

Abstract
Biotin synthase (BS) is an AdoMet-dependent radical enzyme that catalyzes the insertion of sulfur into saturated C6 and C9 atoms in the substrate dethiobiotin. To facilitate sulfur insertion, BS catalyzes the reductive cleavage of AdoMet to methionine and 5′-deoxyadenosyl radicals, which then abstract hydrogen atoms from the C6 and C9 positions of dethiobiotin. The enzyme from Escherichia coli is purified as a dimer that contains one [2Fe-2S]2+ cluster per monomer and can be reconstituted in vitro to contain an additional [4Fe-4S]2+ cluster per monomer. Since each monomer contains each type of cluster, the dimeric enzyme could contain one active site per monomer, or could contain a single active site at the dimer interface. To address these possibilities, and to better understand the manner in which biotin synthase controls radical generation and reactivity, we have examined the binding of AdoMet and DTB to reconstituted biotin synthase. We find that both the [2Fe-2S]2+ cluster and the [4Fe-4S]2+ cluster must be present for tight substrate binding. Further, substrate binding is highly cooperative, with the affinity for AdoMet increasing >20-fold in the presence of DTB, while DTB binds only in the presence of AdoMet. The stoichiometry of binding is ca. 2:1:1 AdoMet:DTB:BS dimer, suggesting that biotin synthase has a single functional active site per dimer. AdoMet binding, either in the presence or in the absence of DTB, leads to a decrease in the magnitude of the UV-visible absorption band at ∼400 nm that we attribute to changes in the coordination environment of the [4Fe-4S]2+ cluster. Using these spectral changes as a probe, we have examined the kinetics of AdoMet and DTB binding, and propose an ordered binding mechanism that is followed by a conformational change in the enzyme-substrate complex. This kinetic analysis suggests that biotin synthase is evolved to bind AdoMet both weakly and slowly in the absence of DTB, while both the rate of binding and the affinity for AdoMet are increased in the presence of DTB. Cooperative binding of AdoMet and DTB may be an important mechanism for limiting the production of 5′-deoxyadenosyl radicals in the absence of the correct substrate.
Biotin synthase (BS)1 catalyzes formation of the thioether ring in the essential vitamin biotin (for reviews, see refs 1 and 2). Thioether ring formation requires substitution of sulfur for hydrogen atoms at the saturated C6 and C9 positions of the substrate dethiobiotin. Functionalization of these relatively unreactive carbon atoms in dethiobiotin is likely initiated by hydrogen atom abstraction (3), generating carbon radicals at one or both positions. Biotin synthase is purified from Escherichia coli as a dimeric iron-sulfur protein (4) that requires S-adenosyl-l-methionine (AdoMet) and reduced flavodoxin for maximal activity (4-8), suggesting that biotin synthase is an AdoMet-dependent radical enzyme that couples reductive cleavage of AdoMet to hydrogen atom abstraction from the substrate (3). Consistent with this assignment, biotin formation is accompanied by production of the AdoMet reductive cleavage products 5′-deoxyadenosine and methionine (9, 10).
AdoMet-dependent radical enzymes comprise a newly identified protein superfamily (11), the members of which catalyze the formation of protein and/or substrate radicals through reductive cleavage of AdoMet (12, 13); the growing list of confirmed superfamily members includes biotin synthase as well as pyruvate formate-lyase (14), class III ribonucleotide reductase (15), lysine 2,3-aminomutase (16), benzylsuccinate synthase (17), lipoyl synthase (18), and spore photoproduct lyase (19). Minimally, these enzymes share a common iron-sulfur cluster binding motif, CxxxCxxC, that binds a [4Fe-4S]2+ cluster under anaerobic conditions (20-22). The [4Fe-4S]2+ cluster interacts directly with bound AdoMet (23-25), and electron transfer into the AdoMet-[4Fe4S]2+ complex leads to reductive cleavage of AdoMet, generating methionine and a transient 5′-deoxyadenosyl radical. In all AdoMet-dependent radical enzymes, this high-energy radical then initiates catalysis by abstraction of a hydrogen atom either directly from the substrate (3, 26) or from an intermediate protein residue (14, 27).
