

Abstract
Ferredoxin (flavodoxin):NADP+ oxidoreductase (FNR) is an essential enzyme that supplies electrons from NADPH to support flavodoxin-dependent enzyme radical generation and enzyme activation. FNR is a monomeric enzyme that contains a non-covalently bound FAD cofactor. We report that reduced FNR from Escherichia coli is subject to inactivation due to unfolding of the protein and dissociation of the FADH2 cofactor at 37°C. The inactivation rate is temperature-dependent in a manner that parallels the thermal unfolding of the protein and is slowed by binding of ferredoxin or flavodoxin. Understanding factors that minimize inactivation is critical for utilizing FNR as an accessory protein for S-adenosyl-l-methionine-dependent radical enzymes and manipulating FNR as an electron source for biotechnology applications.
Keywords: Ferredoxin:NADP reductase, Flavin-adenine dinucleotide, Flavoprotein, Oxidoreductase, Enzyme stability
1. Introduction
Ferredoxin (flavodoxin):NADP+ oxidoreductase (FNR, EC 1.18.1.2) from Escherichia coli catalyzes the NADPH-dependent reduction of flavodoxin and ferredoxin [1-3]. Reduced flavodoxin is essential for several cellular processes, including the S-adenosyl-l-methionine (AdoMet)-dependent reductive methylation of cobalamin-dependent methionine synthase [4-6], the reductive cleavage of AdoMet accompanying generation of glycyl radicals in pyruvate formate-lyase [1,7,8] and class III ribonucleotide reductase [9-11], and the reductive cleavage of AdoMet required for substrate oxidation in biotin synthase [12,13] and probably also lipoyl synthase [14]. Ferredoxin [15] may fulfill an as-yet unidentified role in redox processes accompanying iron-sulfur cluster assembly. Thus FNR plays a critical cellular role in distributing low potential electrons largely derived from glucose metabolism, serving as a funnel between the abundant pyridine-nucleotide hydride pool and the largely protein-bound electron pool.
FNR is a member of the broadly conserved ferredoxin reductase family of oxidoreductases [16,17] that are monomeric and contain a single non-covalently bound FAD cofactor. The paradigm for this family has been the FNR from Anabaena [18,19]. Anabaena FNR catalyzes the transfer of electrons from reduced ferredoxin generated during photosynthesis to the pyridine-nucleotide pool. The enzyme has both high activity and prolonged stability. FNR also is present in numerous microorganisms, and domains homologous to FNR have been described in human enzymes including cytochrome P450 reductase [20], nitric oxide synthase [21], and methionine synthase reductase [22]. E. coli FNR and flavodoxin also serve as heterologous reductants for mammalian cytochrome P450 enzymes [23,24] and may therefore have potential value in industrial enzyme applications.
As part of ongoing studies of the role of FNR in supporting the activities of AdoMet-dependent radical enzymes, particularly biotin synthase [25], we have noted that a rapid loss of FNR activity contributes to the very slow kinetics observed with these enzymes. In this paper, we examine factors that accelerate FNR inactivation. Although oxidized FNR is very stable, reduced FNR is subject to loss of activity due to partial unfolding of the protein and dissociation of the FADH2 cofactor. This process is both temperature- and pH-dependent and is slowed by bound ferredoxin or flavodoxin. In light of the important role of the FNR/Fld pair in sustaining the activity of the newly emerging superfamily of AdoMet-dependent radical enzymes [26,27], we hope that this detailed report of the stability of E. coli FNR will facilitate a better understanding of the role of redox enzymes in sustaining these mechanistically and kinetically complex enzyme systems.
2. Materials and methods
2.1. Enzyme preparation
Flavodoxin [28] and ferredoxin [3] were expressed and purified as previously described. Flavodoxin concentration was determined using ε458 = 8500 M-1 cm-1 [29] and ferredoxin concentration was determined using ε468 = 7240 M-1 cm-1 [3]. All other reagents were purchased commercially and used without further purification.
FNR was expressed over 4-5 days from strain pEE1010/C-600 [30]. Cells were lysed by sonication and cell debris removed by centrifugation at 100 000×g for 30 min. The soluble fraction was loaded on a DEAE fast-flow Sepharose column equilibrated with 10 mM Tris, pH 8, and eluted with a linear gradient to 200 mM NaCl in the same buffer. The yellow fractions were pooled, concentrated in centrifugal concentrators to > 10 mg/ml, and dialyzed overnight against 5 mM potassium phosphate, pH 7, followed by a 2 h dialysis against deionized water. The protein was loaded on a hydroxyapatite column (type I, Bio-Rad) equilibrated with 1 mM potassium phosphate, pH 7, and eluted with a linear gradient to 150 mM potassium phosphate, pH 7. Yellow fractions were checked by SDS-polyacrylamide gel electrophoresis for purity. Pure fractions were pooled and concentrated to > 500 μM. FNR concentration is determined using ε452 = 7100 M-1 cm-1 [4].
