

Summary
Yeast prions are protein-based genetic elements that self-perpetuate changes in protein conformation and function. A protein-remodeling factor, Hsp104, controls the inheritance of several yeast prions, including those formed by Sup35 and Ure2. Perplexingly, deletion of Hsp104 eliminates Sup35 and Ure2 prions, whereas overexpression of Hsp104 purges cells of Sup35 prions, but not Ure2 prions. Here, we used pure components to dissect how Hsp104 regulates prion formation, growth, and division. For both Sup35 and Ure2, Hsp104 catalyzes de novo prion nucleation from soluble, native protein. Using a distinct mechanism, Hsp104 fragments both prions to generate new prion assembly surfaces. For Sup35, the fragmentation endpoint is an ensemble of non-infectious, amyloid-like aggregates and soluble protein that cannot replicate conformation. In vivid distinction, the endpoint of Ure2 fragmentation is short prion fibers with enhanced infectivity and self-replicating ability. These advances explain the distinct effects of Hsp104 on the inheritance of the two prions.
Introduction
The breadth of biological phenomena attributable to prion proteins has greatly expanded and now encompasses self-replicating agents of infectivity, inheritance, and potentially long-term memory formation (Shorter and Lindquist, 2005). Prions switch between profoundly different structural and functional conformations. Critically, at least one of these conformations self-replicates by templating the conversion of other conformers of the same protein to the self-replicating form. Prions transmit spongiform encephalopathies in mammals (Shorter and Lindquist, 2005). However, prions also function as protein-based genetic switches. The propagation of functionally distinct conformers produces distinct, often advantageous, heritable phenotypes (True and Lindquist, 2000; True et al., 2004). Such self-replicating states may function to store and transmit information, including long-term memory in neurons (Si et al., 2003; Shorter and Lindquist, 2005).
Different prions have unrelated sequences and functions, but all form self-propagating, amyloid fibers under physiological conditions (Shorter and Lindquist, 2005). These fibers are akin to those connected with several devastating neurodegenerative disorders (Muchowski and Wacker, 2005). However, other amyloids function as specialized cellular nanostructures (Wickner et al., 2004; Shorter and Lindquist, 2005). The conformational transitions of prions and amyloids are profoundly modulated by molecular chaperones and protein-remodeling factors (Tuite and Cox, 2003; Muchowski and Wacker, 2005). However, despite intense scrutiny from many cutting-edge laboratories worldwide, there is still not yet a single example where the mechanistic interplay between chaperones/protein-remodeling factors and amyloid/prion biochemistry and biology is well understood. Here, we address these questions for two prions of Saccharomyces cerevisiae: [PSI+], whose protein determinant is Sup35, and [URE3], whose protein determinant is Ure2. Following standard nomenclature, italicized capital letters denote dominant genetic elements, and brackets indicate a non-Mendelian mode of inheritance. Both properties stem from the ability of prions to store and transmit biological information via alternative, self-perpetuating structures and functions.
The genetic trait conferred by [PSI+] is due to the conversion of a translation termination factor, Sup35, to a nonfunctional prion state (Tuite and Cox, 2003). [PSI+] causes ribosomes to read-through stop codons. This unleashes hidden genetic variation that confers phenotypic diversity and selective advantages in diverse settings (True and Lindquist, 2000; True et al., 2004). The ability of Sup35 to form [PSI+] has been conserved for hundreds of million years (Nakayashiki et al., 2001), and is almost certainly due to the advantageous phenotypic plasticity and evolvability it imparts (Shorter and Lindquist, 2005; but see Nakayashiki et al., 2005).
Sup35 has three functionally distinct domains. The C-terminal GTPase domain (C) (amino acids 254–685) confers translation termination activity (Tuite and Cox, 2003). The N-terminal, glutamine/asparagine-rich domain (N, amino acids 1–123) drives the switch from the soluble, functional [psi−] state to the insoluble [PSI+] prion state (Paushkin et al., 1996; Patino et al., 1996). The highly charged middle domain (M, amino acids 124–253) promotes solubility in the [psi−] state and ensures [PSI+] stability through cell division (Liu et al., 2002).
[URE3] alters the way yeast utilize nitrogen sources (Wickner et al., 2004). Its protein determinant, Ure2, negatively regulates a suite of proteins that import and catabolize secondary nitrogen sources. When Ure2 converts to its dysfunctional prion form, yeast cells use poor nitrogen sources even when rich ones are available (Wickner et al., 2004). Ure2 has a C-terminal functional domain (amino acids 81–354) and an N-terminal asparagine-rich domain (amino acids 1–80) that is necessary and sufficient for prion formation and propagation (Wickner et al., 2004). [URE3] is not as well conserved as [PSI+], but may sometimes be advantageous (Talarek et al., 2005; Shorter and Lindquist, 2005; but see Nakayashiki et al., 2005).
Sup35 and Ure2 form prions once their N-terminal domains switch from soluble, largely unstructured states to β-sheet-rich amyloid forms that rapidly convert soluble protein to the same state (Glover et al., 1997; Taylor et al., 1999). In vitro, the N and M domains of Sup35 (NM) form amyloid fibers after a lag phase. Fibers elongate from both ends, catalyzing the conformational conversion of soluble protein (Serio et al., 2000; Krishnan and Lindquist, 2005). [psi−] cells can be transformed to the [PSI+] state by NM fibers (Tanaka et al., 2004). Thus, fibers harbor prion infectivity. Similarly, Ure2 fibers assemble after a lag phase, seed the assembly of soluble Ure2, and transform [ure-o] cells to the [URE3] state (Taylor et al., 1999; Fay et al., 2003; Zhu et al., 2003; Brachmann et al., 2005).
[PSI+] and [URE3] are faithfully inherited, with a frequency of loss of ~10−5−10−7 (Tuite and Cox, 2003). Their formation and inheritance depends absolutely on Hsp104, a protein-remodeling factor (Chernoff et al., 1995; Moriyama et al., 2000). Hsp104 rescues cells from environmental stress by wresting denatured, aggregated proteins apart and reactivating them (Parsell et al., 1994b; Glover and Lindquist, 1998). Hsp104 is an AAA+ (ATPases Associated with diverse Activities) protein (Hanson and Whiteheart, 2005) that hexamerizes in the presence of ADP or ATP (Parsell et al., 1994a). Each protomer has two AAA+ modules (nucleotide binding domains, NBD1 and NBD2) separated by a coiled-coil middle domain, and flanked by N- and C-terminal domains. During substrate remodeling, the middle domain of Hsp104 undergoes cycles of conformational change driven by the cooperative ATPase activities of both NBDs (Hattendorf and Lindquist, 2002; Cashikar et al., 2002).
Deletion of Hsp104 eliminates [PSI+] and [URE3] (Chernoff et al., 1995; Moriyama et al., 2000), as do specific point mutations in the NBDs of Hsp104, and growth in the presence of 5mM guanidium chloride (GdmCl), an uncompetitive inhibitor of Hsp104 ATPase activity (Patino et al., 1996; Wegrzyn et al., 2001; Hattendorf and Lindquist, 2002; Ripaud et al., 2003; Grimminger et al., 2004). Strikingly, however, overexpression of Hsp104 eliminates [PSI+], but not [URE3] (Chernoff et al., 1995; Moriyama et al., 2000). The molecular basis of this difference remains a mystery.
Hsp104 has different effects on the conformation of NM, depending on the relative concentrations of Hsp104 and NM, the conformational state of NM, and the nature of the available nucleotides (Shorter and Lindquist, 2004). These activities seem to explain how both deletion and overexpression of Hsp104 cures [PSI+] (Chernoff et al., 1995; Paushkin et al., 1996; Patino et al., 1996), but contradictions have arisen (Inoue et al., 2004; Krzewska and Melki, 2006). To resolve them, it is imperative to investigate the effects of Hsp104 on full-length, wild-type Sup35 purified under native conditions, because this is the actual basis of the prion [PSI+]. NM is much more aggregation prone than full-length Sup35 in vivo (Patino et al., 1996; Glover et al., 1997) and earlier analyses only employed previously denatured, N-terminal fragments of Sup35 or proteins with artificial tags (Glover et al., 1997; Serio et al., 2000; King and Diaz-Avalos, 2004; Tanaka et al., 2004; Shorter and Lindquist, 2004; Inoue et al., 2004; Krzewska and Melki, 2006). How Hsp104 affects Ure2 has not been addressed at the biochemical level at all.
Here, we analyze the effects of Hsp104 on both the soluble and prion forms of full-length, untagged, natively structured Sup35 and Ure2. Using diverse conditions and a panel of Hsp104 mutants, we find extraordinary congruencies in the ways that Hsp104 remodels the conformations of these two unrelated proteins, including the mechanisms by which: (a) Hsp104 promotes prion assembly, (b) high Hsp104 concentrations restrict access to the prion state, and (c) Hsp104 deconstructs prions. Despite these remarkable and unexpected similarities in how Hsp104 regulates these different prions, we also elucidate a critical singularity that explains why Hsp104 overexpression cures [PSI+] but not [URE3].