Sulfur insertion between the C6 and C9 positions of dethiobiotin requires abstraction of one hydrogen atom from each of these positions. Studies by Marquet and co-workers have shown that formation of substoichiometric amounts of biotin (∼0.3 equiv of biotin per BioB monomer) is accompanied by production of ∼3 equiv of 5′-deoxyadenosine and methionine (9), and they have proposed that AdoMet is a stoichiometric oxidant of dethiobiotin. When dethiobiotin is labeled with deuterium at either C6 or C9, the 5′-deoxyadenosine that is produced contains ∼50% of the initial deuterium (3). Together, these data suggest that biotin synthase sequentially catalyzes the direct abstraction of a hydrogen atom by transient 5′-deoxyadenosyl radicals from both the C6 and C9 positions in dethiobiotin (3). Biotin synthase will also catalyze reductive cleavage of AdoMet in the absence of dethiobiotin (28). When aerobically purified biotin synthase is reconstituted to contain one [4Fe-4S]2+ cluster per monomer, photoreduced to a [4Fe-4S]+ cluster with deazaflavin, and then incubated with AdoMet, 5′-deoxyadenosine and methionine are produced in a slow reaction that is no longer coupled to biotin formation (28). Reductive cleavage of AdoMet in the absence of substrate will generate a high-energy 5′-deoxyadenosyl radical that likely quenches by abstracting a hydrogen atom from either the protein or a buffer component. Uncoupled 5′-deoxyadenosyl radical generation is thus a potentially damaging side reaction that could lead to oxidative damage comparable to that caused by hydroxyl radicals, and minimizing this reaction in vivo is likely an evolutionary advantage.
If the minimization of uncoupled 5′-deoxyadenosyl radical generation is an evolved feature of AdoMet-dependent radical enzymes, then these enzymes should incorporate structural and/or mechanistic features that can increase the rate of AdoMet cleavage in the presence of the proper second substrate. In biotin synthase, the simplest mechanism for promoting coupled radical generation under in vivo conditions would be to couple the binding of AdoMet and dethiobiotin. We have investigated the binding of both substrates to active and inactive cluster forms of biotin synthase, and find that DTB binds only in the presence of AdoMet; the stoichiometry of binding is ca. 2:1:1 AdoMet:DTB:biotin synthase dimer. In contrast, AdoMet alone will bind weakly to biotin synthase, but the affinity and stoichiometry of binding are greatly improved in the presence of DTB. High-affinity binding of both DTB and AdoMet requires the presence of both [2Fe-2S]2+ and [4Fe4S]2+ clusters. We have observed that AdoMet binding is associated with a slight decrease in the magnitude of the UV-visible absorption band associated with the [4Fe-4S]2+ cluster, and have used this spectral perturbation to investigate the kinetics of substrate binding using a stopped-flow spectrophotometer. AdoMet binding proceeds with two kinetic phases whose rate constants and amplitudes depend on the concentrations of both AdoMet and DTB; we propose a complex ordered binding mechanism that can account for the observed kinetics. Finally, as further evidence of a tightly bound enzyme-substrate complex, we observe that substrate binding results in dramatic stabilization of the [4Fe-4S]2+ cluster to oxidative degradation following exposure to air. We propose that under physiologic concentrations, tight stoichiometric AdoMet binding requires concurrent binding of dethiobiotin, thus decreasing the level of uncoupled radical generation by minimizing productive binding of AdoMet alone.
MATERIALS AND METHODS
Materials. Unless otherwise stated, all reagents were obtained from commercial sources and used without further purification. N-Terminally histidine-tagged BS was purified as previously described (29) and was used for all of the experiments described in this paper. Unless otherwise stated, all protein purification steps were performed under aerobic conditions and all protein reduction, reconstitution, and analyses were performed under an oxygen-free argon or nitrogen atmosphere. For aerobically purified BS, the protein concentration was determined immediately after purification using an ε452 of 8400 M-1 cm-1 per monomer (30), and all protein concentrations are given as the monomer concentration. For other samples, the protein concentration was determined using the Bradford assay with BSA as a standard; the resulting values were divided by a correction factor of 1.10, determined by quantitative amino acid analysis (30).
Reconstitution of [4Fe-4S]2+ Clusters in BioB. Aerobically purified BS contains only [2Fe-2S]2+ clusters. Protein samples were generated that contained [4Fe-4S]2+ clusters by three different methods. BS containing approximately one [2Fe-2S]2+ and one [4Fe-4S]2+ cluster per monomer was generated by incubating the protein with FeCl3, Na2S, and DTT in 50 mM Tris (pH 8) as previously described (30). BS containing approximately one [4Fe-4S]2+ cluster, with no residual [2Fe-2S]2+ clusters, was generated by the method of Johnson and co-workers (22): BS initially containing [2Fe-2S]2+ clusters was incubated with sodium dithionite in 60% ethylene glycol and 50 mM Tris (pH 8) for 4 h. BS containing primarily [4Fe-4S]2+ clusters was also generated by a modification of the method of Fontecave and co-workers (28): BS was stripped of most of the initial [2Fe-2S]2+ clusters by incubation with sodium dithionite (2 mM), methyl viologen (5 μM), and EDTA (100 mM) for 6 h, and then desalted by being passed through a Sephadex G25 column to yield the apoprotein. This apoprotein retained ca. 10% of the original [2Fe-2S]2+ cluster, as judged by the UV-visible spectrum. This protein was then reconstituted with FeCl3, Na2S, and DTT to yield a protein that appeared to contain ∼80-90% [4Fe-4S]2+ and ∼10-20% [2Fe-2S]2+ clusters, as judged by manually fitting the UV-visible spectra. All of the reconstitution steps were performed under argon, and excess reagents were removed after reconstitution by passing the sample through a Sephadex G25 column (1 cm × 25 cm) equilibrated with 100 mM Tris and 100 mM NaCl (pH 8).