2.2. FNR assays
FNR activity was determined using the steady-state rate of flavodoxin or ferredoxin reduction in the presence of excess NADPH [3]. These assays were performed in a stopped-flow spectrophotometer to maximize reproducibility between the fast reactions with ferredoxin and 2,6-dichloroindophenol (DCIP), and the slower reactions with flavodoxin and O2. In addition, the stopped-flow instrument affords improved anaerobicity over prolonged incubation times. FNR (1 μM, 0.5 μM final concentration after mixing) and NADPH (500 μM) were rapidly mixed with flavodoxin or ferredoxin (100 μM) in 50 mM 1,3-bis[tris(hydroxymethyl)methylamino]propane (Bis-Tris Propane), 50 mM NaCl, pH 7.5, and spectral changes were monitored at 580 nm (flavodoxin) or 510 nm (ferredoxin). To minimize oxygen contamination, protocatechuic acid (0.5 mM) and protocatechuate dioxygenase (0.5 mg/ml) were added to all samples [31]. The rate of protein reduction was determined for flavodoxin using Δε580 = 4250 M-1 cm-1 and for ferredoxin using Δε510 = -4070 M-1 cm-1 [3].
Alternatively, FNR activity was determined by monitoring NADPH consumption in the presence of oxygen or DCIP. To measure the residual activity of reduced enzyme, FNR (1 μM) and NADPH (500 μM) in anaerobic 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5, were rapidly mixed with either air-saturated 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5 ([O2]∼200 μM) or anaerobic buffer containing DCIP (100 μM). To measure the residual activity of oxidized enzyme, NADPH was simply moved from the enzyme syringe to the oxygen (substrate) syringe. Oxidized FNR (1 μM) in anaerobic 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5, was rapidly mixed with NADPH (500 μM) in aerobic 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5 ([O2]∼200 μM). NADPH consumption was monitored by observing the decrease in absorbance at 340 nm and the linear (steady-state) portion of this curve converted to a rate using ε340 = 6200 M-1 cm-1 [32].
The pH dependence of the inactivation rate and the activity were fit assuming that there is a single pKa that governs a transition between a protonated state that has decreased activity and a low inactivation rate and a deprotonated state that has higher activity but also a more rapid inactivation rate. Data were fit to:
where kslow is the inactivation rate (or activity) of the low pH state, kfast is the inactivation rate (or activity) of the high pH state, and pKa is the acid dissociation constant for a hypothetical residue that governs conversion between these states.
The temperature dependence of the inactivation rate and of activity were fit to a standard Arrhenius equation, which requires the simplifying assumption that inactivation (unfolding) proceeds through a single transition state. Data were fit to:
where Ea is the activation energy for proceeding through the transition state of the inactivation process and A is a constant of integration.
2.3. Determination of thermal stability
The thermal stability of oxidized and reduced FNR was determined using changes in the fluorescence of the FAD cofactor. Spectra were recorded on an Aviv Model 105 titrating scanning differential/ratio spectrofluorometer with excitation and emission bandwidths set at 10 nm. Maximal excitation and emission wavelengths were determined for free FAD (1 μM) in 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5 to be λex = 450 nm and λem = 520 nm. FNR was diluted to 1 μM in chilled 50 mM Bis-Tris Propane, 50 mM NaCl, 0.5 mM dithiothreitol (DTT), pH 7.5, in a septum-covered fluorescence cuvette and the solution equilibrated with an argon atmosphere. For reduced samples, NADPH (500 μM) or sodium dithionite (1 mM) were added with a syringe. The protein sample was equilibrated at increasing 5°C intervals for 5 min and sample and buffer fluorescence measurements were recorded at each temperature. Since each experiment yields relative fluorescence data on different scales, data was normalized by subtracting the initial fluorescence and dividing by the final fluorescence. Data were fit assuming a simple two-state unfolding model [33], where TM is the midpoint for denaturation and ΔH is the enthalpy of denaturation:
3. Results
3.1. FNR is subject to time-dependent loss of activity
E. coli FNR catalyzes the reduction of several protein and small molecule electron acceptors, including ferredoxin, flavodoxin, cytochrome c, oxygen, ferricyanide, and dichloroindophenol (DCIP) [1-3]. To assess the steady-state activity of FNR, we have developed assays that monitor FNR-catalyzed ferredoxin or flavodoxin reduction by NADPH, or that monitor FNR-catalyzed NADPH oxidation by oxygen, ferricyanide, or DCIP. Since FNR turns over with ferredoxin and DCIP at rates that are much faster than with flavodoxin or oxygen, we performed each assay in a stopped-flow spectrophotometer such that the results with each electron acceptor could be directly compared under identical conditions. To monitor the loss of steady-state activity, FNR was diluted to 1 μM in a chilled anaerobic solution containing NADPH (500 μM), and then warmed by drawing the solution into the stopped-flow syringe pre-equilibrated to 37 ± 0.3°C. Assays were performed by mixing this sample (0.5 μM FNR after mixing) with an electron acceptor (50-100 μM final concentration) in the stopped-flow spectrophotometer at intervals, and monitoring either NADPH oxidation or flavodoxin and ferredoxin reduction to assess the residual activity. In each case, the activity is based upon a measurement of the initial rate of steady-state electron transfer under conditions where less than 20% of the substrates have been consumed and where the monitored absorbance changes remain linear over the monitored time range, thus ensuring that substrate depletion and product inhibition do not contribute significantly to the observed inactivation rates.