Results
Spontaneous assembly of Sup35 and Ure2 fibers
First, we examined whether full-length Sup35 can convert from the soluble to the fibrous state. The C-terminal domain of Sup35, which contains four GTP-binding modules, made the protein prone to amorphous non-prion aggregation (not shown). To reduce this problem the protein was purified under native conditions in the constant presence of GTP, and GTP was retained in all assembly reactions. Sup35 was analyzed immediately after purification, as freezing and thawing caused denaturation. Further, we eliminated proteolytic fragments harboring the prion domain, which influenced assembly kinetics (not shown). Fibrillization of native Sup35 was analyzed by SDS-solubility, Thioflavin-T (ThT) fluorescence, and electron microscopy (EM). All three assays gave very similar results throughout.
Newly prepared Sup35 formed fibers spontaneously after a ~ 2h lag phase, TL (time elapsing before the first detection of amyloid; Fig. 1A, B, Table S1). TL was much longer for Sup35 than for the isolated NM prion domain purified under native conditions (Scheibel and Lindquist, 2001) (Fig. 1A, TL ~0.75h). The assembly phase was also much longer (Fig. 1A, B). AT1/2, the time that elapsed between the first appearance of amyloid and the halfway point of complete assembly, was ~0.5h for NM, but ~2.5h for Sup35. Thus, the C-terminal domain of Sup35 impedes the acquisition of a nucleating conformation and reduces the rate of conformational replication.
Figure 1. Hsp104 catalyzes or abolishes the de novo assembly of Sup35 and Ure2 fibers.
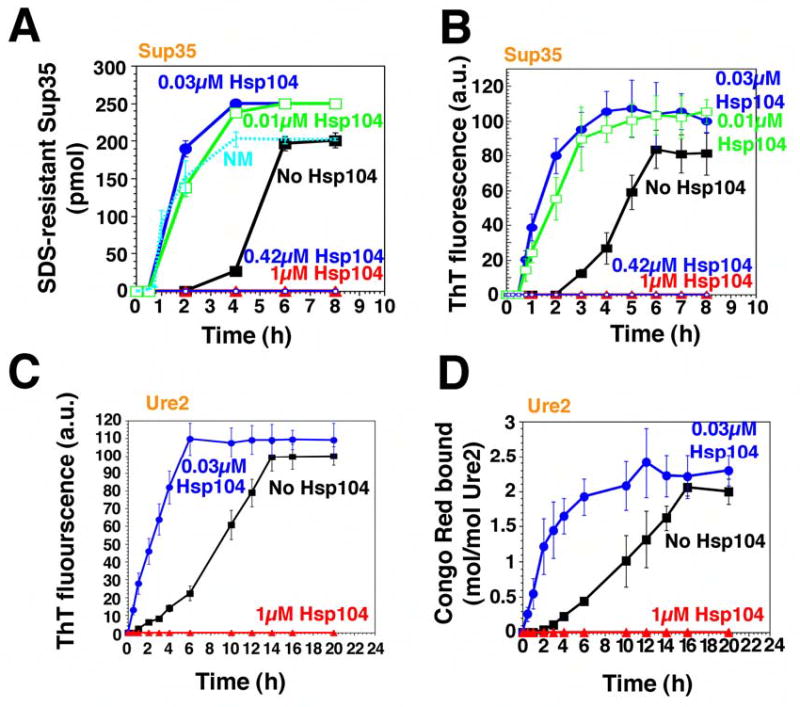
(A, B) Kinetics of unseeded, rotated (80rpm) Sup35 (2.5μM) polymerization without or with Hsp104 (0.01–1μM) plus ATP (5mM). Fiber assembly was assessed by the formation of SDS-insoluble Sup35 (A) and ThT fluorescence (B). In (A), the kinetics of unseeded, rotated (80rpm) NM (2.5μM) fibrillization is also shown. Values represent means±SD (n=5).
(C, D) Kinetics of unseeded, rotated (80rpm) Ure2 (2.5μM) polymerization without or with Hsp104 (0.03μM or 1μM), plus ATP (5mM). Fibrillization was measured by ThT fluorescence (C) or CR binding (D). Values represent means±SD (n=3).
Ure2 forms amyloid fibers spontaneously (Fay et al., 2003; Zhu et al., 2003). Under our reaction conditions, freshly prepared Ure2 formed fibers after a TL of ~1h and AT1/2 of ~9h (Table S2) as determined by ThT fluorescence (Fig. 1C), Congo Red (CR) binding (Fig. 1D), and EM (Fig. 4A).
Figure 4. Hsp104 accelerates the consumption of Ure2 oligomers and formation of Ure2 fibers.
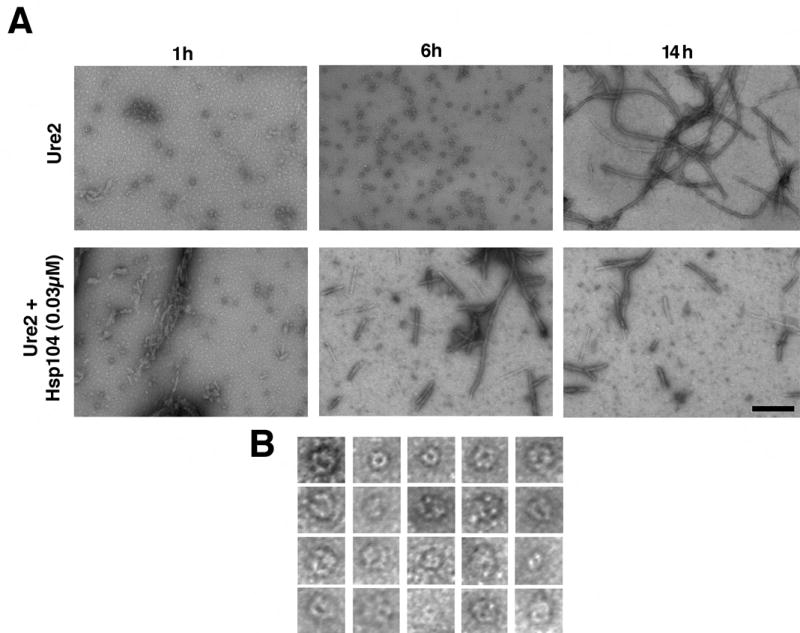
(A) Unseeded, rotated (80rpm) Ure2 (2.5μM) fibrillization reactions were performed without or with Hsp104 (0.03μM) plus ATP (5mM). At various times, reactions were processed for EM. Bar, 0.25μm.
(B) Gallery of twenty separate Ure2 oligomers formed after 6h without Hsp104. EM images are 41nm squares.
Effects of Hsp104 on Sup35 and Ure2 assembly depend upon stoichiometry
To investigate how Hsp104 governs the formation and inheritance of [PSI+] and [URE3], we first examined the effects of Hsp104 on the assembly of soluble protein into amyloid fibers. When Hsp104 was added to newly purified Sup35 at high concentrations (Sup35 monomers: Hsp104 hexamers, 2.5:1 or 6:1), it blocked assembly (Fig. 1A, B). In fact, at a ratio of Sup35 monomers: Hsp104 hexamers of 14:1, which may approximate the in vivo ratio (Ghaemmaghami et al., 2003), Hsp104 also blocked assembly (not shown). In contrast, at low concentrations (Sup35 monomers: Hsp104 hexamers, 250:1 or 83:1) Hsp104 strongly stimulated fibrillization (Fig. 1A, B), reducing the lag phase from ~2h to ~0.5h (Table S1). Hsp104 did not, however, eliminate the lag phase, as it did with NM (Shorter and Lindquist, 2004), indicating that the C-terminal domain imposes an additional remodeling requirement for Hsp104. Low concentrations of Hsp104 also accelerated the assembly phase, reducing AT1/2 from ~2.5h to ~0.9h (Table S1; Fig. 1A, B). The stimulation of both reaction phases by Hsp104 was specific. Cdc48, another protein-remodeling factor of the AAA+ family, which like Hsp104 is hexameric and contains two NBDs per monomer (Hanson and Whiteheart, 2005), did not stimulate Sup35 assembly (Table S1).
Hsp104 exerted similar control over Ure2 fibrillization. At high concentrations (Ure2 monomers: Hsp104 hexamers, 2.5:1 or 1.3:1), which may mimic the in vivo ratio of Ure2:Hsp104 (Ghaemmaghami et al., 2003), Hsp104 eliminated fibrillization (Fig. 1C, D; not shown). At low concentrations (Ure2 monomers: Hsp104 hexamers, 83:1), Hsp104 dramatically accelerated Ure2 fibrillization (Fig. 1C, D; Fig. 4A). As with Sup35, Hsp104 promoted assembly in two ways: it eliminated the lag phase (Fig. 1C, D, Table S2) and accelerated assembly phase (Fig. 1C, D, Table S2). Thus, Hsp104 interacts with Ure2 directly to promote amyloidogenesis. As with Sup35, these effects were specific to Hsp104, and were not conferred by Cdc48 (Table S2).