Equilibrium Binding of Substrates to BioB. Substrate binding was assessed by a microdialysis method (31) carried out in an anaerobic chamber. BS (80 μL of ∼100 μM monomer), either as purified or anaerobically reconstituted, was dialyzed against varying mixtures of DTB, AdoMet, or both substrates (200 μL, 0.01-5 mM) for 16 h at 20 °C. A sample of the dialysis buffer was analyzed to determine the free substrate concentration, while the protein/substrate mixture was analyzed to determine the total substrate (free + bound). Dethiobiotin was assessed by HPLC analysis using a Symmetry C18 column (3 mm × 150 mm, Waters) equilibrated with a 95:5 H2O/CH3CN mixture (5 mM H3PO4) at 35 °C and compared to a standard of commercial DTB (Sigma) prepared gravimetrically. AdoMet was assessed by HPLC analysis using a Symmetry C18 column (3 mm × 150 mm, Waters) equilibrated with 50 mM ammonium acetate (pH 6.8) at 35 °C and compared to a standard of AdoMet determined using an ε260 of 15 400 M-1 cm-1. The extent of AdoMet degradation and racemization was determined by incubating AdoMet alone in the same buffer and analyzing degradation products by HPLC. The activity of reconstituted biotin synthase before and after dialysis was compared in a 60 min assay at 37 °C as previously described (32).
When AdoMet was added to BS containing [2Fe-2S]2+ and [4Fe-4S]2+ clusters in a 1:1 ratio, either in the presence or in the absence of saturating DTB, a small decrease in the UV-visible spectrum at ca. 380-420 nm was observed. Using this spectral change as a probe, accurate binding constants for AdoMet could be determined. BS (50 μM monomer) was reconstituted as previously described and incubated under argon in an anaerobic cuvette. DTB and AdoMet stock solutions were equilibrated with argon in separate vials. DTB (100 μM) was added to selected samples. AdoMet was titrated into the protein sample, and after each addition, the sample was stirred at 25 °C for 10 min and a UV-visible spectrum was recorded. The ratio of absorbance at 410 nm to that at 452 nm was examined as a function of added AdoMet. The Kd was determined by fitting the resulting data to a quadratic binding isotherm:
where α = A410/A452, n is the number of binding sites per BS monomer, E is the BS monomer concentration, L is the concentration of added AdoMet, and Kd is the dissociation constant.
Examination of the Kinetics of AdoMet Binding. The kinetics of AdoMet binding to reconstituted biotin synthase were examined using stopped-flow UV-visible spectrophotometry. All experiments were performed using a HiTech SF-61 DX2 stopped-flow spectrophotometer with the temperature maintained at 20 °C. To ensure complete anaerobiosis, the interior components of the stopped-flow instrument were soaked with and all enzyme solutions contained the enzymatic oxygen-scrubbing system protocatechuic acid dehydrogenase (0.5 mg/mL) and protocatechuic acid (200 μM) (33). Solutions were made anaerobic in glass tonometers by exchanging argon for air using adaptations of previously described techniques (34). Biotin synthase (300 μM) was reconstituted to contain [2Fe-2S]2+ and [4Fe-4S]2+ clusters in a 1:1 ratio and was then diluted to 50 μM monomer in 100 mM Tris and 100 mM NaCl (pH 8) in an anaerobic tonometer. AdoMet (50-8000 μM) was diluted in the same buffer and bubbled with argon for 15 min before being transferred to an anaerobic tonometer. All concentrations are diluted 2-fold after mixing in the stopped-flow instrument has taken place. Rate constants were obtained from multiple exponential fits based on the Marquardt-Levenberg algorithm using KinetAsyst 2 software (HiTech). Simulations of the kinetic data were generated using HopKinSim 1.7.2 (D. Wachsstock, The Johns Hopkins University, Baltimore, MD) and the mechanism as depicted in Figure 9.
Figure 9.
