

Abstract
The contribution of the aryl hydrocarbon receptor (AhR) in induction of a battery of xenobiotic-metabolizing enzymes has been studied extensively. However, no direct proof has been obtained that it plays a role in modulating carcinogenesis. To address the question of whether AhR is required for tumor induction, we have investigated the response of AhR-deficient mice to benzo[a]pyrene (B[a]P), a widely distributed environmental carcinogen. B[a]P treatment induced expression of the cytochrome P450 gene Cyp1a1 in the skin and liver of AhR-positive mice bearing +/+ and +/− genotypes and did not induce expression of the cytochrome P450 gene Cyp1a1 in AhR-null mice in either skin or liver. In contrast, Cyp1a2 gene expression was positive in liver irrespective of the presence or absence of the AhR gene, or B[a]P treatment, although its inducibility was lost in the AhR(−/−) mouse. All AhR-positive male mice of both +/+ and +/− genotypes that received subcutaneous injection of B[a]P (2 mg) on the first and the eighth days had developed subcutaneous tumors at the site of injection at the end of the 18-week experiment. In contrast, no tumors were apparent in any of the AhR-deficient mice. Likewise, topical application of B[a]P (200 μg) at weekly intervals to the skin of female mice for 25 weeks produced skin tumors only in the AhR-positive mice. Thus the carcinogenic action of B[a]P may be determined primarily by AhR, a transcriptional regulator of the gene for CYP1A1. The results of the present study provide direct evidence that AhR is involved in carcinogenesis.
The aryl hydrocarbon receptor (AhR) is the ligand-binding subunit of a dimeric ligand-activated transcription factor that plays critical roles in embryonic development and maintenance of homeostasis, responding to xenobiotic compounds or other stimuli by modifying cell shapes and also influencing the extracellular matrix interactions with other cells. With hazardous environmental chemicals such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and benzo[a]pyrene (B[a]P), the AhR is considered to mediate teratogenic and carcinogenic effects. Its functions have been investigated most thoroughly with regard to induction of a battery of xenobiotic-metabolizing enzymes. On ligand binding the AhR stimulates expression of several genes, including those encoding cytochrome P4501A1-, P4501A2-, and P4501B1-dependent monooxygenases, glutathione S-transferase Ya subunit, quinone oxidoreductase, and UDP-glucuronosyltransferase (1–6). To further explore AhR function, the murine AhR gene has been cloned (7–9), and AhR-deficient mice have been generated by homologous recombination (10–12). Their use in previous studies clearly demonstrated that the AhR is involved in TCDD-induced toxicity in the thymus and liver of adult animals (13) and also in teratogenic effects of TCDD on palate and kidney development (12). However, its contribution to carcinogenesis has remained unclear. B[a]P, a constituent of tobacco smoke and automobile exhaust, exerts potent carcinogenic activity in several animal species, and human beings are no exception (14, 15). Cytochrome P450-dependent metabolic activation appears to be required, and in vitro studies have revealed that benzo[a]pyrene-7,8-diol-9,10-epoxide (BPDE), the ultimate metabolite, forms DNA adducts and thus acts as a mutagen (16). In the first step of metabolism of B[a]P to BPDE, B[a]P binds to AhR and the liganded AhR translocates to nuclei, where it forms a heterodimer with AhR-nuclear translocator (Arnt).
This complex then binds to the xenobiotic-responsive element (XRE) in the upstream region of the Cyp1a1 gene, inducing its expression (17–19). The induced CYP1A1 and cytochrome P450 reductase act on B[a]P to generate two dozen metabolites, including BPDE. However, there is no direct proof that the AhR plays a role in vivo as a mediator of carcinogenesis. Therefore, in the present study we analyzed the response of AhR-deficient mice to skin tumorigenesis induced by long-term treatment with B[a]P.
Materials and Methods
Animals.
