Paradigm
The limited access time (1100–1300 hrs; 2 hr after lights out) was selected based on preliminary findings with continuous access consumption patterns in mice that indicated a 2- to 3-fold increase in ethanol lick responses during the third hour of the dark phase, when compared to light phase activity. Mice were removed from their homecages, weighed, and placed immediately in the lickometer chambers daily. A saccharin fading procedure (adapted from Middaugh et al., 2000) was implemented to initiate mice to consume a 10% v/v ethanol (10E) solution. Mice were provided access to a water sipper and an ethanol sipper containing the following solutions across sessions: 0.2% saccharin (0.2S) for 4 sessions, 10E/0.2S for 3 sessions, 10E/0.1S for 3 sessions, 10E/0.05S for 3 sessions, 10E/0.025S for 3 sessions, 10E/0.01S for 4 sessions, and 10E for 10 sessions. The mean 10E intake at the conclusion of the saccharin fading procedure was 1.16 ± 0.15 g/kg/2-hrs. Ethanol-containing fluid sippers were counterbalanced between the left and right sides across lickometer chambers to control for side preferences.
Injection Baseline Determinations and Neurosteroid Treatments
Following the establishment of stable 10E intakes (less than 10% variability in intake over 3 consecutive sessions) during the 10-day exposure referenced above, all mice were exposed to vehicle (VEH; 20% w/v 2-hydroxypropyl-β-cyclodextrin; β-cyclodextrin; 0.01 ml/g; i.p.; Cerestar USA, Inc., Hammond, IN) injections until stable intake patterns were re-established (12 sessions). Mice were then segregated into two treatment cohorts (n = 12 for each) that were balanced for g/kg of ethanol intake. One cohort received acute systemic injections of ALLO (0, 3.2, 10, 17 or 24 mg/kg) whereas the second cohort was administered EPI (0, 0.15, 1, 3 or 10 mg/kg) immediately prior to the drinking session start. Each mouse was treated once with all doses of respective neurosteroid over a 2-week period. Following each acute neurosteroid dose, mice were administered VEH until the 10E intake baseline (pre-treatment value) was re-established (typically 3–4 sessions). Neurosteroid doses were evaluated in ascending order. At the conclusion of this initial dose series, the two treatment cohorts then received all doses of the second neurosteroid, thereby counterbalancing the order of drug exposure between the cohorts. Neurosteroid doses were chosen based on previous reports demonstrating their respective effectiveness in modulating ethanol-related behaviors (Barbosa and Morato, 2000; Bowen et al., 1999; Sinnott et al., 2002).
Drugs
Ethanol solutions (10% v/v; Pharmco Products, Brookfield, CT) were prepared by dilution of a 200 proof stock in tap water. Saccharin solutions (w/v saccharin sodium salt; Sigma Chemical Co., St. Louis, MO) were similarly prepared by dilution into tap water. ALLO was synthesized by and purchased from Dr. R. H. Purdy (Veterans Medical Research Foundation, San Diego, CA) whereby EPI was purchased from Steraloids Inc (Newport, RI). Each neurosteroid was solubilized in 20% w/v β-cyclodextrin with the aid of a stir plate at a concentration that permitted an injection volume of 0.01 ml/g body weight.
Ethanol Consumption Criterion
Although B6 mice typically consume large quantities of ethanol under continuous access conditions (Middaugh et al., 1999a), three mice in the current study failed to acquire and maintain consistent intakes ≥ 0.50 g/kg subsequent to the saccharin fading procedure. Based on previous experience (Ford et al., 2002), independent variables assessed during consumption pattern microanalysis tend to be less reliable at this low intake level. Furthermore, the influence of a neurosteroid treatment would likely be masked by a ‘floor’ effect due to the absence of detectable behavioral endpoints. For this reason, low intake mice, as determined by the above criterion during the establishment of baseline 10E intake prior to each neurosteroid treatment, were excluded from all statistical analyses.
Statistical Analysis
Ethanol intakes (g/kg; g ethanol per kilogram body weight) were calculated based on the 10E volume depleted (to the nearest 1/20 ml) throughout the 2-hour sessions. Daily lick responses on the ethanol-containing and water tubes were recorded by MED-PC IV software (MED Associates, Inc.) and compiled by a custom data analysis program that determined several behavioral endpoints: total lick responses, bout frequency, bout size, bout lick rates (licks/minute), and latency to first bout (minutes). The B6 mice in this study typically exhibited ethanol preference ratios ranging between 0.85–1.00, and the low number of total water lick responses recorded (≅ 100) precluded analysis of bout dynamics and consumption patterns for water. Based upon our previous work in a rat self-administration model (Ford et al., 2002), an ethanol bout was experimentally defined as a minimum of 20 licks with no more than a 60 second pause between successive licks. More than 95% of 10E lick responses typically were incorporated within bouts utilizing this criterion. The reported lick rates were derived from the average rate of all bouts expressed, and did not reflect session time falling outside defined bouts. All statistical analyses were performed with the SigmaStat version 2.03 software package (SPSS Inc., Chicago, IL), all figures and cumulative records were depicted with the Sigma Plot 2001 software program (SPSS Inc.), and temporal distribution analysis of lick responses was facilitated by the SoftCR for Windows program (MED Associates, Inc.). The SigmaPlot linear regression function (based on least squares fit of data points) was utilized to calculate the correlation between lick responses versus g/kg of ethanol (see Figure 1 below). Treatment effects on drinking pattern parameters were assessed by one-way repeated measures ANOVA (factor: Dose). A two-way repeated measures ANOVA was used to identify a potential Dose × Time Interval interaction for each neurosteroid on the temporal distribution of total session licks (20- and 5-minute intervals). If a Dose × Time Interval interaction was detected, an analysis of simple main effects for neurosteroid dose within each interval was conducted. When appropriate, pair-wise differences were determined by the Fisher’s least significant difference multiple comparisons procedure. For all analyses, statistical significance was set at p ≤ 0.05. On limited occasions an obvious discordance between total licks and g/kg ethanol consumption was apparent, indicating that either a computer detection error occurred or a mouse pawed/grabbed a drinking sipper, leading to an unrepresentative total lick count. A licks per milliliter (licks/ml) screening criterion was imposed to pinpoint and eliminate these data points. Drinking pattern data with licks/ml values falling outside 1302 ± 678 (mean ± 2 standard deviations) were excluded from further analyses. Under this criterion, one mouse was excluded from all analyses of the ALLO dose series.
Figure 1. Positive correlation between 10E licks and ethanol dose (g/kg).
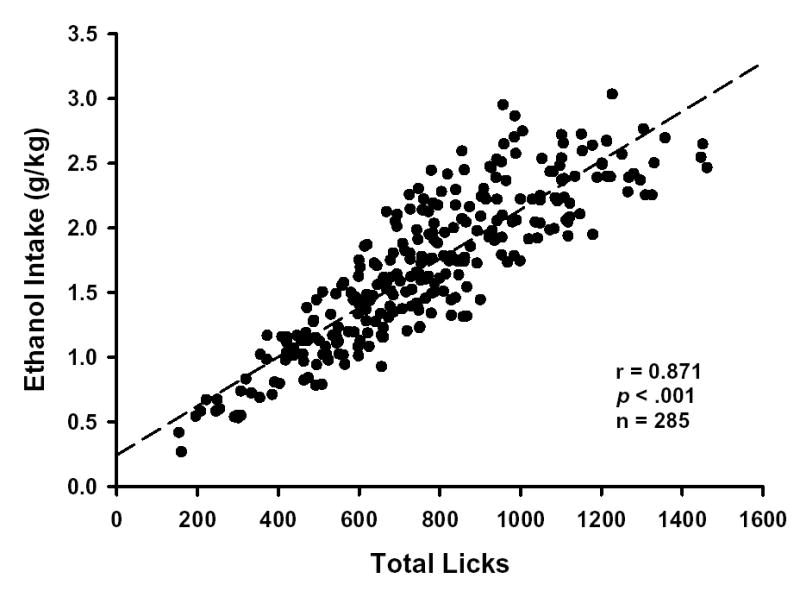
Graphed points were determined by total session 10E licks and total session ethanol dose (g/kg) for each mouse (n = 19) during all baseline and neurosteroid pretreatment sessions. The dashed line denotes a linear regression of the data. p ≤ 0.001 denotes significant positive correlation between variables as determined by Pearson product moment correlation analysis.