FNR reduced in the presence of NADPH is slowly inactivated under all conditions, although the observed rate of inactivation differs depending upon the electron acceptor (Fig. 1). Reactivity towards flavodoxin is lost most rapidly (kinact = 0.031 min-1) with less than 10% of the original activity remaining after 30 min. Curiously, reactivity towards ferredoxin is not as rapidly affected (kinact = 0.0065 min-1) with ∼80% of the original activity remaining after 30 min. Reactivity towards the unnatural electron acceptor DCIP is lost at a rate comparable to ferredoxin (kinact = 0.0064 min-1), while reactivity towards oxygen is lost slightly more rapidly (kinact = 0.019 min-1). To verify that inactivation occurs only in the reduced enzyme, we mixed an anaerobic sample of oxidized FNR (1 μM) with a sample of NADPH (250 μM) and oxygen (∼100 μM) after incubation at 37°C. In this case, oxidized FNR is very stable, with ∼98% of the original activity towards oxygen remaining after 30 min (kinact = 0.0005 min-1). Thus the inactivation process clearly affects only FNR containing reduced FAD, and the observed differences in the residual activity with alternate electron acceptors suggest that different structural factors within FNR affect reactivity with each species.
Fig. 1.
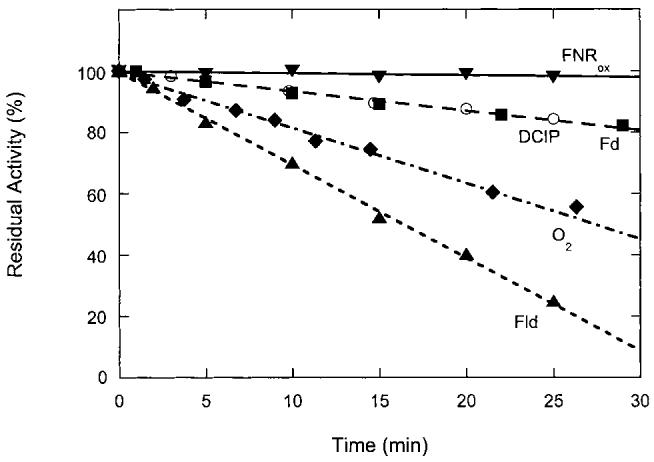
Loss of FNR activity towards various electron acceptors. FNR (1 μM) was reduced with NADPH (500 μM) in 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5, under argon and incubated at 37°C. At various intervals, the reduced FNR solution was mixed 1:1 with Fd (■), DCIP (○), oxygen (◆), or Fld (▲) in a stopped-flow spectrophotometer and activity monitored as described in Section 2. Alternately, oxidized FNR (▼, 1 μM) was incubated in anaerobic buffer at 37°C and mixed with an aerobic solution of NADPH (500 μM). Each data point is based upon an average of the initial slope of five experiments with an average standard deviation of ±5%. The initial activity (kcat) for each electron acceptor varies widely: flavodoxin, 0.023 s-1; ferredoxin, 0.34 s-1; O2, 0.064 s-1; DCIP, 9.8 s-1.
To further investigate conditions that could limit inactivation of FNR under steady-state assay conditions, we examined how pH and temperature affect the rate of inactivation. To examine the effect of pH on the rate of inactivation, we prepared a series of buffers by titrating 50 mM Bis-Tris Propane, 50 mM NaCl with 6 N HCl, and removing portions of buffer equilibrated at each pH from 9.5 to 6.5. FNR (1 μM) was reduced at 10-15°C by adding to NADPH (500 μM) in each buffer under argon. The sample was introduced into the stopped-flow syringe at 37°C and mixed at intervals with aerobic buffer at the same pH. The rate of inactivation was determined from the slope of a plot of activity vs. time, similar to plots shown in Fig. 1. As shown in Fig. 2A, the rate of inactivation varied from 0.025 min-1 at pH 6.5 to 0.09 min-1 at pH 9.5. The data is well fit to a single pKa = 8.4 ± 0.1, suggesting that deprotonation of a specific protein residue contributes to inactivation. Interestingly, the initial activity towards oxygen behaves similarly (Fig. 2A, inset), rising from kcat = 0.055 s-1 at pH 6.5 to 0.1 s-1 at pH 9.5 with an apparent pKa = 9.0 ± 0.5. Within the error of our data, this is identical to the pKa determined for inactivation, and suggests that deprotonation of the same protein residue contributes both to increased activity (at least towards oxygen) but potentially also to more rapid inactivation of the enzyme.
Fig. 2.