AhR-deficient mice were generated by using a homologous recombination method as previously described (12). Briefly, a genomic DNA fragment containing the 5′-flanking region to the second intron of the AhR gene was isolated and fused in-frame with the lacZ cassette (HindIII-SmaI fragment of pENL) containing the simian virus 40 T antigen intron and polyadenylation signal (20) so that β-galactosidase expression could mimic the mode of AhR expression in the homologous recombinant. The construct was introduced into 129/SV mouse-derived embryonic stem cells, which were then subjected to double selection with G418 and ganciclovir. Clones with a legitimate homologous recombination were isolated and microinjected into C57BL/6J blastocysts to give rise to chimeric animals, as judged from coat color. Male AhR(−/−) mice were back-crossed to C57BL/6J AhR(+/+) females to give rise to heterozygotes. The AhR(+/−) mice were interbred to yield AhR(+/+), AhR(+/−), and AhR(−/−) mice. Among 100 offspring obtained from heterozygous matings, the relative frequencies of AhR(+/+), AhR(+/−), and AhR(−/−) mice were approximately 1:2:1, as expected from Mendelian law. The genotype of each pup was determined by analyzing the presence of the mutant AhR allele by the polymerase chain reaction (PCR) with genomic DNA taken from the tail. The sense primer for wild-type amplification targeted the 5′ region of the AhR gene: 5′-CGCGGGCACCATGAGCAG-3′. The antisense primer for intron 1 was 5′-GAGACTCAGCTCCTGGATGG-3′. The same 5′ primer was used for the amplification of the mutant type, and the antisense primer for the lacZ gene was 5′-CGCCGAGTTAACGCCATCAA-3′. PCR amplification was carried out for 35 cycles under the following conditions; 94°C for 30 sec, 60°C for 60 sec, and 72°C for 10 sec. The PCR products were electrophoresed on 1.5% agarose gels in 1× TBE (90 mM Tris/90 mM boric acid/2.0 mM EDTA, pH 8.0) buffer and visualized by ethidium bromide staining.
Chemicals.
B[a]P was obtained from Daiichi Pure Chemicals (Tokyo), and solutions for treatment were prepared immediately before use as follows. For topical application, 10 mg of B[a]P was dissolved in 10 ml of acetone. For subcutaneous injection the B[a]P was ground in an agate mortar and suspended in olive oil at a concentration of 10 mg/ml.
Tumor Induction Study.
Mice were housed in steel cages in a specific-pathogen-free facility with a filtered-air environment, controlled for temperature, humidity, and day and night cycles. Sterilized rodent chow (NMF, Oriental Yeast, Tokyo) and autoclaved water were provided ad libitum. The right flank skin of male mice 15 weeks of age was shaved and 2 mg of B[a]P in 200 μl of olive oil was subcutaneously injected on the 1st and the 8th day of the experiment. Groups of female mice 15 weeks of age were treated on an area of shaved dorsal skin with 200 μg of B[a]P dissolved in acetone once a week. The treatment was repeated for 25 weeks. All the animals were weighed once per week and were examined for development of neoplastic lesions at least once per week. The numbers of palpable firm nodules larger than 5 mm in diameter appearing on the flanks of male mice were recorded throughout the experimental period. In female mice, numbers of skin papillomatous lesions larger than 2 mm in size were counted. Male animals were sacrificed in the 18th week and females in the 28th week. Tumors were dissected out, fixed in neutralized 10% formalin, embedded in paraffin, sectioned at 5-μm thickness, stained with hematoxylin/eosin, and examined microscopically. The B[a]P-treated skin region of non-tumor-bearing mice was also dissected out and was examined microscopically.
Isolation and Analysis of RNA.