RESULTS
Lickometer System Validation
The sensitivity of lick detection and the precision of fluid volume measurements are critical variables in optimizing a lickometer system. Estimates of fluid volume consumed were recorded to the nearest 0.05 ml by implementing custom-built graduated reservoir tubes. The predictive validity of the drinking pattern microanalysis is dependent upon a concordant relationship between the total number of licks and the change in fluid volume. From the volume depleted during the session, total ethanol dose (g/kg) can be estimated. As illustrated in Figure 1, a significant positive correlation between ethanol licks and ethanol dose was found (r = 0.87, p < .001, n = 285). The goodness of fit (r2) of the regression line was 0.76, indicating that 76% of the variation in 10E dose (g/kg) could be accounted for by the variation in 10E licks. Thus, cumulative licks represent a reliable and non-intrusive measure for estimating 10E intake on a sub-second time scale within this system.
Dose-Response Evaluation of Acute ALLO Effects on Ethanol Intake Patterns
Ethanol intake (g/kg) over the 2-hr session was significantly influenced by ALLO pretreatment dose [F(4,66) = 10.45; p < .001]. Whereas the 3.2 mg/kg ALLO dose significantly enhanced intake by 19% (p < .05), the 24 mg/kg dose significantly attenuated consumption by 43% (p < .001), when compared to VEH pretreatment (Table 1). A significant main effect of ALLO dose [F(4,66) = 7.67; p < .001] also was found for the 10E licks. The 17 and 24 mg/kg doses significantly decreased cumulative 10E licks by 18% (p < .05) and 30% (p < .001), respectively, versus VEH administration. ALLO pretreatment dose had no significant influence on either the total number of water licks detected throughout the sessions or the preference ratio (determined by the proportion of 10E licks to all fluid licks). It was observed that the 17 mg/kg and 24 mg/kg ALLO doses attenuated both 10E and water licks proportionally by approximately 15–20% and 30–35%, respectively, when compared to VEH pretreatment (Table 1).
Table 1. Acute ALLO effects on ethanol intake patterns.
Values represent the mean ± SEM for ALLO-treated mice (n = 18). Each mouse received all ALLO doses, and the 0 mg/kg dose represents the average of baseline values (from sessions immediately preceding each ALLO pretreatment day). One mouse was excluded as a statistical outlier (outside 2 standard deviations of the mean) from the analysis of latency to start following pretreatment with 17 mg/kg ALLO.
| ALLO Dose (mg/kg) | 0 | 3.2 | 10 | 17 | 24 |
|---|---|---|---|---|---|
| Total Session Intake | |||||
| Intake (g/kg) | 1.74 ± .11 | 2.06 ± .15* | 1.82 ± .10 | 1.53 ± .15 | 1.22 ± .10*** |
| 10E Licks | 813 ± 47 | 883 ± 61 | 741 ± 48 | 665 ± 74* | 572 ± 47*** |
| Water Licks | 132 ± 28 | 102 ± 15 | 99 ± 16 | 110 ± 19 | 88 ± 14 |
| Preference Ratio | .87 ± .02 | .90 ± .01 | .89 ± .02 | .86 ± .02 | .87 ± .02 |
| Total Session Patterns | |||||
| Bout Frequency | 13.7 ± 0.8 | 15.5 ± 1.2 | 11.9 ± 1.1 | 9.9 ± 1.1*** | 10.1 ± 0.8** |
| Bout Size (g/kg) | .102 ± .005 | .114 ± .006 | .137 ± .013** | .122 ± .011 | .096 ± .006 |
| Lick Rate (licks/min) | 61 ± 11 | 71 ± 14 | 63 ± 11 | 72 ± 17 | 58 ± 10 |
| Bout Length (min) | 2.37 ± .26 | 2.33 ± .28 | 2.93 ± .48 | 2.51 ± .39 | 2.14 ± .25 |
| Inter-Bout Interval | 6.56 ± 0.73 | 5.18 ± 0.51 | 9.26 ± 1.42 | 9.39 ± 1.40 | 13.43 ± 2.84** |
| First Bout Dynamics | |||||
| Bout Size (g/kg) | .118 ± .011 | .136 ± .021 | .273 ± .064** | .188 ± .037 | .183 ± .031 |
| Lick Rate (licks/min) | 88 ± 21 | 100 ± 38 | 47 ± 11 | 80 ± 28 | 78 ± 31 |
| Latency to Start (min) | 7.51 ± 1.16 | 5.38 ± 1.19 | 6.41 ± 1.66 | 7.38 ± 3.54 | 2.95 ± 0.91 |
p ≤ 0.05,
p ≤ 0.01 and
p ≤ 0.001 versus within-subject baseline (0 mg/kg)
ALLO additionally altered the microarchitecture of ethanol self-administration. Most notably, bout frequency was significantly affected by ALLO dose [F(4,66) = 9.33; p < .001]. The two highest ALLO doses (17 and 24 mg/kg) significantly reduced bout frequency by 28% (p < .001) and 26% (p < .01), respectively, when compared to VEH pretreatment (Table 1). Mean bout size was likewise significantly influenced by ALLO dose [F(4,66) = 4.10; p < .01], but in a dose-dependent manner that was discordant from ALLO effects on bout frequency. The 10 mg/kg ALLO dose significantly enhanced mean bout size by 34% (p < .005) versus VEH administration (Table 1). Although a significant main effect of ALLO dose on inter-bout interval [F(4,66) = 9.33; p < .001] was detected, this effect was restricted to the highest ALLO pretreatment dose examined. Consistent with its reduction of bout frequency, the 24 mg/kg ALLO dose significantly extended the inter-bout interval by 105% (p < .01; Table 1) when compared to VEH pretreatment. ALLO did not influence mean lick rate or bout length, indicating that ethanol bout parameters were sensitive to modulation by ALLO in a discriminative as well as a dose-responsive manner.
In an attempt to dissociate the overall effects of ALLO on the maintenance of ethanol consumption throughout the 2-hr session from a potential role for this neurosteroid in the onset of self-administration, the dynamics of the first bout were assessed. Consistent with pretreatment effects on the mean bout size, a significant main effect of ALLO dose on the size of the first bout [F(4,66) = 2.70; p < .05] also was observed. The 10 mg/kg ALLO dose significantly augmented the size of the first bout by 131% (p < .01) versus VEH pretreatment (Table 1). Although not approaching statistical significance, a concomitant suppression was observed in the lick rate of the first bout by 47% following administration of the 10 mg/kg ALLO dose. Thus, although the onset time of consumption (latency to first bout) was not altered by ALLO pretreatment (Table 1), the size of the first drinking episode was augmented within a limited dose range.