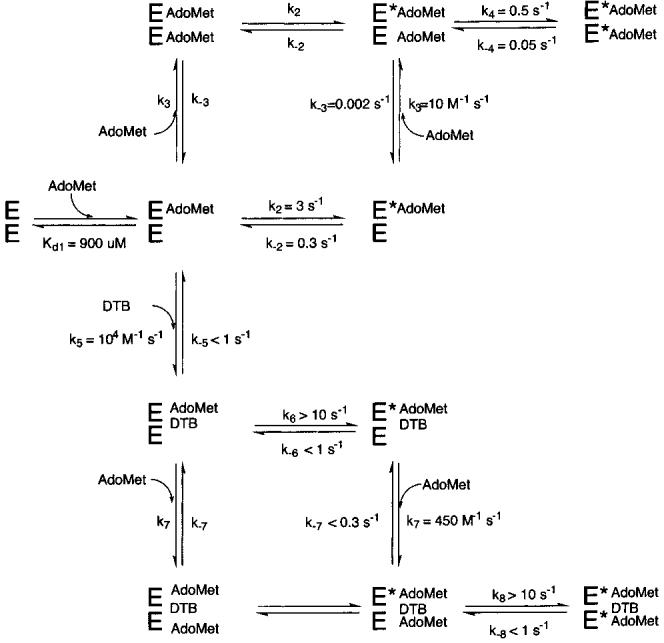
Proposed kinetic scheme for substrate binding to biotin synthase. In each case, substrate binding steps are assumed to be unobservable, while the observed spectral change is assumed to be due to an undefined conformational change (E → E*) that results in a decrease in the extinction coefficient of the [4Fe-4S]2+ cluster. In the absence of DTB (top), biotin synthase can initially bind 1 equiv of AdoMet with a Kdl of 900 μM and undergo a slow, reversible conformational change (k2 = 3 s-1). A second equivalent of AdoMet is bound much slower (k3 = 10 M-1 s-1), inducing a similar conformational change at a slightly slower rate (k4 = 0.5 s-1). Alternately, after the first molecule of AdoMet binds, 1 equiv of DTB can bind at a moderate rate (k5 = 104 M-1 s-1), leading to a rapid conformational change (k6 > 10 s-1). Now in the presence of bound DTB, a second equivalent of AdoMet binds slowly (k7 = 450 M-1 s-1), but the subsequent conformational change is again rapid (k8 > 10 s-1), leading to formation of the active enzyme-substrate complex.
Oxidative Degradation of the [4Fe-4S]2+ Cluster. The air sensitivity of the [4Fe-4S]2+ cluster was examined in the presence and absence of substrates for BS containing [2Fe-2S]2+ and [4Fe-4S]2+ clusters in a 1:1 ratio. BS (final concentration of ∼50 μM) was reconstituted and anaerobically repurified as previously described (30). The protein was split into two portions, and DTB (50 μM) and AdoMet (100 μM) were added to one sample. Each sample was removed from the anaerobic chamber, and 700 μL of each protein mixture was diluted with 700 μL of air-saturated 100 mM Tris and 100 mM NaCl (pH 8.0). Spectra were recorded at intervals, and the ratio of absorbance at 410 nm to that at 452 nm was plotted over time. After 2 h, the samples were returned to the anaerobic chamber and assayed in the absence of DTT, FeCl3, and Na2S as previously described (32).
RESULTS
Substrate Binding Is Optimal in the Presence of both [2Fe-2S]2+ and [4Fe-4S]2+ Clusters. Biotin synthase catalyzes a complex multistep reaction that requires the simultaneous presence of dethiobiotin, AdoMet, a sulfur donor, and a low-potential electron within the enzyme. The reaction sequence that we have proposed is initiated by electron transfer into the [4Fe-4S]2+ cluster, triggering reductive cleavage of AdoMet to yield a 5′-deoxyadenosyl radical. This transient high-energy radical then abstracts a hydrogen atom from dethiobiotin, generating a C9-centered dethiobiotin radical, which is followed by trapping of this substrate radical by a sulfur atom from the [2Fe-2S]2+ cluster (32). A second equivalent of AdoMet would then be required for formation of the second C-S bond. This reaction sequence would optimally require the presence of both [2Fe-2S]2+ and [4Fe-4S]2+ clusters as well as dethiobiotin and AdoMet prior to initiation of the reaction. Other mechanisms that require only the [4Fe-4S]2+ cluster have been proposed (28, 40). As a first step toward resolving these potentially conflicting mechanisms, we have explored the ability of biotin synthase to bind both substrates following reconstitution of the iron-sulfur clusters by various previously described methods.