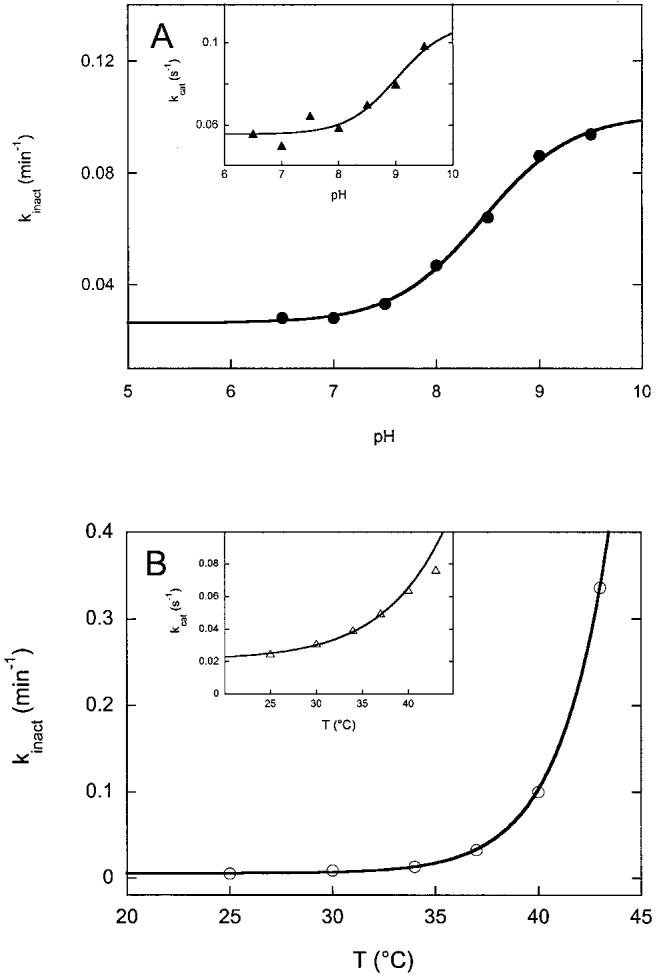
Effect of pH and temperature on the rate of FNR inactivation and FNR activity. FNR was reduced with NADPH under argon and incubated at each respective pH and temperature, then mixed with oxygen while the rate of NADPH oxidation was monitored at 340 nm. A: The pH of the assay buffer was adjusted from 6.5 to 9.5 in 50 mM Bis-Tris Propane, 50 mM NaCl. The rate of the loss of activity towards oxygen was monitored by measuring activity at various times after warming to 37°C. The data are fit with a single pKa = 8.5. A (inset): The initial activity (kcat) is shown at each pH. The data are fit with a single pKa = 9.0. B: The equilibration temperature was varied from 25 to 45°C, all in 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5. The rate of loss of activity was monitored by measuring activity at various times after warming from ∼10°C to each respective temperature. Above 43°C, the protein rapidly precipitated and no activity was measurable after 2 min. The data are fit to an Arrhenius equation with EA = 80 kcal/mol. B (inset): The initial activity (kcat) is shown at increasing temperatures. The data from 25-40°C are fit to an Arrhenius equation with EA = 30 kcal/mol. Each data point in A and B is an average of two determinations of the inactivation rate, where the inactivation rate is based upon the slope of an inactivation curve similar to those shown in Fig. 1.
To further probe conditions that could limit inactivation, we varied the incubation temperature and measured the initial activity and rate of inactivation. Not surprisingly, the rate of inactivation rises dramatically at increased temperature (Fig. 2B), varying from 0.006 min-1 at 25°C to 0.34 min-1 at 43°C. The data are fit to an Arrhenius equation, assuming a purely thermal inactivation process, and yield an apparent activation energy of 80 ± 10 kcal/mol. This large activation energy suggests that enzyme inactivation was the result of the loss of numerous interactions within the protein, most likely the result of cooperative global unfolding of the protein. We also examined the temperature dependence of the initial enzyme activity (Fig. 2B, inset). Although the activity increased with temperature, this increase was not nearly as dramatic, with kcat rising from 0.025 at 25°C to 0.076 at 43°C. The data from 25 to 40°C was fit to an Arrhenius equation and indicates an activation energy of 30 ± 4 kcal/mol for the rate limiting event in catalysis. Together with the pH data above, comparison of the inactivation rates with the initial activity suggests that one can optimize the stability of the enzyme by working at low pH (6.5-7.5) and at low temperature (25-30°C). However, by restricting conditions to favor stability in this way, the initial activity of FNR is decreased ∼6-fold as compared to the optimal initial activity achieved at pH 8.5-9 and 37-40°C.
3.2. Inactivation is due to irreversible protein unfolding
The dramatic increase in the rate of inactivation at high temperature suggested that inactivation could be due to global protein unfolding or at least minimally unfolding of the polypeptide chain near the flavin cofactor. It seemed surprising, however, that this would be observed only for the reduced enzyme. We followed the thermal unfolding of FNR by monitoring the fluorescence of the FAD cofactor; the free cofactor is strongly fluorescent in the visible region with λex = 450 nm and λem = 520 nm, and this fluorescence is quenched by ∼80% when FAD is bound to the protein. When oxidized FNR (0.5 μM) is heated slowly in anaerobic 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5, containing 0.5 mM DTT under argon, the fluorescence is stable to 50°C, and then undergoes a sharp increase as the temperature is increased from 60 to 70°C with a TM of 66°C and ΔH of 100 ± 15 kcal/mol (Fig. 3, squares). However, when the protein is reduced with excess NADPH, under otherwise identical conditions, a similar fluorescence increase is observed at much lower temperature (Fig. 3, circles). For reduced FNR, denaturation occurs between 30 and 45°C with a TM of 41°C, a decrease of 25°C in the stability of the enzyme upon reduction. The enthalpy of unfolding is also decreased to ΔH = 80 ± 10 kcal/mol. Since this sample was reduced in the presence of excess NADPH, the reduced enzyme was likely bound with an additional equivalent of NADPH, and it was possible that a steric clash between reduced pyridine nucleotide and reduced FAD cofactor caused this destabilization. However, when FNR was reduced under identical conditions with dithionite (1 mM), the same melting curve (Fig. 3, triangles) was obtained within the error of the fluorescence data, again with a TM of 41°C. Thus the destabilization of the reduced protein is clearly due to a poor interaction between the reduced cofactor and protein, independent of the source of reducing equivalents.