Normal and AhR-deficient male mice were injected subcutaneously with 2 mg of B[a]P, and 3 days thereafter subcutaneous tissues from the B[a]P-treated flank region and liver tissue were dissected out. Total RNA was extracted from these tissues by using an Isogen kit (Nippon Gene, Tokyo), and 1-μg aliquots were reverse-transcribed to cDNA with Superscript-II reverse transcriptase (Life Technologies, Gaithersburg, MD) and random hexamers at 37°C for 1 hr. The resulting cDNAs were subjected to 20 and 22 cycles of PCR using the specific primers for the genes for AhR (5′ primer, 5′-CGCGGGCACCATGAGCAG-3′; 3′ primer, 5′-CTGTAACAAGAACTCTCC-3′), CYP1A1 (5′ primer, 5′-CAGATGATAAGGTCATCACGA-3′; 3′ primer, 5′-TTGGGGATATAGAAGCCATTC-3′), CYP1A2 (5′ primer, 5′-CAGTATCCAAGACATCACAAG-3′; 3′ primer, 5′-TGTGTATCGGTAGATCTCCAG-3′), and β-actin (5′ primer, 5′-ATGGATGACGATATCGCT-3′; 3′ primer, 5′-ATGAGGTAGTCTGTCAGGT-3′). The reaction was performed under the following conditions: 94°C for 45 sec, 55°C for 45 sec, and 72°C for 2 min. The resultant products were analyzed by electrophoresis on agarose gels.
Results
AhR-null mice appeared normal at birth, but their growth was slightly slower than that of wild-type mice during the first few weeks of life. Thereafter they caught up and no difference was apparent in the animals over 12 weeks of age used for the experiment. AhR-heterozygous (AhR(+/−)) mice showed no significant difference in growth rate and in appearance compared with wild-type mice. Reverse transcription (RT)-PCR analysis of the expression of the gene for AhR in the skin and livers of AhR +/+, +/−, and −/− mice demonstrated a lack of the expression in AhR(−/−) mice, consistent with the gene loss (see Fig. 1). The basal level of Cyp1a1 gene expression was very low; RT-PCR experiments with 2 μg of tissue RNA and 45 cycles of amplification failed to detect any specific mRNA signals. In the two groups of AhR-positive animals, +/+ and +/−, however, subcutaneous injection of B[a]P induced expression of Cyp1a1 in both skin and liver. Essentially no expression was observed in the AhR-null mice. Expression of the Cyp1a2 gene was not observed in the skin, but it was constitutive even in the liver of AhR-null mice. A slightly enhanced expression of Cyp1a2 was observed in AhR(+/+) and AhR(+/−) mice in response to B[a]P treatment, in line with its constitutive gene regulation.
Figure 1.

Cyp1a1, Cyp1a2, and AhR gene expression in the skin and liver of AhR(+/+), AhR(+/−), and AhR(−/−) mice, with and without B[a]P treatment. One-microgram aliquots of RNA extracted from skin and liver of control and B[a]P-treated mice of the three genotypes were reverse-transcribed and analyzed by PCR using specific primers for the Cyp1a1, Cyp1a2, and AhR and β-actin genes.
No significant differences in body weight change of male and female mice, among the three groups of AhR(+/+), AhR(+/−), and AhR(−/−) mice were observed during the experimental period.
In B[a]P-treated males, the first subcutaneous tumor was identified 12 weeks after the treatment. The subsequent time course of subcutaneous tumor development in the mice is shown in Fig. 2. By the end of the 17th week after the first treatment, all AhR-positive mice, both +/+ and +/−, bore tumors (17/17 and 17/17). All tumors were solitary. In contrast, no tumors were found in AhR-null mice throughout the experiment (0/16). Statistically, using a χ2 test with Yates' correction, there was significant difference in tumor incidence between AhR(+/+) and AhR(−/−) mice (P < 0.01). There was also significant difference in tumor incidence between AhR(+/−) and AhR(−/−) mice (P < 0.01). Close microscopic examination of the non-tumor-bearing skin from B[a]P-treated AhR-null mice also revealed no signs of neoplastic change. The right flanks of mice treated by subcutaneous injections for 17 weeks are illustrated in Fig. 3. Data for numbers and classification of the tumors are summarized in Table 1. Histologically most of them were fibrosarcomas, while the others were 3 squamous cell carcinomas and 3 rhabdomyosarcomas. There was no notable difference in the incidence or histology between the AhR +/+ and +/− genotypes. The histological appearance of a representative fibrosarcoma, rhabdomyosarcoma, and squamous cell carcinoma is shown in Fig. 4 A, B, and C, respectively. All the neoplastic lesions tested formed tumors when transplanted into the back skin of nude mice, indicating their neoplastic character.