Sup35 and Ure2 fibers are bona fide prions
If the acceleration of Sup35 and Ure2 fibrillization by Hsp104 is relevant to prion propagation, then the resulting fibers should: (a) seed fibrillization of the corresponding unpolymerized proteins; and (b) induce the corresponding prion when transformed into prion-minus cells. To test these predictions, Hsp104-generated fibers were depleted of Hsp104. For these experiments, we used an N-terminally His6-tagged Hsp104 whose assembly activities were indistinguishable from those of the wild-type protein (not shown). A weak physical interaction between Hsp104 and Sup35 or Ure2 was observed (Fig. 2A, C). However, consistent with the transient nature of Hsp104-substrate interactions (Cashikar et al., 2002), His6-Hsp104 was readily separated from Sup35 and Ure2 (Fig. 2A, C). Hsp104-generated fibers were shorter than those produced in the absence of Hsp104 (Fig. 3A, 4A). Because polymerization occurs at fiber ends (Serio et al., 2000), short fibers seed more efficiently than long fibers. Therefore, for comparison of prion status, fibers formed spontaneously and fibers generated with Hsp104 were sonicated to reduce them to the same average size.
Figure 2. Hsp104 accelerates or abolishes de novo Sup35 and Ure2 prion formation.
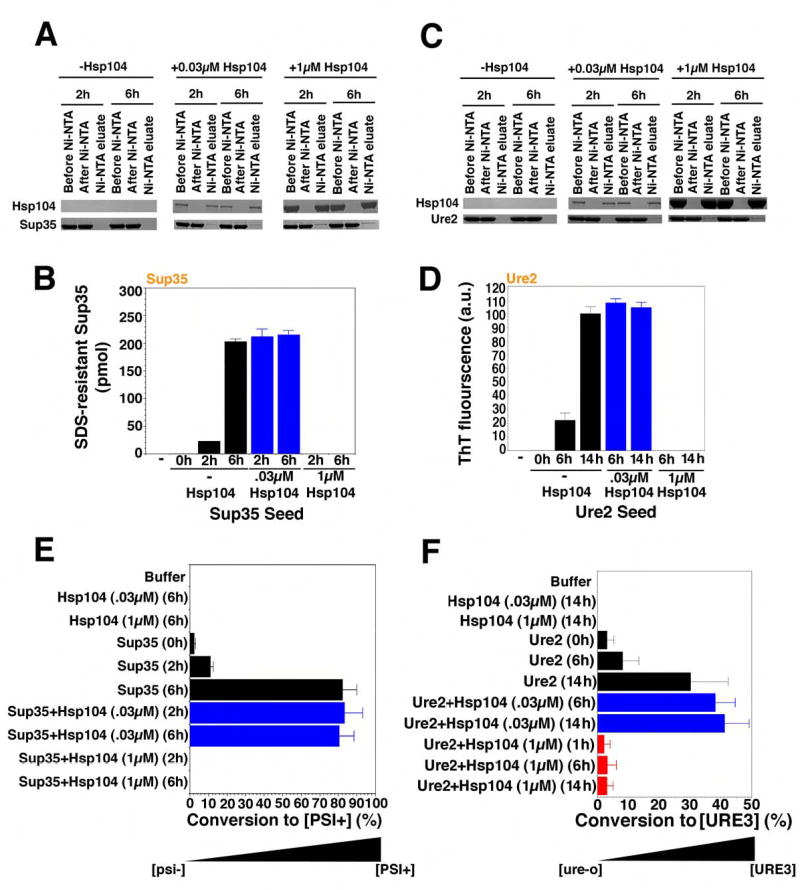
(A, B) Sup35 (2.5μM) was incubated with rotation (80rpm) for either 0h, 2h, or 6h without His6-Hsp104, 2h or 6h with His6-Hsp104 (1μM), or 2h or 6h with His6-Hsp104 (0.03μM). Reactions were depleted of His6-Hsp104 using magnetic Ni-NTA, and the extent of depletion was determined by immunoblot (A). Following His6-Hsp104 depletion, reactions were sonicated, and used to seed (2% wt/wt) unpolymerized Sup35 (2.5μM) for 4h in unrotated reactions (B). Values represent means±SD (n=3). (C, D) Ure2 (2.5μM) was incubated with rotation (80rpm) without or with His6-Hsp104 (0.03μM or 1μM) for 0h, 6h or 14h. Reactions were then depleted of His6-Hsp104 using magnetic Ni-NTA, and the extent of depletion was determined by immunoblot (C). Following His6-Hsp104 depletion, reactions were sonicated, and used to seed (2% wt/wt) unpolymerized Ure2 (2.5μM) for 4h in unrotated reactions (D). Values represent means±SD (n=3).
(E) Reactions were performed as in (A, B) and reaction products were concentrated, sonicated, and transformed into [psi−] cells. The number of [PSI+] colonies relative to total transformants was then determined. Values represent means±SD (n=3). (F) Reactions were performed as in (C, D) and reaction products were concentrated, sonicated, and transformed into [ure-o] cells. The number of [URE3] colonies relative to total transformants was then determined. Values represent means±SD (n=3).
Figure 3. Hsp104 catalyzes or reverses the formation of Sup35 oligomers that nucleate prion assembly.
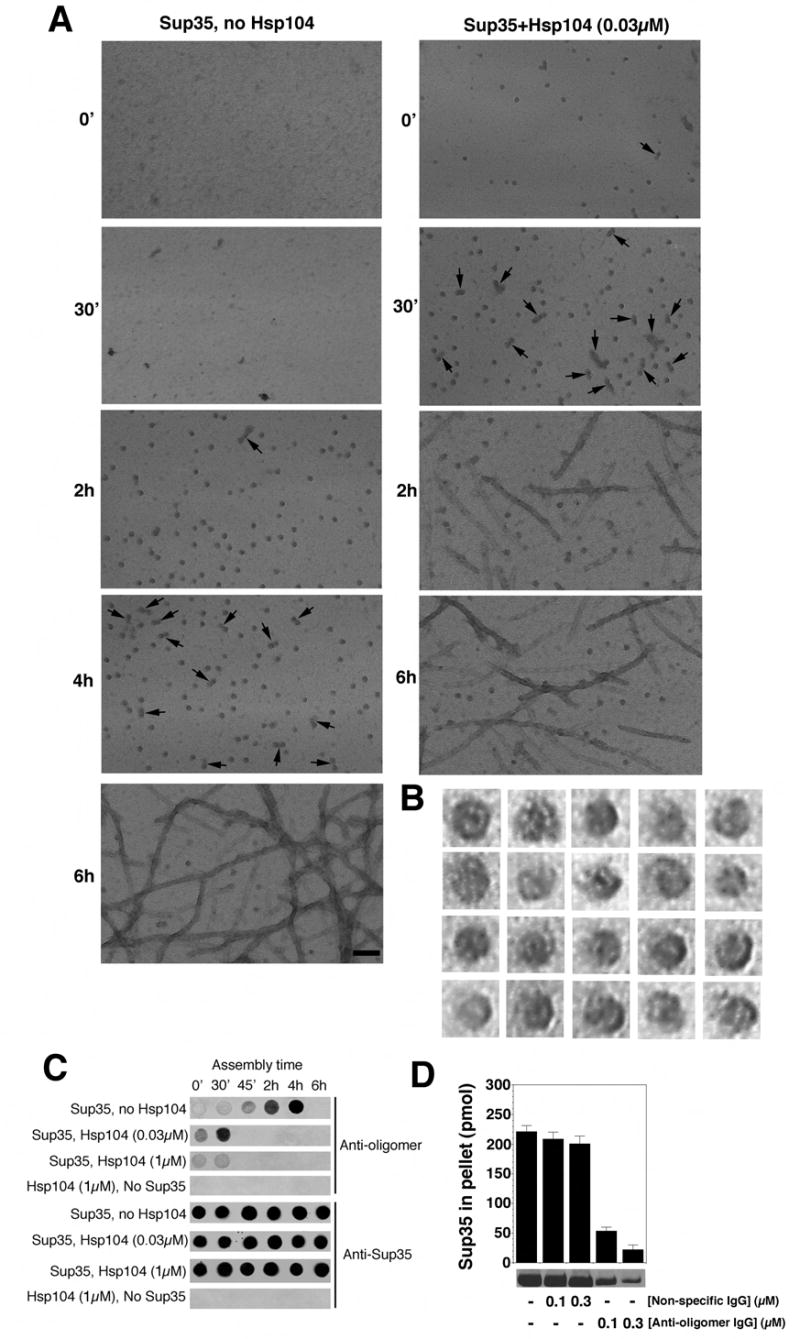
(A) Unseeded, rotated (80rpm) Sup35 (2.5μM) fibrillization reactions were performed without or with Hsp104 (0.03μM) plus ATP (5mM). At various times, reactions were processed for EM. Arrows denote close appositions of spherical oligomers. These oligomers assembled with Hsp104 did not correspond to Hsp104 because they were not observed in the absence of Sup35 (not shown). Although Hsp104 was hexameric in our conditions (not shown), visualization of Hsp104 hexamers by negative stain typically requires higher concentrations of Hsp104 and fixation with glutaraldehyde (Parsell et al., 1994a). Bar, 0.1μm.