Fig. 3.
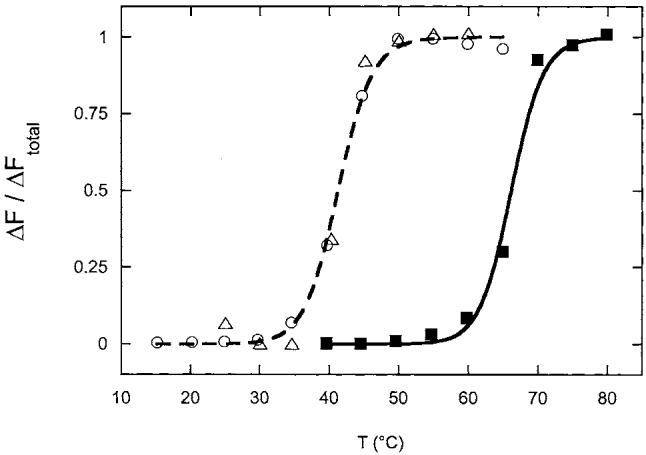
Thermal unfolding of oxidized and reduced FNR. Oxidized FNR (■, 0.5 μM) was incubated in 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5, under argon in an anaerobic fluorescence cuvette. NADPH (○, 500 μM) or sodium dithionite (△, 1 mM) were added to reduced samples. The temperature was raised in 5°C increments, with 5 min equilibration at each temperature prior to recording the fluorescence. The reduced enzyme samples were initially ∼2-3-fold less fluorescent than the oxidized enzyme, but all samples underwent a four-fold increase in fluorescence upon unfolding. Relative fluorescence data was normalized and fit as described in Section 2, yielding values for TM: oxidized, 66°C (solid curve); reduced, 41°C (dashed curve). Each experiment was performed three times and a representative data set is shown.
Since we detected protein unfolding by monitoring flavin fluorescence, it was possible that we were observing only partial unfolding of the protein that exposed the flavin to increased solvent interactions. However, two observations suggest this is not the case. First, if the protein was partially unfolding to an intermediate metastable state, then we should be able to reverse the unfolding reaction by lowering the temperature and observe quenching of the flavin fluorescence as the protein refolds. This was not observed; rather we observe that the final fluorescence is maintained as the protein is slowly cooled back to 20°C. Second, when we attempted to perform unfolding experiments at higher concentrations of FNR (2-10 μM), we were not able to monitor fluorescence due to the formation of a flocculent white precipitate, most likely an aggregate of the apoprotein. When this precipitate was pelleted by centrifugation (14 000×g for 5 min), a white protein pellet was obtained while all of the FAD was found free in solution, as judged by the residual fluorescence of the buffer. We conclude that the protein is undergoing a global unfolding process, and that at high concentration this unfolded protein interacts irreversibly to form a precipitate while the dissociated FAD cofactor remains in solution.
3.3. Inactivation is slowed by binding of ferredoxin or flavodoxin
FNR catalyzes the reduction of flavodoxin to the semiquinone oxidation state; this reaction is essential in supporting the activity of AdoMet-dependent radical enzymes such as class III ribonucleotide reductase and biotin synthase. Since assays of these enzymes require prolonged incubation (1-4 h), we thought it essential to find conditions that could prolong the lifetime of reduced FNR. Reduced FNR is clearly inactivated by an unfolding process, and since both ferredoxin and flavodoxin bind with high affinity to FNR, addition of these proteins might result in the formation of a binary complex with increased stability. To examine this hypothesis, we measured the initial flavodoxin reductase activity of reduced FNR alone (1 μM), and reduced FNR (1 μM) mixed with flavodoxin or ferredoxin (40 μM each) in 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5. We then retested the activity after 30 min incubation at 37°C. We found that addition of flavodoxin or ferredoxin slightly increased the initial activity from 0.023 s-1 to 0.032 and 0.027 s-1 (20-40% increase). Further, both proteins stabilized reduced FNR against loss of activity (Fig. 4). While FNR alone loses 94% of its initial activity in 30 min, in the presence of flavodoxin this loss is reduced to 53% and in the presence of ferredoxin to 18%. The increased protection afforded by ferredoxin correlates with increased affinity; FNR binds ferredoxin (Kd∼0.5 μM) about four-fold tighter than flavodoxin (Kd∼2 μM) [3].
Fig. 4.
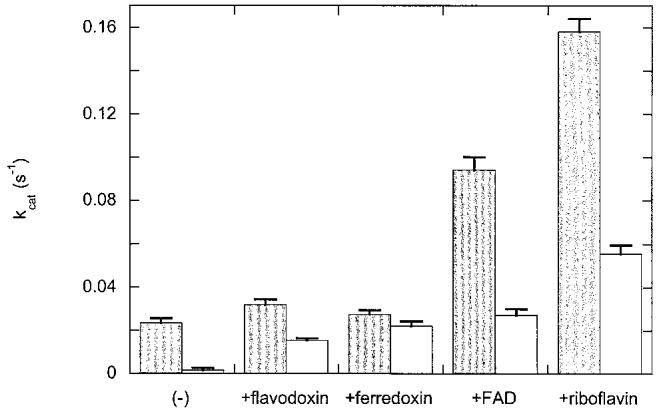
Inactivation of the flavodoxin reductase activity of FNR in the presence of various effectors. FNR (1 μM) alone and in the presence of flavodoxin, ferredoxin, FAD, or riboflavin (40 μM each) was reduced with NADPH (500 μM) in chilled 50 mM Bis-Tris Propane, 50 mM NaCl, pH 7.5, under argon. The initial flavodoxin reductase activity (dark bar) was determined by mixing with flavodoxin (100 μM) in a stopped-flow spectrophotometer at 37°C and following the formation of flavodoxin semiquinone at 580 nm. The samples were then incubated at 37°C for 30 min and the residual activity (light bar) was determined. Each bar represents the average of five trials.