An evaluation of the temporal distribution of licks on the ethanol sipper was conducted to provide complementary insights to the alterations in ethanol intake and bout microarchitecture that were observed following ALLO pretreatment. Lick patterns were significantly affected by ALLO as indicated by a dose × interval interaction [F(20,300) = 2.93; p < .001]. Simple main effect analyses of dose at each session interval indicated significant differences among dose groups for the 20–40 [F(4,66) = 19.91; p < .001], 40–60 [F(4,66) = 6.29; p < .001], 60–80 [F(4,66) = 2.74; p < .05], and 80–100 [F(4,66) = 2.57; p < .05] minute intervals. The 3.2 mg/kg ALLO dose significantly increased 10E licks 20–40 minutes into the session by 21% (p < .01; Fig. 2A); a finding that was congruent with the overall elevation in ethanol intake (g/kg) that occurred throughout the entire session (Table 1). With the exception of the lowest pretreatment dose, ALLO dose-dependently decreased ethanol licks throughout the middle portion of the session (minutes 20–80; Fig. 2A), with the 17 mg/kg and 24 mg/kg doses persistently eliciting a significant suppression of this behavioral endpoint (p < .05, all cases) and the 10 mg/kg dose significantly decreasing licks (p < .05, all cases) more periodically. Notably, the dose-dependent effects of ALLO pretreatment on 10E lick patterns had dissipated by the conclusion of the session (100–120 minutes), suggesting a limited time-frame of efficacy for ALLO in its alteration of ethanol lick responses.
Figure 2. Effects of ALLO pretreatment on the temporal distribution of 10E licks.
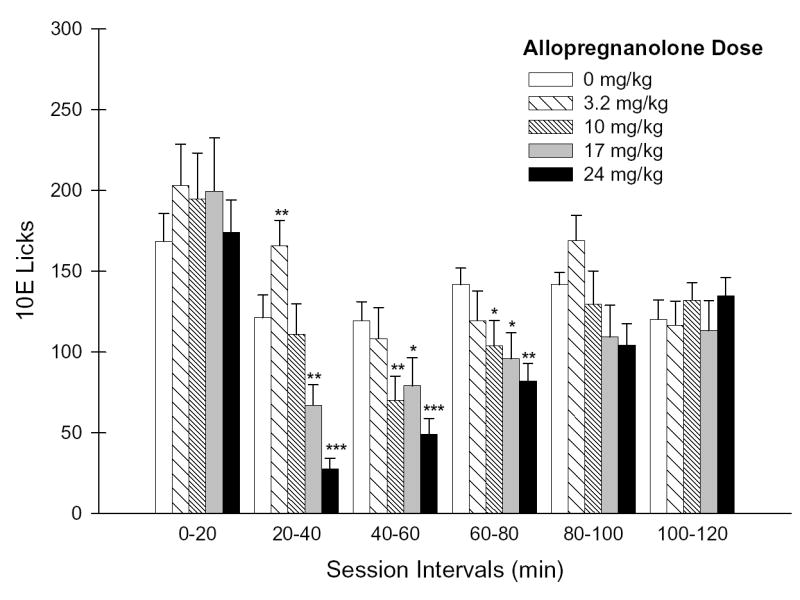
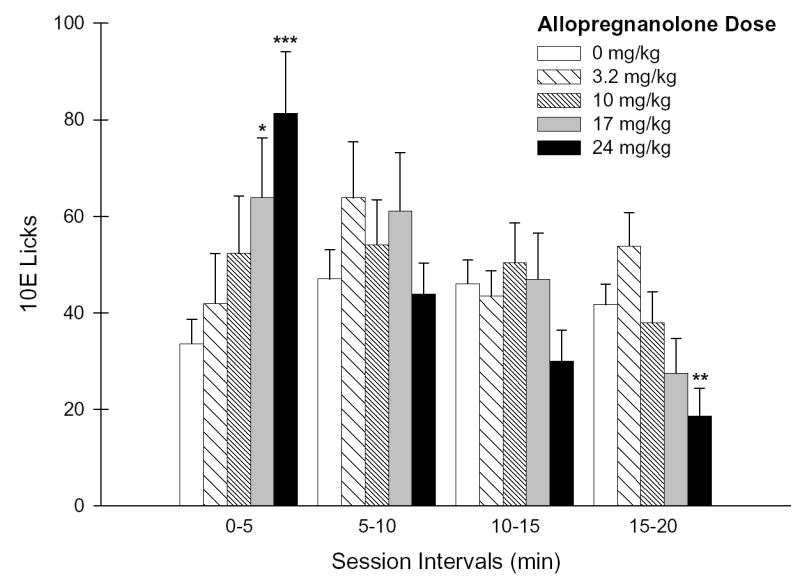
Panel A depicts the number of 10E licks that occurred during each 20-minute session interval throughout the 2-hr drinking session. Panel B illustrates the lick distribution during the initial 20 minutes of the session as 5-minute increments. Vertical bars in both panels represent the mean ± SEM of licks (n = 18 mice). *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001 versus within-subject baseline (0 mg/kg) of respective session interval.
Further examination of lick distributions throughout the initial 20-minutes as 5-minute increments revealed a significant ALLO dose × interval interaction [F(12,180) = 5.11; p < .001]. ALLO was found to dose-dependently increase ethanol licks only during the initial 5 minutes [F(4,66) = 5.25; p < .001; simple main effect of dose during this interval], with the 17 mg/kg (p < .05) and 24 mg/kg (p < .001) doses significantly augmenting 10E licks by 90% and 142%, respectively (Fig. 2B). This initial dose-dependent increase in 10E licks throughout the first 5 minutes contrasted the dose-dependent attenuation in 10E licks patterns that occurred during minutes 15–20 [F(4,66) = 5.80; p < .001; simple main effect of dose during this interval] with the 24 mg/kg dose significantly suppressing 10E licks by 55% (Fig. 2B).
Dose-Response Evaluation of Acute EPI Effects on Ethanol Intake Patterns
In contrast to ALLO, EPI failed to significantly influence any bout parameter examined, whether for the total session intake, total session bout patterns or for the first bout (Table 2). Neither an effect of EPI dose nor a dose × interval interaction were found for the temporal distribution of 10E licks at any time increment examined (Fig. 3A–B), a finding that was consistent with the overall absence of significant EPI effects on ethanol intake and bout microarchitecture.
Table 2. Acute EPI effects on ethanol intake patterns.