(B) Gallery of twenty separate Sup35 oligomers formed after 2h without Hsp104. EM images are 29nm squares.
(C) Unseeded, rotated (80rpm) Sup35 (2.5μM) fibrillization reactions were performed without or with Hsp104 (0.03μM or 1μM) plus ATP (5mM). At various times, reactions were applied to nitrocellulose and probed with anti-oligomer antibody or anti-Sup35 antibody. Reactions lacking Sup35 were also assessed.
(D) Unseeded, rotated (80rpm) Sup35 (2.5μM) fibrillization reactions were performed without or with non-specific IgG (0.1–0.3μM) or anti-oligomer IgG (0.1–0.3μM). After 8h reactions were processed for sedimentation analysis. Values represent means±SD (n=3).
To test seeding capacity in vitro, we added a small quantity (2% wt/wt) of the products of Hsp104 reactions to an excess of unpolymerized protein. The Ure2 and Sup35 proteins that had been incubated with high concentrations of Hsp104 did not seed polymerization (Fig. 2B, D). Thus, the absence of ThT fluorescence or SDS-resistant material in these reactions corresponded to the absence of self-replicating conformers.
Spontaneously assembled Sup35 fibers seeded the assembly of soluble Sup35 (Fig. 2B). Therefore, like native NM (Scheibel and Lindquist, 2001), full-length Sup35 spontaneously populates a self-replicating conformation in vitro. Spontaneously generated Ure2 fibers also seeded the assembly of soluble Ure2 (Fig. 2D). Most importantly, fibers of both Sup35 and Ure2, whose assembly had been catalyzed by Hsp104, seeded polymerization of soluble Sup35 and Ure2 as efficiently as spontaneously assembled fibers (Fig. 2B, D, blue bars).
To test prion-seeding capacity in vivo, we removed the cell wall from yeast cells and introduced reaction products by polyethylene glycol treatment (King and Diaz-Avalos, 2004). As expected, Sup35 and Ure2 proteins that had been incubated with high concentrations of Hsp104 induced neither prion when transformed into prion-minus cells (Fig. 2E, F). Sup35 fibers induced [PSI+], but not [URE3] (Fig. 2E, not shown), and Ure2 fibers induced [URE3], but not [PSI+] (Fig. 2F, not shown). As for in vitro seeding assays, Hsp104-generated fibers were as effective as spontaneously assembled fibers at inducing heritable new prion elements (Fig. 2E, F, blue bars). Thus, the products of Hsp104 assembly reactions are bona fide prions.
Hsp104 appears to act as a true catalyst
An extremely enigmatic aspect of prion biology is the ability of single prion proteins to form multiple types of amyloid fibers that have distinct self-perpetuating structures and functions (Tuite and Cox, 2003). These variants are termed prion “strains”. For the mammalian prion PrP, these variants confer distinct pathologies (Tuite and Cox, 2003). For yeast prions, they confer distinct heritable phenotypes (Tuite and Cox, 2003). [PSI+] variants are readily distinguished: (a) they produce different colony colors on rich media, and (b) grow at different rates on adenine-deficient media (Tuite and Cox, 2003).
Using both criteria, we assessed the [PSI+] variants formed in cells transformed with Hsp104-generated Sup35 fibers. The same spectrum of [PSI+] variants was obtained as with spontaneously assembled Sup35 fibers (~40% strong, 60% weak) (not shown). Hence, in stimulating prion assembly Hsp104 appears to act as a true catalyst. It accelerates the rate of the reaction without being consumed by the reaction or influencing the nature of the product.
Hsp104 catalyzes the assembly of critical oligomeric intermediates of Sup35
To gain a better understanding of prion assembly, we examined Sup35 at various times by EM. Distinct spherical oligomers ~10–20nm in diameter accumulated towards the end of the lag phase (Fig. 3A). At higher magnification, a subpopulation appeared to have pores (Fig. 3B), reminiscent of oligomers formed by disease-associated amyloids (Lashuel et al., 2002). Thus, this morphology is another feature of assembly that is shared by diverse amyloidogenic proteins. Towards the end of lag phase, oligomers increasingly juxtaposed to form binary complexes and even short strings (Fig. 3A, arrows). Importantly, high concentrations of Hsp104, which inhibited assembly, also inhibited oligomer formation (not shown). Low concentrations of Hsp104, which accelerated assembly, accelerated the appearance of oligomers (Fig. 3A).
Next, we exploited an oligomer-specific antibody that recognizes prefibrillar oligomers, but not fibers or monomers, of Aβ40 and several other amyloidogenic proteins, including NM (Kayed et al., 2003; Shorter and Lindquist, 2004). At various times during Sup35 assembly, aliquots were withdrawn, spotted onto nitrocellulose and probed with antibodies. A Sup35-specific antibody recognized the protein at all stages in all reactions (Fig. 3C). In the absence of Hsp104, reactivity with the oligomer-specific antibody was strongest towards the end of the lag phase and disappeared when fibers assembled (Fig. 3C). Thus, maximum oligomer-specific immunoreactivity (Fig. 3C) was coincident with time points at which oligomers were most prevalent by EM (Fig. 3A).
When the oligomer-specific antibody was added to assembly reactions at concentrations 25-fold lower than Sup35, it had no effect on fibrillization seeded with preformed fibers (not shown). However, it prevented spontaneous fibrillization of soluble Sup35 (Fig. 3D). Thus, the oligomeric species of Sup35 recognized by this antibody plays a crucial role in the transition from lag phase to assembly phase.
In reactions containing high concentrations of Hsp104, the small degree of oligomer-specific antibody reactivity observed at time zero gradually disappeared (Fig. 3C). In marked contrast, oligomers assembled much more rapidly in reactions containing low concentrations of Hsp104 (Fig. 3C). As with spontaneous assembly, oligomers assembled by Hsp104 exhibited pores and, towards the end of lag phase, were increasingly juxtaposed in binary complexes and short strings (not shown; Fig. 3A, arrows). Hence, at high concentrations Hsp104 blocks assembly by remodeling preformed oligomers and blocking the formation of new ones. At low concentrations, Hsp104 catalyzes Sup35 prion formation by accelerating the formation of amyloidogenic oligomers. Thus, Hsp104 does not appear to change the nature of the assembly pathway. Rather, its effects on amyloidogenic oligomers confirm the importance of these critical intermediates.
Ure2 likely assembles via similar oligomeric nuclei
Ure2 also populated spherical oligomeric forms (Fig. 4A, 1h, 6h) prior to fiber formation. Because these species did not react with the oligomer-specific antibody (not shown), we could not establish whether they were crucial in nucleating prion formation. Presumably, some distinction in the conformations of Ure2 and Sup35 oligomers precludes recognition by the oligomer-specific antibody. However, Ure2 oligomers appeared remarkably similar to Sup35 oligomers, and were ~15–30nm in diameter with a substantial fraction possessing a pore-like morphology (Fig. 4B). Also, the disappearance of oligomers correlated with fiber assembly, which was accelerated by Hsp104 (Fig. 4A). It seems likely that Ure2 oligomers play a role in fiber assembly similar to that of Sup35 oligomers and that Hsp104 promotes their formation (Fay et al., 2003; Zhu et al., 2003).
Figure 6. Hsp104 couples ATP hydrolysis to Ure2 prion fragmentation.