Since changes in fluorescence indicate that FNR unfolding is accompanied by release of FAD, we postulated that if this process was initially reversible, then addition of FAD would also protect FNR against inactivation. When FAD (40 μM) is added to reduced FNR (1 μM), we observe that the flavodoxin reductase activity is initially increased ∼4-fold (kcat = 0.094 s-1). This is likely due to the very slow turnover of FNR with flavodoxin (kcat = 0.01-0.02 s-1), which allows free FAD to function as a mediator for transferring electrons from FNR to flavodoxin. After 30 min, much of this activity (71%) is lost, but the residual activity is still about the same as the initial activity in the absence of FAD. Although this might be interpreted as stabilization of FNR, we feel that it is more likely due to the ability of free FAD to act as a redox mediator that accepts electrons from FNR and donates them to flavodoxin. Flavodoxin is reduced slowly by FNR [3], and both FMN and riboflavin are reduced by FNR with second order rate constants that are more rapid than for flavodoxin alone [3], suggesting that low concentrations of free flavin could promote more rapid electron transfer between flavodoxin and FNR. To further demonstrate this was likely the case, we repeated the protection experiment with riboflavin, which lacks the adenine nucleotide and will not bind to FNR. We see a similar effect, with the initial activity (kcat = 0.158 s-1) increased seven-fold compared to FNR alone, while the residual activity after 30 min is decrease 65%. Thus the apparent protective effect of FAD or riboflavin is likely due not to a reversal of FNR unfolding, but is instead due to the ability of free flavin to act as a redox mediator and buffer for flavodoxin reduction.
4. Discussion
E. coli FNR is utilized in many laboratories as a general NADPH-dependent reductant of flavodoxin for sustaining the activity of E. coli-derived AdoMet-dependent radical enzymes [11,34,35], and for supplying electrons to heterologous cytochrome P450 enzymes [23,24]. These roles require prolonged stability of FNR. For example, biotin synthase utilizes electrons from reduced flavodoxin to reductively cleave AdoMet [36], generating a 5′-deoxyadenosyl radical that is proposed to be the oxidant of dethiobiotin [37]. Biotin synthase has been reported to turn over once in ∼30 min [25], and typical assays involve incubation under anaerobic conditions for 2-4 h [35]. Class III ribonucleotide reductase also catalyzes reductive cleavage of AdoMet to generate 5′-deoxyadenosine, in this case as an oxidant for a polypeptide glycine residue, generating a glycyl radical that is required for initiating ribonucleotide reduction [38]. This activation process also requires incubation for ∼30-60 min. In the case of biotin synthase, assay conditions typically include 0.1-1 μM FNR, 5-20 μM flavodoxin, and 0.5-1 mM NADPH. Depending upon the concentration of flavodoxin present, FNR will lose 50-90% of its original activity within 30 min (Fig. 4). In vivo this may be of little concern, since more enzyme is continuously generated to replace inactivated enzyme, but in a 4 h in vitro assay, active FNR may represent the limiting reagent after 30 min at 37°C.
The inactivation of FNR appears to be related to the thermal unfolding of the protein. Although the oxidized enzyme is quite stable, reduced FNR unfolds at temperatures above 35°C (Fig. 3). The extent of unfolding appears to correlate well with the effect of temperature on the rate of inactivation (Fig. 2B). The pH dependence of the inactivation rate suggests that deprotonation of a protein residue with a pKa of 8.5-9 contributes not only to the inactivation process, but potentially also contributes to a general increase in activity. Although there are numerous residues that could have pKas in this range, the structure of E. coli FNR [39] shows that Tyr247 is in a particularly important and vulnerable location. Tyr247 is coplanar and in hydrophobic contact with the flavin and makes hydrogen bonds to a backbone carbonyl and a tightly bound water that is in turn hydrogen bonded to the flavin C4 carbonyl. Thus, this residue is in a position that likely influences the reactivity of the flavin; indeed, Ingelman, et al., suggest that rotation or movement of this residue may be required prior to hydride transfer from NADPH [39]. Further, Tyr247 and Trp248 are the C-terminal residues in E. coli FNR, and contacts made by these residues may be essential in solidifying the C-terminal structure within the NADP-binding domain. E. coli FNR contains a novel two-stranded β-sheet extension formed by residues 232-239 in the NADP-binding domain near the C-terminus of the protein; this extension contains several positively charged residues and is proposed to form a portion of the flavodoxin-binding surface [39]. Thus we propose that C-terminal fraying of the E. coli FNR structure, which may be promoted by deprotonation of Tyr247, could be an initial step in global unfolding that leads to loss of activity.