Using a simple microdialysis procedure originally developed for assessing binding of AdoMet to methionine synthase (31), we screened the aerobically purified protein containing only [2Fe-2S]2+ clusters, the protein reconstituted to contain only [4Fe-4S]2+ clusters, either by the method of Duin et al. (22) or by the method of Ollagnier-De Choudens et al. (28), and the protein reconstituted to contain one [2Fe-2S]2+ and one [4Fe4S]2+ cluster per monomer (30, 36). Each of these proteins (∼100 μM) was equilibrated in 100 mM Tris and 100 mM NaCl (pH 8.0) by being passed through an anaerobic desalting column, and then equilibrated against either AdoMet alone, DTB alone, or AdoMet and DTB (all at 200 μM) in an anaerobic chamber for 15 h at 20 °C. At this point, the protein with bound substrates and the buffer with excess unbound substrates were separated and analyzed for substrate content by HPLC. To our initial surprise, we found that only enzyme containing both [2Fe-2S]2+ and [4Fe-4S]2+ clusters would bind either substrate with measurable affinity (Table 1). When both substrates are incubated together with protein containing [2Fe-2S]2+ and [4Fe-4S]2+ clusters in an ∼1:1 ratio, we observe the presence of 0.77 ± 0.1 equiv of AdoMet and 0.51 ± 0.1 equiv of dethiobiotin per BS monomer. Under otherwise identical conditions, but in the absence of DTB, the reconstituted protein will bind 0.18 equiv of AdoMet. In the absence of AdoMet, DTB does not bind at all. Protein missing either the [2Fe-2S]2+ or [4Fe-4S]2+ clusters binds substrates either much weaker or not at all. In our hands, biotin synthase reconstituted by the method of Ollagnier-De Choudens et al. (28) initially contains ∼80% [4Fe-4S]2+ cluster and ∼20% [2Fe-2S]2+ cluster, as estimated from the UV-visible spectrum, and we find that in the presence of both AdoMet and DTB, this protein binds ∼0.07 equiv of AdoMet and ∼0.04 equiv of DTB per BS dimer. The observed low binding stoichiometry is likely due to a relatively high dissociation constant, as suggested by spectral titrations described below. Aerobically purified BS containing only [2Fe-2S]2+ clusters also does not bind either substrate. We conclude that the presence of both [2Fe-2S]2+ and [4Fe-4S]2+ clusters is essential for optimal substrate binding.
Table 1.
Substrates Binding to Biotin Synthase Containing Different Iron–Sulfur Clustersa
| [substrate]bound/[BS monomer] |
||||
|---|---|---|---|---|
| cluster state | AdoMet | AdoMet (+DTB) | DTB | DTB (+AdoMet) |
| [2Fe-2S]2+ | 0 | 0 | 0 | 0 |
| [4Fe-4S]2+ (22) | 0 | 0 | 0 | 0 |
| [4Fe-4S]2+ (28) | 0 | 0.07 | 0 | 0.04 |
| [2Fe-2S]2+ and [4Fe-4S]2+ | 0.18 | 0.77 | 0 | 0.51 |
Biotin synthase (100 μM monomer) initially contained one [2Fe-2S]2+ cluster per monomer; this was reconstituted using the alternate methods of Duin et al. (22) and Ollagnier-De Choudens et al. (28) to contain approximately one [4Fe-4S]2+ cluster per monomer, and by the method of Ugulava et al. (30) to contain approximately one [2Fe-2S]2+ cluster and one [4Fe-4S]2+ cluster per monomer. Proteins were dialyzed against DTB (200 μM) and AdoMet (200 μM), alone or in combination, in an anaerobic chamber for 15 h at 20 °C. The amounts of total and free substrate were determined by HPLC analysis, and the amount of bound substrate was inferred as the difference between these values; values are given as equivalents of bound substrate per BS dimer. Values are averages of three samples.
Equilibrium Dialysis Demonstrates that Optimal Substrate Binding Requires Both Substrates. Since a preliminary screen of substrate binding to biotin synthase containing only [2Fe-2S]2+ clusters, only [4Fe-4S]2+ clusters, or both [2Fe-2S]2+ and [4Fe-4S]2+ clusters suggested that only the latter bound both substrates tightly, we focused our further binding studies on this form of the enzyme. Biotin synthase (100 μM monomer) was reconstituted to contain [2Fe-2S]2+ and [4Fe4S]2+ clusters in a 1:1 ratio, and was then dialyzed against buffer that contained varying amounts of DTB (0-500 μM) in the presence or absence of AdoMet (200 μM), or varying amounts of AdoMet (0-500 μM) in the presence or absence of DTB (200 μM). Following equilibration for 15 h at 20 °C, the buffer containing excess free substrate was removed, and the protein sample (containing both bound and free substrate) was precipitated by addition of 5 M sodium acetate (pH 4). The final concentration of biotin synthase was determined by the Bradford assay of an unprecipitated sample, while the concentration of DTB and AdoMet in each sample was determined by HPLC analysis; the ratio of bound or total substrate concentration to the BS monomer concentration is plotted in panels A and B of Figure 1.
Figure 1.