Values represent the mean ± SEM for EPI-treated mice (n = 19). Each mouse received all EPI doses, and the 0 mg/kg dose represents the average of baseline values (from sessions immediately preceding each EPI pretreatment day). One mouse was excluded as a statistical outlier (outside 2 standard deviations of the mean) from the analysis of water licks and preference following pretreatment with 10 mg/kg EPI.
| EPI Dose (mg/kg) | 0 | 0.15 | 1 | 3 | 10 |
|---|---|---|---|---|---|
| Total Session Intake | |||||
| Intake (g/kg) | 1.66 ± .10 | 1.70 ± .16 | 1.70 ± .11 | 1.76 ± .12 | 1.78 ± .16 |
| 10E Licks | 762 ± 51 | 795 ± 71 | 741 ± 53 | 824 ± 60 | 798 ± 70 |
| Water Licks | 97 ± 26 | 133 ± 36 | 86 ± 19 | 72 ± 17 | 147 ± 44 |
| Preference Ratio | .90 ± .02 | .86 ± .04 | .90 ± .02 | .93 ± .01 | .85 ± .04 |
| Total Session Patterns | |||||
| Bout Frequency | 12.8 ± 0.8 | 13.3 ± 1.3 | 13.2 ± 1.2 | 12.7 ± 0.9 | 14.3 ± 1.3 |
| Bout Size (g/kg) | .104 ± .005 | .140 ± .038 | .114 ± .009 | .122 ± .008 | .105 ± .006 |
| Lick Rate (licks/min) | 86 ± 18 | 100 ± 29 | 89 ± 26 | 80 ± 20 | 79 ± 25 |
| Bout Length (min) | 2.17 ± .27 | 2.59 ± .45 | 2.17 ± .37 | 2.81 ± .38 | 2.04 ± .28 |
| Inter-Bout Interval | 7.38 ± .61 | 6.08 ± .72 | 7.29 ± .85 | 5.82 ± .54 | 6.20 ± .79 |
| First Bout Dynamics | |||||
| Bout Size (g/kg) | .125 ± .015 | .136 ± .039 | .118 ± .012 | .171 ± .023 | .120 ± .014 |
| Lick Rate (licks/min) | 86 ± 24 | 115 ± 46 | 92 ± 35 | 45 ± 12 | 100 ± 37 |
| Latency to Start (min) | 10.23 ± 2.04 | 10.53 ± 3.42 | 14.02 ± 3.17 | 14.15 ± 5.59 | 11.61 ± 5.01 |
Figure 3. Effects of EPI pretreatment on the temporal distribution of 10E licks.
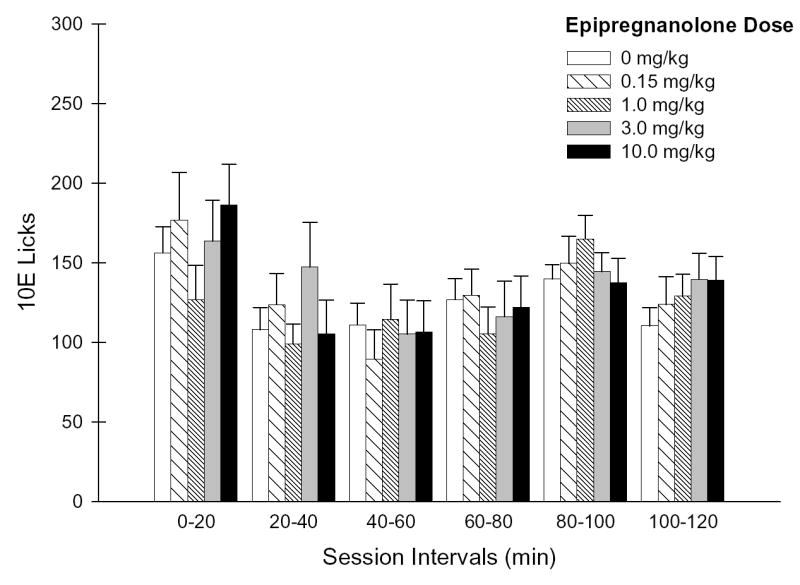
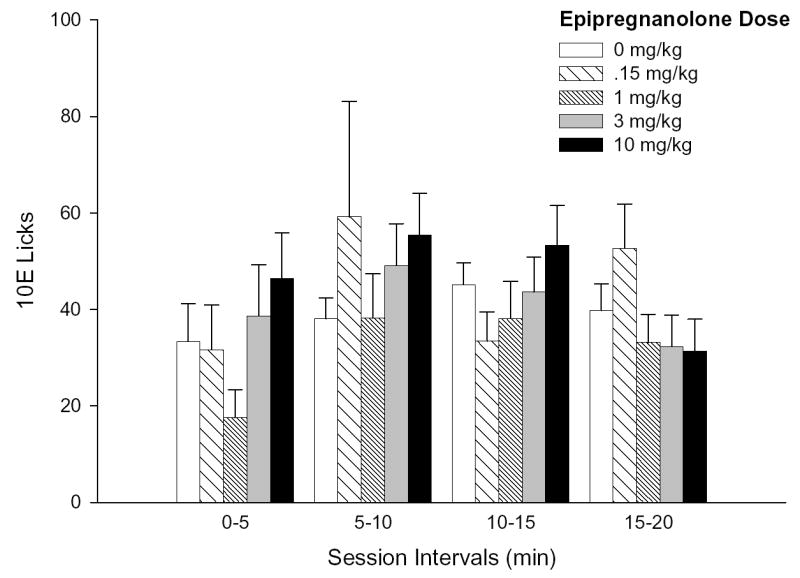
Panel A depicts the number of 10E licks that occurred during each 20-minute session interval throughout the 2-hr drinking session. Panel B illustrates the lick distribution during the initial 20 minutes of the session as 5-minute increments. Vertical bars in both panels represent the mean ± SEM of licks (n = 19 mice).
DISCUSSION
The present study validated the use of lickometers to assess the microarchitecture of ethanol drinking patterns and demonstrated that total session 10E licks and ethanol dose (g/kg) were significantly positively correlated. Thus, ethanol self-administration patterns in mice can be reliably assessed on a sub-second time scale by employing this microanalysis procedure on ethanol bout dynamics. Notably, ALLO and EPI exhibited divergent efficacies in the modulation of ethanol intake patterns in male B6 mice. The GABAA receptor positive modulator (agonist), ALLO, was found to dose-dependently modulate overall ethanol intake throughout the 2-hr session, a finding that corresponded with a dose-dependent decrease in bout frequency. ALLO also dose- and time-dependently altered the temporal distribution of ethanol licks, with a significant dose-dependent elevation occurring during the first 5 minutes, followed by a subsequent significant dose-dependent reduction in 10E licks across minutes 20–80 of the session. In contrast, EPI, a GABAA receptor partial agonist/antagonist, had no discernable effect on ethanol intake or the pattern of consumption. Collectively, the present observations suggest that the pharmacological profile of each neurosteroid, purportedly at GABAA receptors, may underlie their propensity to modulate ethanol self-administration patterns. These findings also implicate a potential interaction of ALLO with the regulatory processes underlying the control of ethanol intake.
Ethanol preference drinking procedures, in which ethanol solution and water are concurrently available, are commonly employed in the assessment of potential therapeutic drugs for the treatment of alcoholism and alcohol abuse (Johnson and Ait-Daoud, 2000). However, in both limited and continuous access paradigms, the total ethanol dose consumed is typically the only quantitative measure determined. In a recent study evaluating the implications of drinking patterns among multiple populations of human males and the commensurate rates of ethanol-related problems, it was reported that overall ethanol consumption alone was an insufficient indicator for the basis of developing preventive strategies (Bobak et al., 2005; Gmel et al., 2001). Perhaps more relevant to the current study, the efficacy of naltrexone administered to human alcoholics has been demonstrated to be contingent upon the drinking routine assigned to the subjects. Although an earlier study documented that naltrexone inhibited the progression (maintenance) of drinking in alcoholics when a 45-minute delay was imposed following the initial drink (O’Malley et al., 2002), a recent comparison of immediate versus delayed access to subsequent drinks revealed that naltrexone had no affect on drinking patterns in alcoholics unless the delay was imposed (Anton et al., 2004). This finding suggests that pharmacotherapeutic effectiveness may depend on the expression of a responsive drinking pattern or repertoire.