This structural model may also explain one of the apparent complexities in our data: the reactivity of flavodoxin with FNR is lost at a much faster rate than the reactivity of ferredoxin or DCIP. This suggests that unfolding may be a stepwise process and that the region of the protein that interacts with flavodoxin may be more affected at lower temperature and earlier incubation time, while the core of FNR initially remains intact and active. As mentioned above, several residues that are proposed to contact flavodoxin are located within the C-terminal region of the NADP-binding domain [39]. In contrast, based upon the co-crystal structures of ferredoxins with homologous FNRs from Anabaena [40] and maize leaf [41], contacts between ferredoxin and FNR are predicted to be concentrated in the N-terminal FAD-binding domain. Thus the initial rapid loss of flavodoxin reductase activity may reflect a partial unfolding of the protein near the C-terminus within the NADP-binding domain, which may initially have little effect on the ferredoxin reductase activity. Eventually global unfolding and subsequent protein aggregation lead to loss of all activity, especially at elevated temperature and pH.
Our studies suggest several factors that could be useful in stabilizing FNR towards thermal inactivation. First, inactivation is clearly more rapid at elevated pH, but is relatively slow at pH 7. Further, since inactivation appears to linked to thermal denaturation, the rate of inactivation is significantly slowed at temperatures below 30°C. High concentrations of both ferredoxin and flavodoxin bind to FNR and stabilize the protein towards inactivation, thus it may be critical to keep reduced FNR in the presence of one of its partner proteins. Finally, since the flavodoxin reductase activity of FNR is more sensitive than the ferredoxin reductase activity, we would propose that wherever possible ferredoxin should be substituted for flavodoxin as an accessory protein in enzyme assays. Although clearly the high specificity of flavodoxin for some E. coli enzymes [16] may not allow this beneficial substitution, in situations where specificity is low, for instance electron transfer to heterologous enzymes such as cytochrome P450 reductases [24], ferredoxin may provide electrons with significantly higher turnover rates and sustained catalytic activity.
Acknowledgements
We would like to thank Dr. Marcos Milla (University of Pennsylvania) for the use of the Aviv spectrofluorometer. We thank Dr. Andrew Munro (University of Leicester) and Dr. Rowena Matthews (University of Michigan) for providing improved FNR expression strains. This work was supported in part by a Grant from NIGMS (GM59175).
Footnotes
- AdoMet
- S-adenosyl-l-methionine
- Bis-Tris Propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- DCIP
- 2,6-dichloroindophenol
- DTT
- dithiothreitol
- Fd
- ferredoxin
- Fld
- flavodoxin
- FNR
- ferredoxin (flavodoxin):NADP+ oxidoreductase
Edited by Vladimir Skulachev
References
- 1.Blaschkowski HP, Neuer G, Ludwig-Festl M, Knappe J. Eur. J. Biochem. 1982;123:563–569. [PubMed] [Google Scholar]
- 2.McIver L, Leadbeater C, Campopiano DJ, Baxter RL, Daff SN, Chapman SK, Munro AW. Eur. J. Biochem. 1998;257:577–585. doi: 10.1046/j.1432-1327.1998.2570577.x. [DOI] [PubMed] [Google Scholar]
- 3.Wan JT, Jarrett JT. Arch. Biochem. Biophys. 2002;406:116–126. doi: 10.1016/s0003-9861(02)00421-6. [DOI] [PubMed] [Google Scholar]
- 4.Fujii K, Huennekens FM. J. Biol. Chem. 1974;249:6745–6753. [PubMed] [Google Scholar]
- 5.Fujii K, Galivan JH, Huennekens FM. Arch. Biochem. Biophys. 1977;178:662–670. doi: 10.1016/0003-9861(77)90238-7. [DOI] [PubMed] [Google Scholar]
- 6.Jarrett JT, Hoover DM, Ludwig ML, Matthews RG. Biochemistry. 1998;37:12649–12658. doi: 10.1021/bi9808565. [DOI] [PubMed] [Google Scholar]
- 7.Wagner AF, Frey M, Neugebauer FA, Schafer W, Knappe J. Proc. Natl. Acad. Sci. USA. 1992;89:996–1000. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frey M, Rothe M, Wagner AF, Knappe J. J. Biol. Chem. 1994;269:12432–12437. [PubMed] [Google Scholar]
- 9.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, Reichard P. Biochem. Biophys. Res. Commun. 1993;197:792–797. doi: 10.1006/bbrc.1993.2548. [DOI] [PubMed] [Google Scholar]
- 10.Sun X, Ollagnier S, Schmidt PP, Atta M, Mulliez E, Lepape L, Eliasson R, Graslund A, Fontecave M, Reichard P, Sjoberg BM. J. Biol. Chem. 1996;271:6827–6831. [PubMed] [Google Scholar]