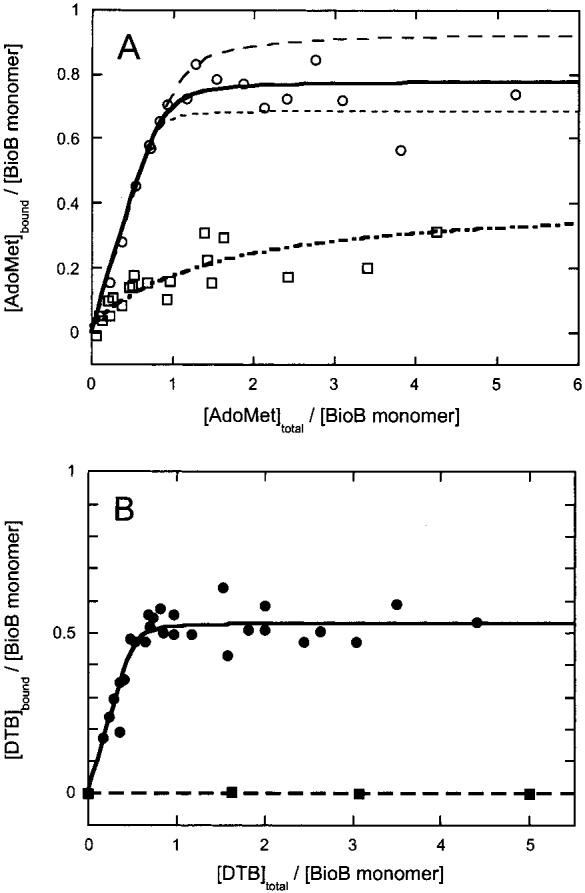
Determination of the affinity of biotin synthase for AdoMet and DTB by equilibrium dialysis. Biotin synthase (100 μM) was reconstituted to contain one [2Fe-2S]2+ and one [4Fe-4S]2+ cluster per monomer (32) and dialyzed against AdoMet and/or DTB in 100 mM Tris and 100 mM NaCl (pH 8.0) using 12 kDa cutoff dialysis membranes. (A) Biotin synthase is dialyzed against AdoMet (0-500 μM) in the presence of DTB [200 μM (○)] or in the absence of DTB (□). In the presence of DTB, the data are fit with a quadratic binding isotherm holding the enzyme concentration constant at 100 μM, yielding an apparent dissociation constant of 2.3 μM and a binding stoichiometry of 0.78 (solid curve). For comparison, curves with 0.69 site/monomer and a Kd of 1 μM or 0.93 site/monomer and a Kd of 4 μM are shown that encompass the scatter of the data (dashed curves). In the absence of DTB, the data are fit holding the enzyme concentration constant at 100 μM, yielding an apparent Kd of 100 μM and a binding stoichiometry of 0.35 site/monomer. (B) Biotin synthase (100 μM) was dialyzed against DTB (0-500 μM) in the presence of AdoMet [200 μM (•)] or in the absence of AdoMet (■). In the presence of AdoMet, the data are fit to a quadratic binding isotherm holding the BS monomer concentration constant at 100 μM, yielding an apparent Kd of 1 μM and a binding stoichiometry of 0.51 site/monomer. In the absence of AdoMet, binding was not observed, and we estimate a lower limit to the dissociation constant of ∼10 mM.
In the absence of DTB, AdoMet is bound to biotin synthase with low affinity [Figure 1A (□)]. Up to an AdoMet concentration of 500 μM we observe binding of only ∼0.35 equiv of AdoMet per BS monomer with a Kd of 100 ± 20 μM. In the presence of DTB, AdoMet binds with higher affinity and stoichiometry. We observe saturation of ∼0.78 ± 0.1 binding sites per BS monomer with a slight excess of AdoMet, with an estimated Kd of 2.3 ± 2 μM [Figure 1A (○ and —)]. A major concern in assessing AdoMet binding by equilibrium dialysis is the instability of AdoMet toward both hydrolytic degradation and racemization at the chiral sulfonium (37). To minimize the effect of these processes on our binding results, we performed dialysis at 20 °C to slow racemization and used HPLC analysis to separate AdoMet from the various degradation products. In a representative control experiment, after dialysis for 15 h at 20 °C, we observe by HPLC ∼70% of the initially added (S,S)-AdoMet, with the remainder present as 5′-methylthioadenosine (MTA, ∼20%), adenosylhomocysteine (AdoHcy, ∼5%), and the unnatural (R,S)-AdoMet (∼5-11%; see the Supporting Information). Both 5′-MTA and AdoHcy are easily separated by HPLC analysis and do not bind to BS (J. Jarrett, unpublished data), and therefore do not affect our analysis. However, the unnatural (R,S)-AdoMet isomer is not well resolved from the natural (S,S) isomer, and both isomers are summed together in analyzing the binding data in Figure 1A. To account for the affect of this on our analysis, we modeled the data assuming that AdoMet is 10% racemized in all of the samples and assuming the unnatural (R,S)-AdoMet does not bind to BS, obtaining the values of 0.78 binding site and a Kd of 2.3 μM reported above. However, the data can be accommodated by modeled curves with 0.69-0.93 binding site per monomer with dissociation constants of 1-4 μM, respectively [Figure 1A (dashed curves)].
In contrast to AdoMet, DTB binding is not detected in the absence of AdoMet at a DTB concentration of up to 1 mM [Figure 1B (■)]. In the presence of AdoMet, DTB binding is very tight, with apparent saturation of the binding sites at a ratio of 0.53 DTB per BS monomer [Figure 1B (•)]. We do not observe further DTB binding up to 1 mM substrate, and we therefore conclude that there is only a single DTB binding site per dimer; perhaps this site lies at the dimer interface. The dissociation constant for DTB is difficult to accurately determine due to the high protein concentrations required in the dialysis experiment, but we estimate a KdDTB of 1 μM, with a range of 0.5-5 μM being consistent with the scatter of the data.