In an effort to improve current understanding of the regulatory processes underlying therapeutic-elicited changes in ethanol intake outcomes, a set of additional quantitative indices should be implemented. The microanalysis of ethanol self-administration patterns permits an evaluation of the regulatory processes underlying ethanol intake (Samson, 2000) via assessment of ethanol bout frequency (number of drinking episodes per session), bout size (quantity consumed per episode), lick rates, and other consumption pattern parameters. Furthermore, evaluation of consumption patterns can aid in determining whether ethanol pharmacology is a probable motivation for the self-administration observed (Cunningham et al., 2000). Alterations in ethanol bout parameters may be predictive of associative changes in endogenous states that govern the regulatory processes that control the onset, maintenance, or termination of drinking episodes (Samson and Hodge, 1996). For instance, one recent report demonstrated the occurrence of estrous cycle phase-related changes in ethanol consumption patterns exhibited by female rats (Ford et al., 2002). In the present study, two pregnane neurosteroids were assessed for their influence on the regulatory processes governing ethanol consumption.
ALLO pretreatment was found to elicit time- and dose-dependent effects on multiple measures of intake, bout microarchitecture and lick patterns throughout the 2-hr limited access session. One major finding was that similar ALLO dose-response profiles were determined for ethanol dose, total 10E licks, and bout frequency (refer to Table 1). These parallel profiles would suggest that an alteration in bout frequency was integral to ALLO’s modulatory influence on total session intake. ALLO notably altered inter-bout interval in a dose-dependent manner that was consistent with changes in bout frequency (Table 1). When interpreting these findings in conjunction with the dose-dependent reduction in 10E licks observed during session minutes 20–80, it could be concluded that ALLO enhanced (in the case of 3.2 mg/kg) or disrupted (in the case of 10–24 mg/kg) the maintenance of ethanol self-administration throughout the drinking session.
A second major finding was that ALLO pretreatment dose-dependently modulated the average ethanol bout size. The size of the first bout notably exhibited a similar dose-dependent profile in response to ALLO pretreatment, and likely comprised the observed significant change in average bout size following administration of 10 mg/kg ALLO (refer to Table 1). Curiously, the dose-dependent profiles stemming from ALLO’s modulation of bout frequency and bout size failed to overlap. A rightward shift in ALLO dose-response curve in regards to bout size occurred (a significant increase at 10 mg/kg followed by a dose-dependent reduction with the two highest doses) when compared to bout frequency (a trend towards an increase at 3.2 mg/kg dose followed by dose-dependent decline with the three highest doses). Although it is difficult to reconcile this disparity in responsiveness, one possible explanation is that bout frequency and bout size were each regulated by distinct GABAA receptor populations that exhibited differential sensitivity to ALLO modulation. Multiple mechanisms for differential neurosteroid sensitivity among brain regions have been proposed, and include GABAA receptor subunit composition (Follesa et al.2004; Lambert et al., 2003), state of localized neurosteroid metabolism (Belelli & Herd, 2003), and intracellular balance between endogenous phosphatase and kinase activities (Brussard & Koksma, 2003). The apparent differences in sensitivities in ALLO modulation of bout frequency and bout size may reflect one or a combination of these mechanisms.
The time-dependent aspect of ALLO’s modulatory effects on drinking patterns was assessed via the temporal distribution of 10E licks. One key finding from this analysis was the identification of a bimodal response to ALLO pretreatment: ALLO initially elicited a dose-dependent, fleeting elevation in 10E licks within the first 5 minutes of the session (Fig. 2B), and then subsequently led to a dose-dependent, sustained suppression in 10E licks for approximately 60–80 minutes (Fig. 2A). The early onset of ALLO’s effects was suggestive of a rapid distribution into the brain from the peripheral injection site. It is tempting to speculate that as ALLO accumulated centrally it exceeded a threshold concentration that consequently resulted in a suppression of 10E licks commensurate with the dose magnitude. Some earlier reports have suggested that nM concentrations of ALLO potentiated GABA-evoked chloride flux whereas μM concentrations of this neurosteroid directly enhanced chloride current through the GABAA receptor ion channel (Majewska, 1992; Puia et al., 1990). This threshold transition in functional activation of evoked chloride current is one possible explanation (albeit speculative) for the observed bimodal effects on ethanol intake and consumption patterns across the broad ALLO dose range examined. Within the boundaries of this interpretation, the 3.2 mg/kg ALLO pretreatment dose apparently produced brain ALLO levels that remained below this theoretical threshold (i.e., 34 ng/ml; Finn et al., 1997), hence supporting an increase in 10E licks during session minutes 20–40 when compared to VEH (Fig. 2A). Pretreatment with 17 and 24 mg/kg ALLO doses (presumably supra-physiologic) significantly decreased total ethanol licks in the absence of significant changes in water licks. However, these ALLO doses proportionally reduced both ethanol and water licks by 17–18% and 30–33%, respectively, versus VEH treatment (Table 1), suggesting that a generalized suppression of total fluid intake occurred. Thus, the propensity of ALLO to selectively alter ethanol intake might be limited to a dose that previously was demonstrated to produce physiologically relevant ALLO concentrations.
The present findings with GABAA receptor active neurosteroids complement an existing body of literature that portends GABAergic involvement in the regulatory processes governing control of ethanol drinking behavior and reward (Koob et al., 1998; Laviolette and van der Kooy, 2001; Samson and Hodge, 1996). Neurosteroids exhibiting GABAA receptor positive modulator properties have previously been shown to enhance ethanol self-administration and/or ethanol-reinforced responding (Janak et al., 1998; Janak and Gill, 2003; Sinnott et al., 2002), reinstate extinguished operant responding on an ethanol-paired lever (Nie and Janak, 2003), and substitute for ethanol’s discriminative stimulus properties (Engel et al., 2001; Grant et al., 1996; Hodge et al., 2001). In contrast, antagonist-like neurosteroids, albeit poorly characterized, typically fail to substitute for ethanol in drug discrimination paradigms (Bowen et al., 1999). In a previous report from our laboratory (Sinnott et al., 2002), a 10 mg/kg ALLO pretreatment dose administered to male B6 mice failed to significantly alter the total intake during a 2-hr session, but did shift the temporal distribution of intake (increased intake by 110% during the first hour and decreased 10E intake by 36% during the second hour) in a manner consistent with the current findings. It has also been demonstrated that a 3 mg/kg, but not a 10 mg/kg, pretreatment dose of ALLO significantly elevated both ethanol-reinforced lever responding and ethanol intake (g/kg) in male Long-Evans rats during a 30-minute operant session (Janak & Gill, 2003; Janak et al., 1998). Thus, the current findings regarding the regulatory influence of ALLO on ethanol self-administration patterns in male B6 mice were largely consistent with earlier reports of neurosteroid interactions with ethanol self-administration.
The differential effects of ALLO and EPI on the pattern of ethanol intake may reflect an alteration in GABAergic inhibitory tone within neurocircuitry fundamental to the regulatory processes underlying ethanol intake that is likely due to their pharmacological profile at GABAA receptors. Tentative conclusions can be drawn regarding the stereo-selectivity of pregnane neurosteroids at GABAA receptors and their influence on the regulatory processes underlying ethanol-drinking behavior. The current findings clearly indicated that ALLO (3-position hydroxyl group in the α orientation; reduced steroid A-ring in the 5α configuration) influenced multiple measures of ethanol intake and consumption patterns whereas EPI (3-position hydroxyl group in the β orientation; reduced steroid A-ring in the 5β configuration) had no effect on any ethanol consumption measure assessed. It has been well established that two crucial structure-activity requirements for neurosteroid stereo-specificity at GABAA receptors are 1) a hydroxyl group in the α orientation at the 3-position of the steroid A-ring and 2) a reduced steroid A-ring at the 5-position in either the α or β configuration (Belelli et al., 1990). Pregnane steroids that adhere to these structure-activity criteria (i.e., ALLO) potently displace [35S]-TBPS from the steroid binding site on the GABAA receptor complex (Belelli et al., 1990) and potentiate GABA-mediated chloride current (Kokate et al., 1994). Neurosteroids not meeting these criteria (i.e., EPI) fail to efficaciously modulate GABAA receptor activity. Furthermore, the efficacy of ALLO and other structurally-related agonists at GABAA receptors were highly correlated with their respective sedative, hypnotic, anesthetic, anxiolytic, and anticonvulsant pharmacological profiles (Rupprecht & Holsboer, 2001). Thus, the current results with ALLO and EPI are consistent with a GABAA receptor-mediated mechanism for neurosteroid modulation of ethanol consumption patterns.