- 11.Mulliez E, Padovani D, Atta M, Alcouffe C, Fontecave M. Biochemistry. 2001;40:3730–3736. doi: 10.1021/bi001746c. [DOI] [PubMed] [Google Scholar]
- 12.Ifuku O, Koga N, Haze S, Kishimoto J, Wachi Y. Eur. J. Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 13.Guianvarc'h D, Florentin D, Tse Sum Bui B, Nunzi F, Marquet A. Biochem. Biophys. Res. Commun. 1997;236:402–406. doi: 10.1006/bbrc.1997.6952. [DOI] [PubMed] [Google Scholar]
- 14.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Marletta MA. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 15.Ta DT, Vickery LE. J. Biol. Chem. 1992;267:11120–11125. [PubMed] [Google Scholar]
- 16.Hall DA, Vander Kooi CW, Stasik CN, Stevens SY, Zuiderweg ER, Matthews RG. Proc. Natl. Acad. Sci. USA. 2001;98:9521–9526. doi: 10.1073/pnas.171168898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karplus PA, Bruns CM. J. Bioenerg. Biomembr. 1994;26:89–99. doi: 10.1007/BF00763221. [DOI] [PubMed] [Google Scholar]
- 18.Serre L, Vellieux FM, Medina M, Gomez-Moreno C, Fontecilla-Camps JC, Frey M. J. Mol. Biol. 1996;263:20–39. doi: 10.1006/jmbi.1996.0553. [DOI] [PubMed] [Google Scholar]
- 19.Hurley JK, Weber-Main AM, Stankovich MT, Benning MM, Thoden JB, Vanhooke JL, Holden HM, Chae YK, Xia B, Cheng H, Markley JL, Martinez-Julvez M, Gomez-Moreno C, Schmeits JL, Tollin G. Biochemistry. 1997;36:11100–11117. doi: 10.1021/bi9709001. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Proc. Natl. Acad. Sci. USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 22.Leclerc D, Wilson A, Dumas R, Gafuik C, Song D, Watkins D, Heng HH, Rommens JM, Scherer SW, Rosenblatt DS, Gravel RA. Proc. Natl. Acad. Sci. USA. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jenkins CM, Waterman MR. J. Biol. Chem. 1994;269:27401–27408. [PubMed] [Google Scholar]
- 24.Jenkins CM, Waterman MR. Biochemistry. 1998;37:6106–6113. doi: 10.1021/bi973076p. [DOI] [PubMed] [Google Scholar]
- 25.Ugulava NB, Sacanell CJ, Jarrett JT. Biochemistry. 2001;40:8352–8358. doi: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frey PA, Booker SJ. Adv. Protein Chem. 2001;58:1–45. doi: 10.1016/s0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- 27.Cheek J, Broderick JB. J. Biol. Inorg. Chem. 2001;6:209–226. doi: 10.1007/s007750100210. [DOI] [PubMed] [Google Scholar]
- 28.Hoover DM, Ludwig ML. Protein Sci. 1997;6:2525–2537. doi: 10.1002/pro.5560061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoover DM, Jarrett JT, Sands RH, Dunham WR, Ludwig ML, Matthews RG. Biochemistry. 1997;36:127–138. doi: 10.1021/bi961693s. [DOI] [PubMed] [Google Scholar]
- 30.Bianchi V, Reichard P, Eliasson R, Pontis E, Krook M, Jornvall H, Haggard-Ljungquist E. J. Bacteriol. 1993;175:1590–1595. doi: 10.1128/jb.175.6.1590-1595.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patil PV, Ballou DP. Anal. Biochem. 2000;286:187–192. doi: 10.1006/abio.2000.4802. [DOI] [PubMed] [Google Scholar]
- 32.Dawson RMC, Elliot DC, Elliot WH, Jones KM. Data for Biochemical Research. Oxford University Press; Oxford: 1986. [Google Scholar]
- 33.Fersht A. Structure and Mechanism in Protein Science. W. H. Freeman and Co.; New York: 1999. pp. 509–513. [Google Scholar]
- 34.Birch OM, Fuhrmann M, Shaw NM. J. Biol. Chem. 1995;270:19158–19165. doi: 10.1074/jbc.270.32.19158. [DOI] [PubMed] [Google Scholar]
- 35.Sanyal I, Gibson KJ, Flint DH. Arch. Biochem. Biophys. 1996;326:48–56. doi: 10.1006/abbi.1996.0045. [DOI] [PubMed] [Google Scholar]
- 36.Ollagnier-de Choudens S, Sanakis Y, Hewitson KS, Roach P, Munck E, Fontecave M. J. Biol. Chem. 2002;277:13449–13454. doi: 10.1074/jbc.M111324200. [DOI] [PubMed] [Google Scholar]
- 37.Escalettes F, Florentin D, Tse Sum Bui B, Lesage D, Marquet A. J. Am. Chem. Soc. 1999;121:3571–3578. [Google Scholar]
- 38.Padovani D, Thomas F, Trautwein AX, Mulliez E, Fontecave M. Biochemistry. 2001;40:6713–6719. doi: 10.1021/bi002936q. [DOI] [PubMed] [Google Scholar]
- 39.Ingelman M, Bianchi V, Eklund H. J. Mol. Biol. 1997;268:147–157. doi: 10.1006/jmbi.1997.0957. [DOI] [PubMed] [Google Scholar]
- 40.Morales R, Charon MH, Kachalova G, Serre L, Medina M, Gomez-Moreno C, Frey M. EMBO Rep. 2000;1:271–276. doi: 10.1093/embo-reports/kvd057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurisu G, Kusunoki M, Katoh E, Yamazaki T, Teshima K, Onda Y, Kimata-Ariga Y, Hase T. Nat. Struct. Biol. 2001;8:117–121. doi: 10.1038/84097. [DOI] [PubMed] [Google Scholar]