AdoMet Binding Is Accompanied by Changes in the UV-Visible Spectrum of the [4Fe-4S]2+ Cluster. When BS initially containing one [2Fe-2S]2+ cluster per monomer is reconstituted to contain an additional [4Fe-4S]2+ cluster per monomer (30), the UV-visible spectrum of the resulting protein displays a broad spectrum with poorly defined shoulders at ca. 410 and 452 nm (Figure 2A). When AdoMet is added to this protein, a decrease in absorbance at 410 nm is observed (Δε410 = -1400 ± 200 M-1 cm-1 per monomer) and the corresponding band becomes more sharply defined. The resulting difference spectrum (Figure 2A inset) broadly mimics the spectrum of the [4Fe-4S]2+ cluster alone, suggesting that this may reflect a general decrease in the extinction coefficient due to changes in the ligation state or coordination geometry of the [4Fe-4S]2+ cluster. Although these spectral changes are induced by the binding of AdoMet, the changes in the spectrum could be due to either protein conformational changes or the covalent interaction of AdoMet with the cluster (substrate conformational change). There is precedent for the direct interaction of AdoMet with the [4Fe-4S]2+ cluster in pyruvate formate-lyase activase (23, 25, 38), and for interaction of methionine with the [4Fe-4S]2+ cluster in lysine 2,3-aminomutase (24). The kinetics of AdoMet binding to biotin synthase (see below) suggest that the observed spectral change is due to an event that occurs after initial AdoMet binding, and we will generically refer to this event as a conformational change. The shape and magnitude of these spectral changes are not a function of the presence or absence of DTB.
Figure 2.
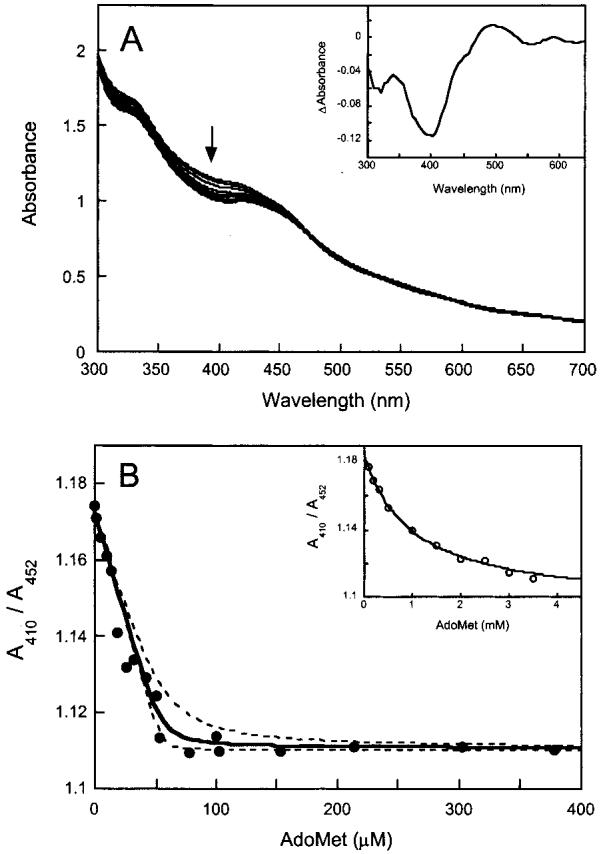
UV-visible spectral changes observed following addition of AdoMet to reconstituted biotin synthase. (A) Biotin synthase (70 μM) was reconstituted to contain [2Fe-2S]2+ and [4Fe-4S]2+ clusters in a ratio of ∼1:1 per monomer (32), mixed with DTB (200 μM), and AdoMet (0-400 μM) was added in small aliquots. Following each addition, the protein sample was stirred for 5 min at 20 °C and a UV-visible spectrum recorded. In the inset, the difference spectrum has a maximal decrease at 400 nm, suggesting a decrease in absorption by the [4Fe-4S]2+ cluster. (B) The absorbance at 410 nm is decreased following addition of AdoMet. The ratio of absorbance at 410 nm to that at 452 nm reflects the relative intensity of overlapping absorbance bands due to [4Fe-4S]2+ and [2Fe-2S]2+ clusters. In the presence of DTB (200 μM), AdoMet binds with high affinity (•). The data are fit to a quadratic binding isotherm holding the enzyme concentration fixed, with the solid curve showing an optimal fit with a Kd of 2 μM and a binding stoichiometry of 0.8, while the dashed curves show corresponding fits with Kd values of 0.2 and 5 μM. In the inset, in the absence of DTB, AdoMet binding is much weaker (O). Although a similar decrease in the A410/A452 ratio is observed, saturation of these spectral changes requires >5 mM AdoMet. The data are fit to a hyperbolic binding isotherm with a Kd of 1 mM (—).