Figure 2.

Subcutaneous tumor induction in wild-type (▵) and AhR-deficient male mice (+/−, □; −/−, ○) injected with B[a]P.
Figure 3.
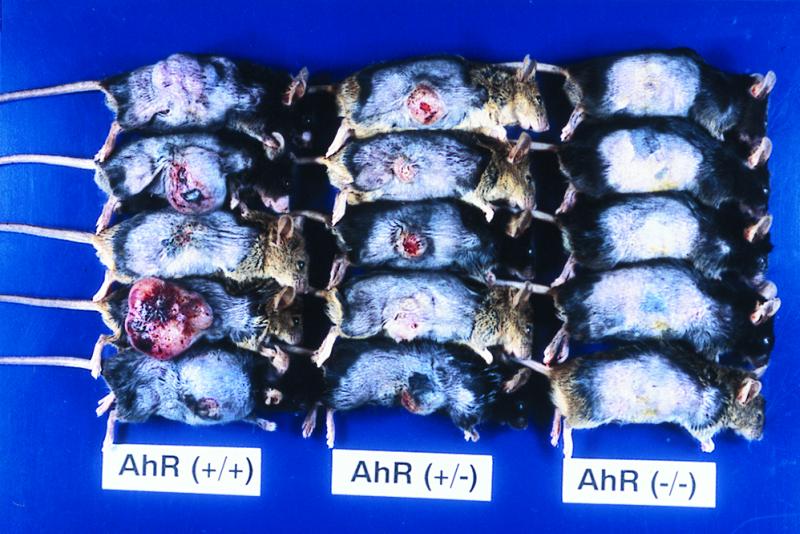
Gross appearance of flank skins in AhR-wild-type mice (+/+), AhR-heterozygous mice (+/−), and AhR-deficient mice (−/−) injected subcutaneously with B[a]P.
Table 1.
Incidence and histology of the tumors induced in three genotypes of mice subcutaneously injected with B[a]P
| Tumor | No. of tumor-bearing mice with AhR genotype
|
||
|---|---|---|---|
| +/+ | +/− | −/− | |
| Fibrosarcoma | 13 | 15 | 0 |
| Rhabdomyosarcoma | 2 | 1 | 0 |
| Squamous cell carcinoma | 2 | 1 | 0 |
| Total no. (%) | 17/17 (100%)* | 17/17 (100%)* | 0/16 (0%) |
*Significant difference in comparison with AhR(−/−) mice (P < 0.01).
Figure 4.
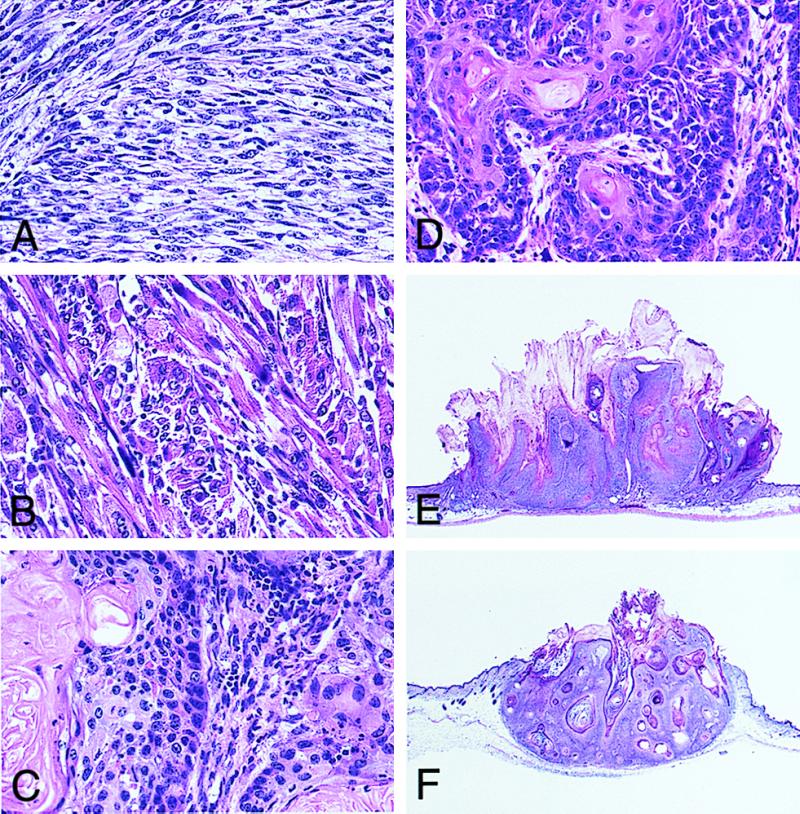
(A–C) Histological appearance of a fibrosarcoma (A), a rhabdomyosarcoma (B), and a squamous cell carcinoma (C) induced in AhR(+/+) male mice by subcutaneous injection of B[a]P. (Hematoxylin/eosin staining, ×100.) (D–F) Histological appearance of a squamous cell carcinoma (D), papilloma (E), and keratoacanthoma (F) induced in AhR(+/+) female mice by topical application of B[a]P. (Hematoxylin/eosin staining, ×100 for D, ×5 for E and F.)
Tumor formation in the skin of the female mice was observed from around the 16th week after the first application of the carcinogen. Sequential data for incidence are shown in Fig. 5. The majority of AhR-positive mice bore tumors: 15 of 16 AhR(+/+) and 13 of 14 AhR (+/−) mice. Tumors were multiple, and