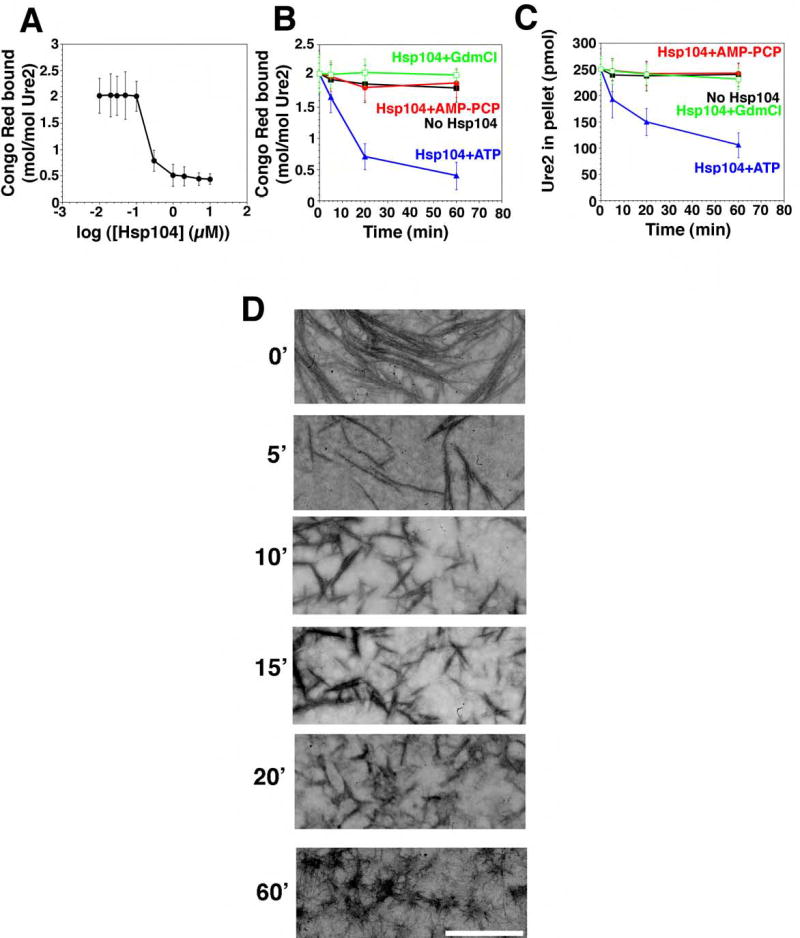
(A) Ure2 (2.5μM) was incubated for 16h with rotation (80rpm) to generate fibers and then incubated with Hsp104 (0.01–10μM) plus ATP (5mM) for 60min at 25°C. Disassembly was assessed by CR binding. Values represent means±SD (n=3). (B, C) Kinetics of Hsp104 (2μM) induced Ure2 fiber (2.5μM monomer) disassembly in the presence of either ATP (5mM), ATP (5mM) plus GdmCl (20mM), or the non-hydrolyzable analog AMP-PCP (5mM) (an alternative to AMP-PNP). Disassembly was monitored by CR binding (B) or sedimentation analysis (C). Values represent means±SD (n=3).
(D) Ure2 fibers (2.5μM monomer) were incubated with Hsp104 (2μM) plus ATP (5mM) for 0–60min, and at various times reactions were processed for EM. Scale bar, 0.25μm.
Hsp104-catalyzed prion assembly: effects of nucleotides, GdmCl, and NBD mutations
To gain further mechanistic insight into Hsp104-catalyzed Sup35 prion formation, we examined the nucleotide requirements of the reaction (Table S1). ATP binding but not hydrolysis was required for Hsp104-mediated nucleation. Hsp104 reduced lag phase equally well in the presence ATP or the non-hydrolyzable analog AMP-PNP, but not in the absence of nucleotide or in the presence of ADP. Hsp104 accelerated assembly phase by a distinct mechanism that required not only ATP binding but also hydrolysis. No acceleration of assembly was observed in the absence of nucleotide, with ADP, or with AMP-PNP. The prion-curing agent, GdmCl (20mM), an uncompetitive inhibitor of Hsp104 ATPase activity, also prevented Hsp104 from stimulating either the nucleation step or the assembly phase. GdmCl itself had no effect on spontaneous prion formation, demonstrating that both effects were exerted through inhibition of Hsp104.
To examine the contributions of each of the Hsp104 NBDs we exploited a series of Hsp104 mutants (Table S1) that harbor various single amino-acid substitutions in their Walker-A or sensor-1 motifs (Parsell et al., 1994a; Hattendorf and Lindquist, 2002). The Walker-A motif contains a lysine residue that directly contacts the phosphates of ATP, and mutation to threonine eliminates nucleotide binding (Parsell et al., 1994a; Hanson and Whiteheart, 2005). The sensor-1 motif contains a threonine or asparagine that interacts with the γ-phosphate of ATP, and mutation to alanine impairs ATP hydrolysis, but not ATP binding (Hattendorf and Lindquist, 2002).
An Hsp104 protein carrying Walker-A mutations in both NBDs (Hsp104 K218T:K620T), is defective in hexamerization and ATP binding at both NBDs (Parsell et al., 1994a). It failed to catalyze Sup35 prion formation, as did an NBD2 Walker-A mutant defective in hexamerization and nucleotide binding at NBD2 alone (Hsp104 K620T; Parsell et al., 1994a). Further, Hsp104 with an NBD1 Walker-A mutation, defective in ATP binding at NBD1 (Hsp104 K218T; Parsell et al., 1994a), but fully capable of hexamerization, also failed to catalyze Sup35 assembly.
In contrast, an NBD2 sensor-1 mutant, that is able to bind ATP at NBD2 but is defective in hydrolysis (Hsp104 N728A; Hattendorf and Lindquist, 2002), catalyzed the nucleation of Sup35 prions. This was also observed for an NBD1 sensor-1 mutant that is able to bind ATP, but is defective in hydrolysis (Hsp104 T317A; Hattendorf and Lindquist, 2002). However, both mutants failed to accelerate assembly phase. Thus, both NBD1 and NBD2 must be able to bind ATP for Hsp104 to reduce the lag phase of Sup35 prion formation. The catalysis of assembly phase, however, requires ATP binding and hydrolysis at both NBDs. Therefore, as suggested by the different effects of ATP, AMP-PNP, and ADP, these two aspects of Hsp104-stimulated prion formation, nucleation and assembly, are mechanistically distinct.
Next, we examined the effects of different nucleotides and of Hsp104 NBD mutations on the Hsp104-catalyzed assembly of Ure2 prions. In every case examined, results were similar for Ure2 and Sup35 (Table S2). The ability of Hsp104 to shorten the lag phase of Ure2 prion formation required ATP binding but not hydrolysis. Accelerating the assembly phase, however, did require ATP hydrolysis. Further, GdmCl had no effect on its own, but inhibited the ability of Hsp104 to promote nucleation or to accelerate assembly. Thus, the mechanism by which Hsp104 catalyzes the assembly of the two prions is remarkably similar.
Effects of nucleotides and GdmCl on the inhibition of prion assembly by high Hsp104 concentrations
In vivo, Hsp104 is required for the formation and propagation of both prions, but Hsp104 overexpression eliminates [PSI+] but not [URE3]. To investigate this phenomenon, we first compared the mechanism by which high concentrations of Hsp104 block the formation of prions from soluble Sup35 and Ure2 (Fig. 1; Table S1, S2). For both, Hsp104 inhibited with ATP or the non-hydrolyzable analogs AMP-PNP or AMP-PCP. In contrast, in the presence of GdmCl and ATP, high concentrations of Hsp104 extended the lag phase, but did not block Sup35 or Ure2 fiber assembly. A similar effect was seen in the presence of ADP. This most likely reflects reduced affinity of Hsp104 for either prion protein in the presence of ADP or ATP+GdmCl (Shorter and Lindquist, 2004). In the absence of nucleotide, high Hsp104 concentrations extended the lag phase for Ure2 more than for Sup35, but this is unlikely to explain the different effects that Hsp104 overexpression has on the inheritance of the two prions. Although ATP:ADP ratios will vary in cells, depending upon conditions, total nucleotide concentrations should remain in the millimolar range, the concentration required to saturate Hsp104 binding sites (Hattendorf and Lindquist, 2002). Therefore, we next asked if there is a more profound difference in the effects of Hsp104 on Sup35 and Ure2 after they have acquired their prion conformations.
Hsp104 deconstructs Sup35 and Ure2 fibers by a similar mechanism
When Hsp104 was added to preformed Sup35 fibers at 25°C, it reduced the amount of SDS-resistant Sup35 by ~60% (Fig. 5A–C). Strikingly, this remodeling activity was very inefficient until a threshold concentration of Hsp104 was breached (Fig. 5A). Such behavior indicates a highly co-operative reaction mechanism. EM revealed that at high concentrations Hsp104 rapidly fragmented networks of Sup35 fibers into shorter and shorter species (Fig. 5D, E). After 20min, some amyloid fibers persisted, but an ensemble of larger amorphous aggregates and smaller oligomeric fragments were more apparent (Fig. 5D, E). By 30–60min fibers were rare, ~40% of the protein remained pelletable by centrifugation (not shown), but it was not obviously fibrous (Fig. 5E), even though it remained SDS-resistant (Fig. 5A) and ThT reactive (not shown), suggesting an amyloid-like conformation.
Figure 5. Hsp104 couples ATP hydrolysis to Sup35 prion fragmentation.