Since sedation and incoordination are common side effects of GABAergic drugs (Johnson et al., 2005), it could be argued that the significant reduction in total ethanol intake and 10E licks associated with ALLO pretreatment was attributable to a suppression of locomotor behavior, especially since a non-significant decline in total session water licks simultaneously occurred (Table 1). However, this interpretation is inconsistent with the 10E lick rates and bout lengths determined in the current study and with a previous study that assessed locomotor activity following an identical ALLO pretreatment dose. Ethanol lick rates and bout lengths were unaltered by any ALLO dose evaluated (Table 1), suggesting that once a bout was initiated, it progressed similarly over time regardless of neurosteroid pretreatment. Furthermore, it has been previously demonstrated that 10 and 17 mg/kg ALLO doses stimulated locomotor activity in male B6 mice, with onset occurring between 5–10 minutes and remaining elevated for more than 30 minutes (Palmer et al., 2002). Notably, the ALLO-elicited modulation in 10E licks observed in the current study (Fig 2A–B; dose-dependent increase in licks during the first 5 minutes followed by dose-dependent decrease beginning at 20 minutes) is discordant with the reported time course of locomotor stimulation. Thus, any ALLO-induced alteration of locomotor activity does not explain ALLO’s dose-dependent reduction of ethanol intake.
Counterbalanced treatment cohorts were run to account for the order of exposure to ALLO and EPI. The practice of carefully re-establishing ethanol intake baselines prior to each acute neurosteroid pretreatment additionally was implemented to minimize potential carry-over effects between exogenous applications. As stated above in the methods, the two treatment cohorts were balanced for ethanol intake and other drinking pattern parameters prior to the initial exposure to neurosteroid. Prior to collapsing ethanol intake baseline measures across cohorts as shown in Tables 1–2, one cohort (ALLO dose series, then EPI series) exhibited baseline intakes of 1.55 ± 0.18 and 1.58 ± 0.14 g/kg during ALLO and EPI pretreatments, respectively, whereas the second cohort (EPI dose series, then ALLO series) had baseline intakes of 1.72 ± 0.16 and 1.80 ± 0.15 g/kg during EPI and ALLO pretreatments, respectively. If a neurosteroid had exerted long-lasting (carry-over) effects on ethanol intake then it would have been expected to impact the intake baseline of the subsequently tested neurosteroid. However, carry-over effects of this nature were not observed for any total session intake variable, total session pattern variable, first bout variable or baseline lick distribution profile evaluated in this study. It also should be noted that the effects of ALLO pretreatments on the temporal distribution of licks indicated that the neurosteroid’s influence (at all doses examined) had dissipated by 100 minutes into the limited access session (refer to Fig. 2A). Thus, we have determined that acute neurosteroid pretreatments appropriately spaced over a several week period did not noticeably confound the effects of subsequent evaluations.
In summary, the present findings demonstrate that acute pretreatment with ALLO dose-dependently modulates ethanol intake and consumption patterns. Preliminary clinical evaluations of neurosteroid concentrations in patients afflicted with premenstrual syndrome, major depression, seizure disorders, and alcoholism have indicated an inverse relationship between neurosteroids and symptoms of these disease states, since restoration of neurosteroids to normal levels alleviates adverse symptomology (Griffin et al., 2001). Taken in conjunction with the recent findings that ethanol self-administration led to significantly elevated plasma ALLO levels in male and female adolescents (Torres and Ortega, 2003; Torres and Ortega, 2004) and brain ALLO concentrations in male mice (Finn et al., 2004), it is tempting to speculate that a treatment strategy that involves manipulation of endogenous levels of ALLO and related agonist neurosteroids could yield beneficial outcomes in the management of ethanol’s abuse-related effects. The purported involvement of GABAergic neurotransmission in the development of ethanol dependence and withdrawal warrants extensive investigation into GABA-active compounds (Johnson et al., 2005). Microanalysis of ethanol bout dynamics may serve as a useful tool to assess the influence of pregnane neurosteroids and other GABAergic drugs on the regulatory processes underlying ethanol consumption behavior, and furthermore, address the issue of loss of intake control.
Acknowledgments
We would like to thank Naomi Yoneyama and Stephen T. Hansen for their technical assistance.
Footnotes
Supported by grants AA015234, AA10760, DA07262, the Department of Veterans Affairs and the N.L. Tartar Research Fund
References
- Anton RF, Drobes DJ, Voronin K, Durazo-Avizu R, Moak D. Naltrexone effects on alcohol consumption in a clinical laboratory paradigm: temporal effects of drinking. Psychopharmacology (Berl) 2004;173:32–40. doi: 10.1007/s00213-003-1720-7. [DOI] [PubMed] [Google Scholar]
- Barbaccia ML, Affricano D, Trabucchi M, Purdy RH, Colombo G, Agabio R, Gessa GL. Ethanol markedly increases “GABAergic” neurosteroids in alcohol-preferring rats. Eur J Pharmacol. 1999;384:R1–R2. doi: 10.1016/s0014-2999(99)00678-0. [DOI] [PubMed] [Google Scholar]
- Barbaccia ML, Serra M, Purdy RH, Biggio G. Stress and neuroactive steroids. Int Rev Neurobiol. 2001;46:243–272. doi: 10.1016/s0074-7742(01)46065-x. [DOI] [PubMed] [Google Scholar]
- Barbosa AD, Morato GS. Effect of epipregnanolone and pregnenolone sulfate on chronic tolerance to ethanol. Pharmacol Biochem Behav. 2000;67:459–464. doi: 10.1016/s0091-3057(00)00372-5. [DOI] [PubMed] [Google Scholar]
- Belelli D, Herd MB. The contraceptive agent Provera enhances GABAA receptor-mediated inhibitory neurotransmission in the rat hippocampus: evidence for endogenous neurosteroids? J Neurosci. 2003;23:10013–10020. doi: 10.1523/JNEUROSCI.23-31-10013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Lan NC, Gee KW. Anticonvulsant steroids and the GABA/benzodiazepine receptor-chloride ionophore complex. Neurosci Biobehav Rev. 1990;14:315–322. doi: 10.1016/s0149-7634(05)80041-7. [DOI] [PubMed] [Google Scholar]
- Biggio G, Follesa P, Sanna E, Purdy RH, Concas A. GABAA-receptor plasticity during long-term exposure to and withdrawal from progesterone. Int Rev Neurobiol. 2001;46:207–241. doi: 10.1016/s0074-7742(01)46064-8. [DOI] [PubMed] [Google Scholar]