the average number of tumors per mouse is shown in Table 2. The histological classification of tumors in Table 2 is that of the largest tumor in each mouse. The backs of mice treated with topical application after 27 weeks are illustrated in Fig. 6. Histologically, the tumors were mainly squamous cell carcinomas (SCC), with papillomas and keratoacanthomas observed in some cases (Table 2). Representative lesions are shown in Fig. 4 D, E, and F, respectively. In the AhR(−/−) mice, tumor formation was completely suppressed and no neoplastic lesion was evident during the experimental period of 27 weeks. Statistically, using χ2 test with Yates' correction, there was significant difference in tumor incidence (percentage of mice with tumor) between AhR(+/+) and AhR(−/−) mice (P < 0.01). There was also significant difference in tumor incidence between AhR(+/−) and AhR(−/−) mice (P < 0.01).
Figure 5.

Skin tumor induction in AhR-wild-type (+/+) (▵) and AhR-deficient female mice (+/−, □; −/−, ○) by repeated topical application of B[a]P.
Table 2.
Incidence and histology of the tumors induced in three genotypes of mice repeatedly painted with B[a]P
| Tumor | No. with AhR genotype
|
||
|---|---|---|---|
| +/+ | +/− | −/− | |
| Squamous cell carcinoma | 13 | 12 | 0 |
| Papilloma | 1 | 1 | 0 |
| Keratoacanthoma | 1 | 0 | 0 |
| Average no. of tumors per mouse ± SD | 4.2 ± 1.9 | 4.6 ± 2.4 | — |
| Total no. of tumor-bearing mice (%) | 15/16 (93.8%)* | 13/14 (92.4%)* | 0/10 (0%) |
*Significant difference in comparison with AhR(−/−) mice (P < 0.01).
Figure 6.