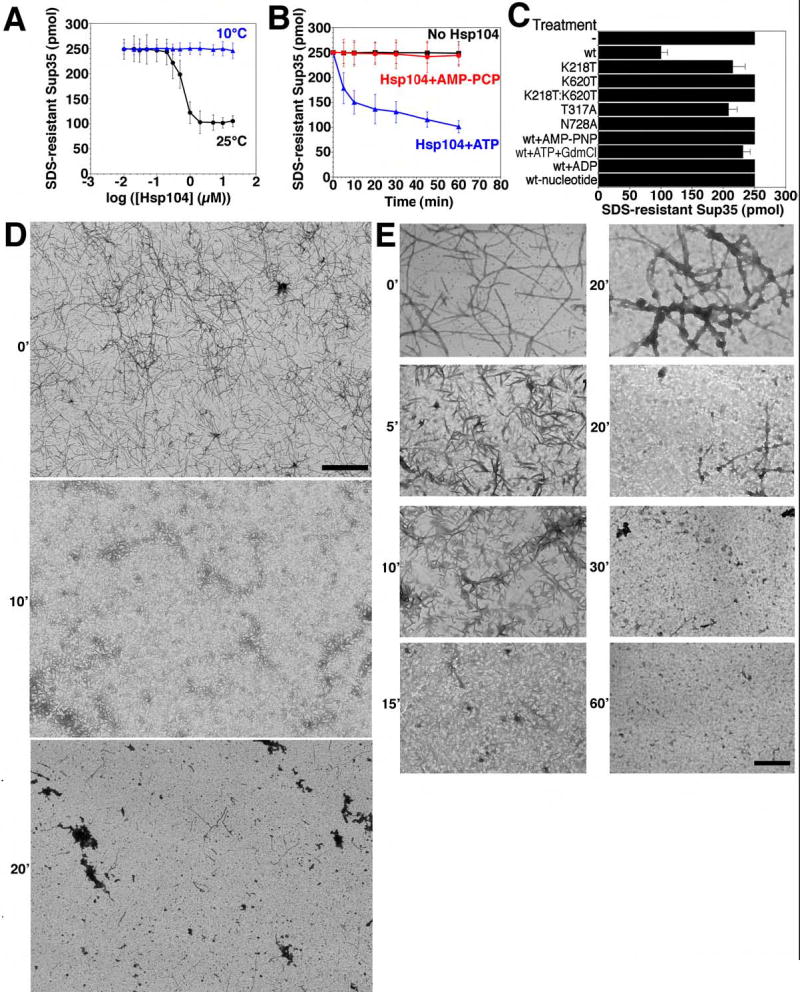
(A) Sup35 (2.5μM) was incubated for 8h with rotation (80rpm) to generate fibers and then incubated with Hsp104 (0.01–20μM) plus ATP (5mM) for 60min at 25°C (black) or 10°C (blue). Disassembly was assessed by the amount of SDS-resistant Sup35. Values represent means±SD (n=3).
(B) Kinetics of Hsp104 (2μM) induced Sup35 fiber (2.5μM monomer) disassembly in the presence of ATP (5mM) or the non-hydrolyzable analog AMP-PCP (5mM) (an alternative to AMP-PNP) at 25°C. Disassembly was monitored as in (A). Values represent means±SD (n=3).
(C) Sup35 fibers (2.5μM monomer) were treated at 25°C with the indicated Hsp104 proteins (2μM) plus ATP (5mM) or Hsp104 plus AMP-PNP (5mM), ATP (5mM) and GdmCl (20mM), ADP (5mM), or no nucleotide. Wt denotes wild type Hsp104. Disassembly was monitored by the amount of SDS-resistant Sup35. Values represent means±SD (n=3).
(D, E) Sup35 fibers (2.5μM monomer) were incubated with Hsp104 (2μM) plus ATP (5mM) for 0–60min at 25°C, and at various times reactions were processed for EM. Scale bars: 2μm (D), 0.25μm (E).
When Hsp104 was added to preformed Ure2 fibers it also rapidly remodeled them. CR binding was reduced by ~80% (Fig. 6A, B) and the amount of Ure2 in the pellet fraction was reduced by ~60% (Fig. 6C). EM revealed that Hsp104 rapidly fragmented Ure2 fibers (Fig. 6D). Most of the fibers were shorter than 0.25μm after 20min (Fig. 6D). After 1h, only tangles of very short Ure2 fibers remained (Fig. 6D).
The final Ure2 and Sup35 reaction products had a distinct appearance (Fig. 5E, 6D). Mechanistically, however, the characteristics of the disassembly reactions were very similar. Both were highly cooperative with an inflection between 0.1 and 1μM Hsp104 (Fig. 5A, 6A). Both required hydrolyzable nucleotide and were inhibited by GdmCl (Table S1; Fig. 5B, C, 6B, C). Analysis of Hsp104 NBD point mutations established that Sup35 disassembly required ATP hydrolysis at both NBDs (Fig. 5C). Furthermore, the co-operative ATPase activity of Hsp104 was profoundly compromised at low temperatures such as 10°C (~20-fold less, not shown), as is Sup35 fiber disassembly (Fig. 5A).
The endpoint of the remodeling activities of Hsp104 is profoundly different for Sup35 and Ure2 prions
Finally, we compared the nature of the reaction products from Sup35 and Ure2 disassembly reactions. To do so, we depleted Hsp104 at various time points and tested the ability of the remaining prion proteins to seed the polymerization of soluble protein in vitro and to transform prion-minus cells to the prion state in vivo. Brief treatments with Hsp104 increased the ability of Sup35 and Ure2 fibers to seed the polymerization of soluble Sup35 and Ure2, respectively (Fig. 7A, C). They also increased their ability to transform prion-minus cells to the corresponding [PSI+] or [URE3] prion state (Fig. 7B, D). In both respects, brief Hsp104 treatments mimicked the effects of fiber sonication (Fig. 7A–D). Sonication increases in vitro seeding activity by producing more ends for fiber polymerization (Serio et al., 2000) and increases the efficiency of in vivo transformation (King and Diaz-Avalos, 2004; Brachmann et al., 2005). Thus, the fiber fragmentation that occurs with brief Hsp104 treatments increases the specific activity of Sup35 and Ure2 prions.
Figure 7. Hsp104 eliminates Sup35 prions but potentiates Ure2 prions.
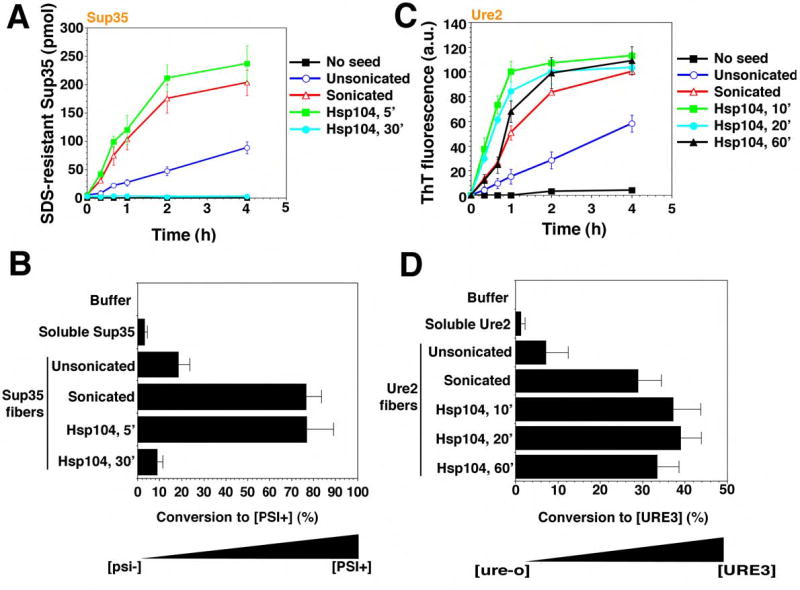
(A, B) Sup35 fibers (2.5μM monomer) were either left untreated, sonicated or treated with His6-Hsp104 (2μM) plus ATP (5mM) for 5–30min. Reactions were then depleted of His6-Hsp104 and used to seed (2% wt/wt) fresh, undisturbed Sup35 (2.5μM) polymerization (A). Values represent means±SD (n=3). Alternatively (B), reaction products were concentrated and transformed into transformed into [psi−] cells. Soluble Sup35 and buffer served as controls. The number of [PSI+] colonies relative to total transformants was then determined. Values represent means±SD (n=3).
(C, D) Ure2 fibers (2.5μM monomer) were either left untreated, sonicated or treated with His6-Hsp104 (2μM) plus ATP (5mM) for 10–60min. Reactions were then depleted of His6-Hsp104 and used to seed (2% wt/wt) fresh, unagitated Ure2 (2.5μM) polymerization (C). Values represent means±SD (n=3). Alternatively (D), reaction products were then concentrated and transformed into [ure-o] cells. Soluble Ure2 and buffer served as controls. The number of [URE3] colonies relative to total transformants was then determined. Values represent means±SD (n=3).