- Bobak M, Room R, Pikhart H, Kubinova R, Malyutina S, Pajak A, Kurilovitch S, Topor R, Nikitin Y, Marmot M. Contribution of drinking patterns to differences in rates of alcohol related problems between three urban populations. J Epidemiol Community Health. 2004;58:238–242. doi: 10.1136/jech.2003.011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen CA, Purdy RH, Grant KA. Ethanol-like discriminative stimulus effects of endogenous neuroactive steroids: Effect of ethanol training dose and dosing procedure. J Pharmacol Exp Ther. 1999;289:405–411. [PubMed] [Google Scholar]
- Boyle AE, Smith BR, Amit Z. A descriptive analysis of the structure and temporal pattern of voluntary ethanol intake within an acquisition paradigm. J Stud Alcohol. 1997;58:382–391. doi: 10.15288/jsa.1997.58.382. [DOI] [PubMed] [Google Scholar]
- Brussaard AB, Koksma JJ. Conditional regulation of neurosteroid sensitivity of GABAA receptors. Ann N Y Acad Sci. 2003;1007:29–36. doi: 10.1196/annals.1286.003. [DOI] [PubMed] [Google Scholar]
- Concas A, Mostallino MC, Porcu P, Follesa P, Barbaccia ML, Trabucchi M, Purdy RH, Grisenti P, Biggio G. Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc Natl Acad Sci U S A. 1998;95:13284–13289. doi: 10.1073/pnas.95.22.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpechot C, Collins BE, Carey MP, Tsouros A, Robel P, Fry JP. Brain neurosteroids during the mouse oestrous cycle. Brain Res. 1997;766:276–280. doi: 10.1016/s0006-8993(97)00749-x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Morrow AL, Criswell H, Breese G. Effects of ethanol on ion channels. Int Rev Neurobiol. 1996;39:283–367. doi: 10.1016/s0074-7742(08)60670-4. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Fidler TL, Hill KG. Animal models of alcohol’s motivational effects. Alcohol Res Health. 2000;24:85–92. [PMC free article] [PubMed] [Google Scholar]
- Czachowski CL, Chappell AM, Samson HH. Effects of raclopride in the nucleus accumbens on ethanol seeking and consumption. Alcohol Clin Exp Res. 2001a;25:1431–1440. doi: 10.1097/00000374-200110000-00005. [DOI] [PubMed] [Google Scholar]
- Czachowski CL, Legg BH, Samson HH. Effects of acamprosate on ethanol-seeking and self-administration in the rat. Alcohol Clin Exp Res. 2001b;25:344–350. [PubMed] [Google Scholar]
- Engel SR, Purdy RH, Grant KA. Characterization of discriminative stimulus effects of the neuroactive steroid pregnanolone. J Pharmacol Exp Ther. 2001;297:489–495. [PubMed] [Google Scholar]
- Feunekes GI, van ‘t Veer P, van Staveren WA, Kok FJ. Alcohol intake assessment: the sober facts. Am J Epidemiol. 1999;150:105–112. doi: 10.1093/oxfordjournals.aje.a009909. [DOI] [PubMed] [Google Scholar]
- Finn DA, Roberts AJ, Lotrich F, Gallaher EJ. Genetic differences in behavioral sensitivity to a neuroactive steroid. J Pharmacol Exp Ther. 1997;280:820–828. [PubMed] [Google Scholar]
- Finn DA, Sinnott RS, Ford MM, Long SL, Tanchuck MA, Phillips TJ. Sex differences in the effect of ethanol injection and consumption on brain allopregnanolone levels in C57BL/6 mice. Neuroscience. 2004;123:813–819. doi: 10.1016/j.neuroscience.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Follesa P, Biggio F, Caria S, Gorini G, Biggio G. Modulation of GABAA receptor gene expression by allopregnanolone and ethanol. Eur J Pharmacol. 2004;500:413–425. doi: 10.1016/j.ejphar.2004.07.041. [DOI] [PubMed] [Google Scholar]
- Ford MM, Eldridge JC, Samson HH. Microanalysis of ethanol self-administration: estrous cycle phase-related changes in consumption patterns. Alcohol Clin Exp Res. 2002;26:635–643. [PubMed] [Google Scholar]
- Gasior M, Carter RB, Witkin JM. Neuroactive steroids: potential therapeutic use in neurological and psychiatric disorders. Trends Pharmacol Sci. 1999;20:107–112. doi: 10.1016/s0165-6147(99)01318-8. [DOI] [PubMed] [Google Scholar]
- Gee KW, Bolger MB, Brinton RE, Coirini H, McEwen BS. Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J Pharmacol Exp Ther. 1988;246:803–812. [PubMed] [Google Scholar]
- Genazzani AD, Luisi M, Malavasi B, Strucchi C, Luisi S, Casarosa E, Bernardi F, Genazzani AR, Petraglia F. Pulsatile secretory characteristics of allopregnanolone, a neuroactive steroid, during the menstrual cycle and in amenorrheic subjects. Eur J Endocrinol. 2002;146:347–356. doi: 10.1530/eje.0.1460347. [DOI] [PubMed] [Google Scholar]
- Gmel G, Klingemann S, Muller R, Brenner D. Revising the preventive paradox: the Swiss case. Addiction. 2001;96:273–284. doi: 10.1046/j.1360-0443.2001.96227311.x. [DOI] [PubMed] [Google Scholar]
- Goldstein DB, Kakihana R. Circadian rhythms of ethanol consumption by mice: a simple computer analysis for chronopharmacology. Psychopharmacology (Berl) 1977;52:41–45. doi: 10.1007/BF00426598. [DOI] [PubMed] [Google Scholar]
- Grant KA, Azarov A, Bowen CA, Mirkis S, Purdy RH. Ethanol-like discriminative stimulus effects of the neurosteroid 3α-hydroxy-5α-pregnan-20-one in female Macaca fascicularis monkeys. Psychopharmacology (Berl) 1996;124:340–346. doi: 10.1007/BF02247439. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Conrad SC, Mellon SH. Current perspectives on the role of neurosteroids in PMS and depression. Int Rev Neurobiol. 2001;46:479–492. doi: 10.1016/s0074-7742(01)46073-9. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Matthews DB, Devaud LL, Morrow AL. The role of GABAA receptors in the acute and chronic effects of ethanol. Psychopharmacology (Berl) 1998;139:2–19. doi: 10.1007/s002130050685. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Physiological roles for the neurosteroid allopregnanolone in the modulation of brain function during pregnancy and parturition. Prog Brain Res. 2001;133:39–47. doi: 10.1016/s0079-6123(01)33003-0. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Nannini MA, Olive MF, Kelley SP, Mehmert KK. Allopregnanolone and pentobarbital infused into the nucleus accumbens substitute for the discriminative stimulus effects of ethanol. Alcohol Clin Exp Res. 2001;25:1441–1447. doi: 10.1097/00000374-200110000-00006. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. Guide for the Care and Use of Laboratory Animals. National Academy Press; Washington, DC: 1996. [Google Scholar]
- Janak PH, Redfern JE, Samson HH. The reinforcing effects of ethanol are altered by the endogenous neurosteroid, allopregnanolone. Alcohol Clin Exp Res. 1998;22:1106–1112. [PubMed] [Google Scholar]
- Janak PH, Gill TM. Comparison of the effects of allopregnanolone with direct GABAergic agonists on ethanol self-administration with and without concurrently available sucrose. Alcohol. 2003;30:1–7. doi: 10.1016/s0741-8329(03)00068-5. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Ait-Daoud N. Neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Psychopharmacology (Berl) 2000;149:327–344. doi: 10.1007/s002130000371. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Swift RM, Addolorato G, Ciraulo DA, Myrick H. Safety and efficacy of GABAergic medications for treating alcoholism. Alcohol Clin Exp Res. 2005;29:248–254. doi: 10.1097/01.alc.0000153542.10188.b0. [DOI] [PubMed] [Google Scholar]
- Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with gamma-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 1994;270:1223–1229. [PubMed] [Google Scholar]
- Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P, Merlo-Pich E, Weiss F. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, HillVenning C, Peters JA. Neurosteroids and GABAA receptor function. Trends Pharmacol Sci. 1995;16:295–303. doi: 10.1016/s0165-6147(00)89058-6. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA. Neurosteroid modulation of GABAA receptors. Prog Neurobiol. 2003;71:67–80. doi: 10.1016/j.pneurobio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, van der Kooy D. GABAA receptors in the ventral tegmental area control bidirectional reward signalling between dopaminergic and non-dopaminergic neural motivational systems. Eur J Neurosci. 2001;13:1009–1015. doi: 10.1046/j.1460-9568.2001.01458.x. [DOI] [PubMed] [Google Scholar]
- Majewska MD. Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol. 1992;38:379–395. doi: 10.1016/0301-0082(92)90025-a. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Kelley BM, Bandy AL, McGroarty KK. Ethanol consumption by C57BL/6 mice: influence of gender and procedural variables. Alcohol. 1999a;17:175–183. doi: 10.1016/s0741-8329(98)00055-x. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Kelley BM, Cuison ER, Jr, Groseclose CH. Naltrexone effects on ethanol reward and discrimination in C57BL/6 mice. Alcohol Clin Exp Res. 1999b;23:456–464. [PubMed] [Google Scholar]
- Middaugh LD, Lee AM, Bandy AL. Ethanol reinforcement in nondeprived mice: effects of abstinence and naltrexone. Alcohol Clin Exp Res. 2000;24:1172–1179. [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Millard WJ, Dole VP. Intake of water and ethanol by C57BL mice: effect of an altered light-dark schedule. Pharmacol Biochem Behav. 1983;18:281–284. doi: 10.1016/0091-3057(83)90377-5. [DOI] [PubMed] [Google Scholar]
- Morrow AL, Suzdak PD, Paul SM. Steroid hormone metabolites potentiate GABA receptor-mediated chloride ion flux with nanomolar potency. Eur J Pharmacol. 1987;142:483–485. doi: 10.1016/0014-2999(87)90094-x. [DOI] [PubMed] [Google Scholar]
- Morrow AL, VanDoren MJ, Penland SN, Matthews DB. The role of GABAergic neuroactive steroids in ethanol action, tolerance and dependence. Brain Res Brain Res Rev. 2001;37:98–109. doi: 10.1016/s0165-0173(01)00127-8. [DOI] [PubMed] [Google Scholar]
- Nie H, Janak PH. Comparison of reinstatement of ethanol- and sucrose-seeking by conditioned stimuli and priming injections of allopregnanolone after extinction in rats. Psychopharmacology (Berl) 2003;168:222–228. doi: 10.1007/s00213-003-1468-0. [DOI] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKC epsilon-deficient mice. Eur J Neurosci. 2000;12:4131–4140. doi: 10.1046/j.1460-9568.2000.00297.x. [DOI] [PubMed] [Google Scholar]
- O’Malley SS, Krishnan-Sarin S, Farren C, Sinha R, Kreek MJ. Naltrexone decreases craving and alcohol self-administration in alcohol-dependent subjects and activates the hypothalamo-pituitary-adrenocortical axis. Psychopharmacology (Berl) 2002;160:19–29. doi: 10.1007/s002130100919. [DOI] [PubMed] [Google Scholar]
- Palmer AA, Moyer MR, Crabbe JC, Phillips TJ. Initial sensitivity, tolerance and cross-tolerance to allopregnanolone- and ethanol-induced hypothermia in selected mouse lines. Psychopharmacology (Berl) 2002;162:313–322. doi: 10.1007/s00213-002-1106-2. [DOI] [PubMed] [Google Scholar]
- Palumbo MA, Salvestroni C, Gallo R, Guo AL, Genazzani AD, Artini PG, Petraglia F, Genazzani AR. Allopregnanolone concentration in hippocampus of prepubertal rats and female rats throughout estrous cycle. J Endocrinol Invest. 1995;18:853–856. doi: 10.1007/BF03349832. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- Prince RJ, Simmonds MA. 5β-pregnan-3β-ol-20-one, a specific antagonist at the neurosteroid site of the GABAA receptor-complex. Neurosci Lett. 1992;135:273–275. doi: 10.1016/0304-3940(92)90454-f. [DOI] [PubMed] [Google Scholar]
- Puia G, Santi MR, Vicini S, Pritchett DB, Purdy RH, Paul SM, Seeburg PH, Costa E. Neurosteroids act on recombinant human GABAA receptors. Neuron. 1990;4:759–765. doi: 10.1016/0896-6273(90)90202-q. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Freeman PA, Rubinstein M, Low MJ, Grandy DK. Lack of operant ethanol self-administration in dopamine D2 receptor knockout mice. Psychopharmacology (Berl) 2000;152:343–350. doi: 10.1007/s002130000548. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Holsboer F. Neuroactive steroids in neuropsychopharmacology. Int Rev Neurobiol. 2001;46:461–477. doi: 10.1016/s0074-7742(01)46072-7. [DOI] [PubMed] [Google Scholar]
- Samson HH. The microstructure of ethanol drinking: genetic and behavioral factors in the control of drinking patterns. Addiction. 2000;95(Suppl 2):S61–S72. doi: 10.1080/09652140050111654. [DOI] [PubMed] [Google Scholar]
- Samson HH, Chappell A. Dopaminergic involvement in medial prefrontal cortex and core of the nucleus accumbens in the regulation of ethanol self-administration: a dual-site microinjection study in the rat. Physiol Behav. 2003;79:581–590. doi: 10.1016/s0031-9384(03)00126-4. [DOI] [PubMed] [Google Scholar]
- Deitrich RA, editor. Neurobehavioral regulation of ethanol intake, in Pharmacological Effects of Ethanol on the Nervous System. CRC Press; New York: 1996. pp. 203–226. [Google Scholar]
- Sinnott RS, Phillips TJ, Finn DA. Alteration of voluntary ethanol and saccharin consumption by the neurosteroid allopregnanolone in mice. Psychopharmacology (Berl) 2002;162:438–447. doi: 10.1007/s00213-002-1123-1. [DOI] [PubMed] [Google Scholar]
- Smith BR, Boyle AE, Amit Z. The effects of the GABAB agonist baclofen on the temporal and structural characteristics of ethanol intake. Alcohol. 1999;17:231–240. doi: 10.1016/s0741-8329(98)00053-6. [DOI] [PubMed] [Google Scholar]
- Tenth Special Report to the U.S. Congress on Alcohol and Health. Bethesda, MD: National Institute on Alcohol Abuse and Alcoholism; 2000. [Google Scholar]
- Torres JM, Ortega E. Alcohol intoxication increases allopregnanolone levels in female adolescent humans. Neuropsychopharmacology. 2003;28:1207–1209. doi: 10.1038/sj.npp.1300170. [DOI] [PubMed] [Google Scholar]
- Torres JM, Ortega E. Alcohol intoxication increases allopregnanolone levels in male adolescent humans. Psychopharmacology (Berl) 2004;172:352–355. doi: 10.1007/s00213-003-1662-0. [DOI] [PubMed] [Google Scholar]