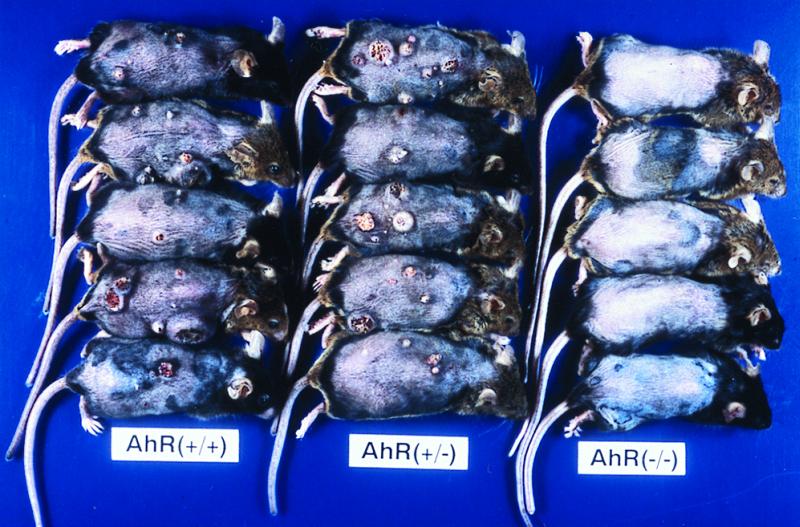
Gross appearance of skin tumors in AhR-deficient (−/−), AhR-heterozygous (+/−), and AhR-wild-type (+/+) female mice after repeated topical application of B[a]P.
For comparison, 4-nitroquinoline 1-oxide, which is known to be metabolically activated by a quite different pathway (21, 22), was repeatedly applied to the skin of AhR(+/+), AhR(+/−), and AhR(−/−) mice. Tumors (papillomas and squamous cell carcinomas) developed equally in AhR(−/−), AhR(+/−), and AhR(+/+) mice (data not shown).
Discussion
AhR-deficient mice have now been generated independently by three different laboratories (10–12), and they manifested somewhat different phenotypes. In the earliest report, Fernandez-Salguero et al. (10) described that half of the deficient mice died shortly after birth, whereas survivors reached maturity and were fertile. In contrast, all of our AhR-null mice appeared healthy at birth and had no obvious phenotypic abnormalities. Although their growth rate was slightly decreased as compared with that of wild-type mice, they survived more than 1 year and were fertile.
In the AhR-positive mice, +/+ and +/− in genotype, B[a]P treatment induced Cyp1a1 gene expression. However, expression of this gene was completely lacking in AhR-null mice, indicating AhR-mediated regulation of the Cyp1a1 gene. In contrast, while inducibility of Cyp1a2 gene expression in response to B[a]P was lost in the AhR-null mouse, the basal expression of Cyp1a2 remained unchanged, consistent with the previous results (12, 23).
The present investigation of skin tumorigenesis by B[a]P treatment, either as subcutaneous injections or as topical application to the skin, revealed tumor induction solely in AhR-positive animals with both methods. The marked difference in the tumor incidence provides strong support for the hypothesis that the carcinogenic action of B[a]P is mediated primarily by AhR.
B[a]P is known to exert its carcinogenic activity by reacting covalently with DNA. In the metabolic activation of B[a]P, three different pathways have been proposed (16). The most widely accepted involves ring oxidation to diol epoxides catalyzed by the monooxygenase, product of the Cyp1a1 gene. While Stansbury et al. (16) have pointed to the existence of a benzylic ester activation mechanism for polycyclic aromatic hydrocarbons, neither topical nor subcutaneous administration was carcinogenic in our AhR-negative animals. Therefore, it is most probable that the AhR plays an essential role in the induction of CYP1A1 and conversion of B[a]P into its active form.
In conclusion, the present study provides direct evidence that AhR is involved in carcinogenesis by B[a]P, a widely distributed environmental carcinogen.
Acknowledgments
This work was supported in part by Core Research for Evolutional Science and Technology, Japan Science and Technology, and Grants-in-Aid for Scientific Research on Special Areas from the Ministry of Education, Science, Sports and Culture of Japan and the Ministry of Health and Welfare of Japan and by a grant from the Smoking Research Foundation.