The final reaction products of Hsp104 disassembly reactions with Sup35 and Ure2, however, had very different behaviors. With longer incubation times, Hsp104 destroyed the ability of Sup35 fibers to seed the polymerization of soluble Sup35 in vitro and virtually eliminated its ability to transform [psi−] cells to the [PSI+] state (Fig. 7A, B). For Ure2 fibers, the final reaction products of Hsp104-mediated disassembly retained a very high capacity to seed the polymerization of soluble Ure2 in vitro, and to transform [ure-o] cells to the [URE3] state in vivo (Fig. 7C, D).
A trivial explanation is that the reactions had simply not gone to an equivalent state of completion for Ure2. However, no further decreases in CR binding or the amount of Ure2 in the pellet fraction could be achieved, even with tenfold higher Hsp104 concentrations or longer incubation times (up to 16h with the repeated addition of ATP and regeneration system), indicating the reaction could go no further (not shown). Moreover, in vitro seeding activity and in vivo transforming activity remained high. Hence, even though there is less fibrous material after a 60min treatment with Hsp104 (Fig. 6A, B), the Ure2 fibers that endure are just as infectious as sonicated Ure2 fibers (Fig. 7D). This implies that there are an approximately equal number of fiber ends (the active sites of conformational replication) in each preparation. Further incubations of Sup35 reaction products did not convert them back to a seeding competent or transformation competent state. Thus, Hsp104 deconstructs Sup35 fibers to create a mixture of non-infectious soluble protein and particulate amyloid-like material. In contrast, Hsp104 only increases the prion-specific activity of Ure2 fibers, producing soluble material and short fibers, with both a physical appearance and a functional state that are distinct from the endpoint of Sup35 reactions.
Discussion
We analyzed the prion remodeling activities of Hsp104 with two completely unrelated prion proteins, Sup35 and Ure2. Their only common feature is that their unstructured N-terminal prion domains are enriched in uncharged, polar residues. For Sup35, glutamines (28%), glycines (17%), tyrosines (16%), and asparagines (16%). For Ure2, asparagines (41%) and serines (12%). Despite these differences, Hsp104 employs several distinct remodeling activities to regulate their conformations in extraordinarily similar ways, with one critical distinction that explains a heretofore baffling aspect of their biology. We established the relevance of these biochemical activities by transforming conformationally altered reaction products into yeast to test their prion converting activities.
When Hsp104 concentrations are low and prion proteins are in the soluble state, Hsp104 catalyzes prion assembly. It does so in two ways, reducing the lag phase and accelerating the assembly phase. Notably, these activities are mechanistically distinct but the same for both prions. Accelerating assembly requires ATP hydrolysis. However, reducing lag phase requires only ATP binding. Such substrate remodeling by AAA+ proteins in the absence of ATPase activity is virtually unprecendented. Two other AAA+ proteins, p97 and NSF, catalyze the folding of select substrates in an ATPase-independent manner (Golbik et al., 1999; Müller et al., 2002), but the substrates were globular proteins. Furthermore, our discovery of Hsp104-catalyzed amyloidogenesis may prove to be an attribute of other AAA+ proteins, because p97 stimulates ataxin-3 fiber assembly by a strikingly similar mechanism (Boeddrich et al., 2006).
Another striking similarity between the two prions is that Hsp104 catalyzes their assembly via a distinctive oligomeric intermediate. For Sup35, at least, this is an obligate intermediate. In its ATP-bound state, Hsp104 has higher affinity for substrates (Shorter and Lindquist, 2004). With ATP-bound, Hsp104 may project a catalytic surface that enables molten prion domains, probably in the context of an oligomer, to establish the requisite intermolecular contacts that drive nucleation (Krishnan and Lindquist, 2005).
When Hsp104 concentrations are high relative to those of the soluble prion proteins, it blocks assembly. The effects of different nucleotides and Hsp104 NBD mutations indicate that here too, the mechanism is remarkably similar for both prions. Again, our results are consistent with AAA+ proteins preferentially engaging substrates when in the ATP-bound conformation (Shorter and Lindquist, 2004; Hanson and Whiteheart, 2005). p97 antagonizes ataxin-3 fiber assembly in a similar manner, implying a more widespread mode of amyloid regulation by select AAA+ proteins (Boeddrich et al., 2006).
Hsp104 has an unusual and powerful remodeling activity on preformed Sup35 or Ure2 prions. No other protein is known to exert such dramatic effects on structures that are otherwise so intractable (Muchowski and Wacker, 2005). Like other amyloids, prions fall at the extremes of structural stability and resist solubilization by anionic surfactants (e.g. SDS) and chaotropes (e.g. urea). Yet, Hsp104 can rapidly and efficiently disassemble amyloid fibers of both. This activity is strongly dependent upon Hsp104 concentrations, indicating a highly cooperative reaction mechanism. Further, fiber disassembly activity absolutely necessitates ATP hydrolysis, as does the acceleration of fiber assembly by low concentrations of Hsp104. In fact, we suggest these are related reactions. That is, acceleration of assembly phase at low Hsp104 concentrations may occur via rare fiber severing events, which liberate new ends for further polymerization. In vivo, when Hsp104 concentrations are low, the extreme concentration dependence of fiber severing would make it rare, perhaps only once per prion fiber per cell cycle. This would promote prion propagation by dividing fibers to stably maintain prion inheritance.
When Hsp104 is overexpressed, the strongly cooperative nature of prion remodeling would greatly increase severing and disassembly. Here, the major distinction in Hsp104’s biochemical activities on Sup35 and Ure2 would create a telling difference in the biological response of the two prions. The endpoint of Hsp104-mediated Sup35 disassembly does not promote polymerization in vitro and is not infectious in vivo. In vivid contrast, the endpoint of Hsp104-mediated Ure2 disassembly is a small fiber that very actively promotes polymerization in vitro and is highly infectious in vivo.
It has been suggested that Hsp104 overexpression might not cure [PSI+] by disassembly (Tuite and Cox, 2003), because some aggregated forms persist in cells that overexpress Hsp104 (Kryndushkin et al., 2003). However, since the products of Sup35 disassembly are a mixture of soluble protein and inactive amyloid-like aggregates, the presence of aggregated forms, even of amyloid-like SDS resistant forms, is a prediction of overexpressing Hsp104.
Deletion or inhibition of Hsp104 would eliminate prion fragmentation activity and block formation of new active sites for conformation replication (i.e. fiber ends). Consequently, prion growth and division would lose synchrony with cell division, and eventually the rate of conformational replication would fall below that required to sustain the prion. Intriguingly, while Sup35 aggregates increase in size in vivo when Hsp104 is inactivated (Wegrzyn et al., 2001; Kryndushkin et al., 2003), Ure2 aggregates do not (Ripaud et al., 2003). We find that Ure2 assembly is considerably slower than Sup35 assembly. Thus, Ure2 prions may require additional assistance from Hsp104 for prion growth to keep pace with cell division.
Hsp104 severs prion fibers and can even completely deconstruct prions without Hsp70 or Hsp40 (Shorter and Lindquist, 2004). This contrasts with the disaggregation of chemically or thermally denatured protein aggregates, which often necessitates Hsp70 and Hsp40 (Glover and Lindquist, 1998). This probably reflects the extremely different architecture of amorphous aggregates and prion fibers. However, Hsp70 and Hsp40 can play a facilitatory, but not essential role, in the disassembly of Sup35 fibers by Hsp104 (Shorter and Lindquist, in preparation). A possible explanation for the discrepancy between our results and other reports is that the amyloid fibers used in those studies (Inoue et al., 2004; Krzewska and Melki, 2006) were not prions, because no transformation experiments were conducted. Moreover, one study used only NM, which was tagged and affixed to surfaces at very high concentrations relative to Hsp104 (Inoue et al., 2004). Our results predict that unassisted Hsp104 severing activity would be precluded under these conditions. Another study used full-length Sup35, however, unlike our Sup35, it was tagged and purified without GTP in an aggregation-prone state (Krzewski and Melki, 2006). We presume this is why many reactions were performed at 10°C (Krzewski and Melki, 2006). Critically, the cooperative ATPase activity that drives Hsp104-mediated fiber disassembly is profoundly compromised at low temperatures and, as a consequence, so is fiber disassembly. Furthermore, in contrast to our studies (Fig. 2A) no direct, physical interaction between Hsp104 and Sup35 could be detected, even with glutaraldehyde crosslinking (Krzewska and Melki, 2006), suggesting the interaction between Hsp104 and Sup35 had been drastically perturbed (see Supplemental Discussion). Finally, in support of our data, Hsp104 disassembles amyloid fibers composed of a short fragment of Sup35 (amino acids 5–26) (Narayanan et al., 2006).