Abbreviations
- AhR
aryl hydrocarbon receptor
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- B[a]P
benzo[a]pyrene
- BPDE
benzo[a]pyrene-7,8-diol-9,10-epoxide
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jones P B C, Galeazzi D R, Fisher J M, Whitlock J P., Jr Science. 1985;227:1499–1502. doi: 10.1126/science.3856321. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez F J, Tukey R H, Nebert D W. Mol Pharmacol. 1984;26:117–121. [PubMed] [Google Scholar]
- 3.Sutter T R, Tang Y M, Hayes C L, Wo Y-Y P, Jabs E W, Li X, Yin H, Cody C W, Greenlee W F. J Biol Chem. 1994;269:13092–13099. [PubMed] [Google Scholar]
- 4.Telakowski-Hopkins C A, Hing R G, Pickett C B. Proc Natl Acad Sci USA. 1988;85:1000–1004. doi: 10.1073/pnas.85.4.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Favreau L V, Pickett C B. J Biol Chem. 1991;266:4556–4561. [PubMed] [Google Scholar]
- 6.Lamb J G, Straub P, Tukey R H. Biochemistry. 1994;33:10513–10520. doi: 10.1021/bi00200a037. [DOI] [PubMed] [Google Scholar]
- 7.Ema M, Sogawa K, Watanabe N, Chujoh Y, Matsushita N, Gotoh O, Funae Y, Fujii-Kuriyama Y. Biochem Biophys Res Commun. 1992;184:246–253. doi: 10.1016/0006-291x(92)91185-s. [DOI] [PubMed] [Google Scholar]
- 8.Burbach K M, Poland A, Bradfield C A. Proc Natl Acad Sci USA. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mimura J, Ema M, Sogawa K, Ikawa S, Fujii-Kuriyama Y. Pharmacogenetics. 1994;4:349–354. doi: 10.1097/00008571-199412000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Salguero P, Pineau T, Hilbert D M, McPhail T, Lee S S T, Kimura S, Nebert D W, Rudikoff S, Ward J M, Gonzalez F J. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt J V, Su G H-T, Reddy J K, Simon M C. Proc Natl Acad Sci USA. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mimura J, Yamashita K, Nakamura K, Morita M, Takagi T N, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M, Fujii-Kuriyama Y. Genes Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Salguero P M, Hilbert D M, Rudikoff S, Ward J M, Gonzalez F J. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 14.Phillips D H. Nature (London) 1983;303:468–472. doi: 10.1038/303468a0. [DOI] [PubMed] [Google Scholar]
- 15.Working Group in Lyon. IARC Monograph. 1973;3:91–136. [Google Scholar]
- 16.Stansbury K H, Flesher J W, Gupta R C. Chem Res Toxicol. 1994;7:254–259. doi: 10.1021/tx00038a019. [DOI] [PubMed] [Google Scholar]
- 17.Swanson H I, Bradfield C A. Pharmacogenetics. 1993;3:213–230. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Fujii-Kuriyama Y, Ema M, Mimura J, Sogawa K. Exp Clin Immunogenet. 1994;11:65–74. doi: 10.1159/000424195. [DOI] [PubMed] [Google Scholar]
- 19.Okey A B, Riddick D S, Harper P A. Trends Pharmacol Sci. 1994;15:226–232. doi: 10.1016/0165-6147(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 20.Gondo Y, Nakamura K, Nakao K, Sasaoka T, Ito K, Kimura M, Katsuki M. Biochem Biophys Res Commun. 1994;202:830–837. doi: 10.1006/bbrc.1994.2005. [DOI] [PubMed] [Google Scholar]
- 21.Tada M, Tada M. Nature (London) 1975;255:510–512. doi: 10.1038/255510a0. [DOI] [PubMed] [Google Scholar]
- 22.Galiegue-Zouitina S, Bailleul B, Loucheux-Lefebvre M H. Cancer Res. 1985;45:520–525. [PubMed] [Google Scholar]
- 23.Zaher H, Yang T J, Gelboin H V, Fernandez-Salguero P, Gonzalez F J. Biochem Pharmacol. 1998;55:235–238. doi: 10.1016/s0006-2952(97)00476-0. [DOI] [PubMed] [Google Scholar]