The roles of Hsp104 in the formation and propagation of another prion, [RNQ+], remain undeciphered. [RNQ+] affects the formation [PSI+] and [URE3] (Shorter and Lindquist, 2005), as do other chaperones whose roles are only beginning to be elucidated (Tuite and Cox, 2003). Nevertheless, our work provides a persuasive molecular explanation for a recalcitrant conundrum of why Hsp104 has distinct effects on [PSI+] and [URE3] inheritance. Further, our data truly clarify how Hsp104 mediates prion inheritance. In both cases, two activities promote the prion state, a capacity to nucleate new prions from soluble proteins and, at low concentrations, a weak capacity to sever fibers. The latter would provide new polymerization surfaces and ensure division and transmission to daughter cells. Hsp104 overexpression cures cells of [PSI+] but not [URE3]. Although Hsp104 rapidly and efficiently disassembles fibers of both proteins at high concentrations, its overexpression cures cells of [PSI+] because Hsp104 destroys the prion nature of Sup35 fibers. By contrast, Hsp104 does not cure cells of [URE3] because it leaves these fibers in a very active prion conformation. In regulating yeast prions, Hsp104 likely specifically recognizes and deconstructs elements of the generic ‘cross-beta’ structure of amyloid fibers as well as the distinct generic structure of their cytotoxic oligomeric precursors (Muchowski and Wacker, 2005), which are common to a plethora of disease-associated amyloids. The unprecedented alacrity at which Hsp104 remodels these structures might have therapeutic applications for many of the deadly and intractable disorders associated with amyloidogenesis.
Experimental Procedures
Proteins
NM, Hsp104, and Cdc48 were purified under native conditions as described (Scheibel and Lindquist, 2001; Shorter and Lindquist, 2004). Ure2 and Sup35 were purified under native conditions as described in Supplemental Experimental Procedures.
Fiber Assembly and Disassembly
Fibrillization was initiated by diluting freshly purified Sup35 or Ure2 to 2.5μM in Assembly Buffer (AB) (40mM Hepes-KOH pH 7.4, 150mM KCl, 20mM MgCl2, 10% (w/v) glycerol, 5mM ATP, and 1mM DTT). For Sup35, AB was supplemented with 1mM GTP. In reactions containing ATP a regeneration system was included comprising creatine phosphate (40mM) and creatine kinase (5μM) (Roche). Unseeded reactions were rotated (80rpm on a Mini-Rotator, Glas-Col) for 0–16h at 25°C. Seeded reactions were unrotated. Hsp104 or Cdc48 were exchanged into AB and were present upon Sup35 or Ure2 addition. Hsp104 and Cdc48 concentrations refer to hexamers. For experiments probing nucleotide requirements, the ATP in AB was replaced with AMP-PNP, AMP-PCP, or ADP (5mM) (Roche). For reactions without nucleotides MgCl2 was omitted and NaEDTA (20mM) was added. Reactions containing AMP-PNP or AMP-PCP were preincubated with hexokinase (0.5U/μl) (Sigma) and glucose (10mM) for 20min at 25°C to consume any contaminating ATP. Hsp104 depletion and dot blots were as described (Shorter and Lindquist, 2004). For disassembly, fibers were assembled as above and then treated as indicated. The extent of Sup35 or Ure2 fiber assembly or disassembly was monitored by EM, SDS-resistance, sedimentation analysis, CR binding or ThT fluorescence (Shorter and Lindquist, 2004; Supplementary Experimental Procedures).
Protein Transformation
Prion-minus yeast cells were transformed with Sup35 or Ure2 proteins as described in the Supplementary Experimental Procedures.
Supplementary Material
Acknowledgments
We thank C. Glabe and R. Wickner for reagents, N. Watson for EM assistance, M. Dünnwald, J. Su, J. Tyedmers, A. Gitler, P. Tessier, A. Steele, R. Krishnan, and K. Matlack for comments on the manuscript. This work was supported by a Charles A. King Trust post-doctoral fellowship and an American Heart Association scientist development grant to J.S., and an NIH grant (no. GM25874) and a DuPont-MIT-Alliance grant to S.L.
References
- Boeddrich A, Gaumer S, Haacke A, Tzvetkov N, Albrecht M, O Evert B, Muller EC, Lurz R, Breuer P, Schugardt N, et al. An arginine/lysine-rich motif is crucial for VCP/p97-mediated modulation of ataxin-3 fibrillogenesis. Embo J. 2006;25:1547–58. doi: 10.1038/sj.emboj.7601043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. Embo J. 2005;24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashikar AG, Schirmer EC, Hattendorf DA, Glover JR, Ramakrishnan MS, Ware DM, Lindquist SL. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol Cell. 2002;9:751–760. doi: 10.1016/s1097-2765(02)00499-9. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- Fay N, Inoue Y, Bousset L, Taguchi H, Melki R. Assembly of the yeast prion Ure2p into protein fibrils. Thermodynamic and kinetic characterization. J Biol Chem. 2003;278:30199–30205. doi: 10.1074/jbc.M303000200. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Golbik R, Lupas AN, Koretke KK, Baumeister W, Peters J. The Janus face of the archaeal Cdc48/p97 homologue VAT: protein folding versus unfolding. Biol Chem. 1999;380:1049–1062. doi: 10.1515/BC.1999.131. [DOI] [PubMed] [Google Scholar]
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Grimminger V, Richter K, Imhof A, Buchner J, Walter S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J Biol Chem. 2004;279:7378–7383. doi: 10.1074/jbc.M312403200. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- Hattendorf DA, Lindquist SL. Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. Embo J. 2002;21:12–21. doi: 10.1093/emboj/21.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Taguchi H, Kishimoto A, Yoshida M. Hsp104 binds to yeast Sup35 prion fiber but needs other factor(s) to sever it. J Biol Chem. 2004;279:52319–52323. doi: 10.1074/jbc.M408159200. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- Krzewska J, Melki R. Molecular chaperones and the assembly of the prion Sup35p, an in vitro study. Embo J. 2006;25:822–833. doi: 10.1038/sj.emboj.7600985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Sondheimer N, Lindquist SL. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+] Proc Natl Acad Sci U S A . 2002;99(Suppl 4):16446–16453. doi: 10.1073/pnas.252652099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Müller JM, Shorter J, Newman R, Deinhardt K, Sagiv Y, Elazar Z, Warren G, Shima DT. Sequential SNARE disassembly and GATE-16-GOS-28 complex assembly mediated by distinct NSF activities drives Golgi membrane fusion. J Cell Biol. 2002;157:1161–1173. doi: 10.1083/jcb.200202082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell. 2001;7:1121–1130. doi: 10.1016/s1097-2765(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci U S A. 2005;102:10575–10580. doi: 10.1073/pnas.0504882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan S, Walter S, Reif B. Yeast Prion-Protein, Sup35, Fibril Formation Proceeds by Addition and Substraction of Oligomers. Chembiochem. 2006;7:757–765. doi: 10.1002/cbic.200500382. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Lindquist S. Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J Biol Chem. 1994a;269:4480–4487. [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994b;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. Embo J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Ripaud L, Maillet L, Cullin C. The mechanisms of [URE3] prion elimination demonstrate that large aggregates of Ure2p are dead-end products. Embo J. 2003;22:5251–5259. doi: 10.1093/emboj/cdg488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibel T, Lindquist SL. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat Struct Biol. 2001;8:958–962. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell. 2003;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- Talarek N, Maillet L, Cullin C, Aigle M. The [URE3] Prion Is Not Conserved Among Saccharomyces Species. Genetics. 2005;171:23–34. doi: 10.1534/genetics.105.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science. 1999;283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature. 2004;431:184–187. doi: 10.1038/nature02885. [DOI] [PubMed] [Google Scholar]
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- Tuite MF, Cox BS. Propagation of yeast prions. Nat Rev Mol Cell Biol. 2003;4:878–890. doi: 10.1038/nrm1247. [DOI] [PubMed] [Google Scholar]
- Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol. 2001;21:4656–4669. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner, R. B., Liebman, S.W., Saupe, S. J. (2004). Prions of Yeast and Filamentous Fungi: [URE3], [PSI+], [PIN+], and [Het-s]. In Prion Biology and Diseases, S. B. Prusiner, ed. (Cold Spring Harbor, New York, Cold Spring Harbor Laboratory Press), pp. 305–372.
- Zhu L, Zhang XJ, Wang LY, Zhou JM, Perrett S. Relationship between stability of folding intermediates and amyloid formation for the yeast prion Ure2p: a quantitative analysis of the effects of pH and buffer system. J Mol Biol. 2003;328:235–254. doi: 10.1016/s0022-2836(03)00249-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.