When these spectral changes are replotted as a function of the added AdoMet concentration, the data can be modeled with standard binding isotherms and the dissociation constant for AdoMet can be extracted (Figure 2B). Although the primary spectral change is a decrease in absorbance at 410 nm, this change is small and is easily masked by scatter in the data arising from experimental artifacts. To minimize this scatter, we examined the change in the ratio of the absorbance for shoulders observed in the spectrum at ∼410 and 452 nm; this ratio is 1.18 ± 0.02 for freshly reconstituted protein and decreases to 1.11 ± 0.02 in the presence of AdoMet. When this ratio is replotted for the addition of AdoMet to biotin synthase in the presence of excess DTB, we observe a decrease in absorbance that saturates abruptly at one AdoMet per BS monomer [Figure 2B (•)]. Because of the observed tight binding, we fit these data to a quadratic binding isotherm, holding the protein concentration fixed at 70 μM, and find an optimal fit with a Kd of 2 μM (solid curve) and 0.8 ± 0.1 binding site per monomer. As shown, the scatter observed in the data could accommodate dissociation constants ranging from 0.2 to 5 μM (dashed curves). This binding curve is consistent with the data obtained from equilibrium dialysis [Figure 1A (○)], indicating that the observed spectral change is indeed due to an event that accompanies substrate binding. In the absence of DTB (Figure 2B inset), we observe similar spectral changes, but with much weaker binding. Although saturation is not observed up to 1 mM AdoMet, we estimate a Kd of ∼1 mM, indicating that concurrent DTB binding results in a 50-fold increase in the affinity for AdoMet. However, on the basis of very slow spectral changes that were observed in kinetic experiments described below, we suspect that this value is artificially high due to very slow equilibration of at least one AdoMet binding site and potentially also due to very slow cluster destruction at high AdoMet concentrations (>1 mM). Thus, in the absence of DTB, neither spectral titrations nor equilibrium dialysis provides a particularly satisfactory account of AdoMet binding, with apparent substoichiometric binding and dissociation constants that range from 100 to 1000 μM.
Although our preliminary binding screen suggested that BS containing one [4Fe-4S]2+ cluster per monomer (and no [2Fe-2S]2+ cluster) bound AdoMet either weakly or not at all (Table 1), a similarly reconstituted preparation of BS has been shown to catalyze reductive cleavage of AdoMet during photoreduction in the presence of deazaflavin (28), suggesting that AdoMet likely interacts, at least weakly, with the [4Fe-4S] cluster in the absence of DTB. We prepared a sample containing approximately one [4Fe-4S]2+ cluster by reducing BS containing one [2Fe-2S]2+ cluster (120 μM) with sodium dithionite (1 mM) in the presence of 2 equiv of FeCl3 and Na2S in 60% ethylene glycol and 100 mM Tris (pH 8) for 4 h, followed by desalting to remove excess reagents and ethylene glycol. The resulting protein (∼60 μM) was titrated with AdoMet (0-2 mM), and spectra were recorded after each addition (Figure 3A). In this case, an increase in absorbance at all wavelengths (and visually observed turbidity) suggests that the enzyme is not completely stable throughout the titration and generates a small amount of precipitated protein. However, in the midst of the general increase due to protein precipitation, we observe a dip in absorbance at 395 nm (Figure 3A inset) that is similar to the spectral changes observed in the presence of both [2Fe-2S]2+ and [4Fe-4S]2+ clusters (Figure 2A inset). Unfortunately, the magnitude of this spectral change is comparable to the observed turbidity, and extracting an accurate binding constant is difficult. However, we have attempted to correct for the turbidity by subtracting the change in absorbance at 520 nm (where Figure 2A indicates no cluster-associated spectral changes) from the data at 395 and 452 nm. The ratio of the residual absorbance at these wavelengths is then plotted as a function of added AdoMet (Figure 3B), and fit to a weak Kd of 1.4 ± 0.3 mM. Addition of DTB did not affect these results. In contrast, when we titrated BS containing one [2Fe-2S]2+ cluster per monomer (but no [4Fe-4S]2+ cluster) with AdoMet (0-2 mM), we observed no spectral changes (see the Supporting Information). This was not unexpected, since this oxidized form of BS was shown to not bind either substrate using equilibrium dialysis. Overall, these data suggest that the [2Fe-2S]2+ cluster is not required for AdoMet binding or for interactions between AdoMet and the [4Fe-4S]2+ cluster; this may prove to be useful in designing experiments that spectroscopically probe the physical nature of this interaction (23, 25, 38). However, tight substrate binding appears to require the simultaneous presence of both AdoMet and DTB bound with BS containing both [2Fe-2S]2+ and [4Fe-4S]2+ clusters.
Figure 3.
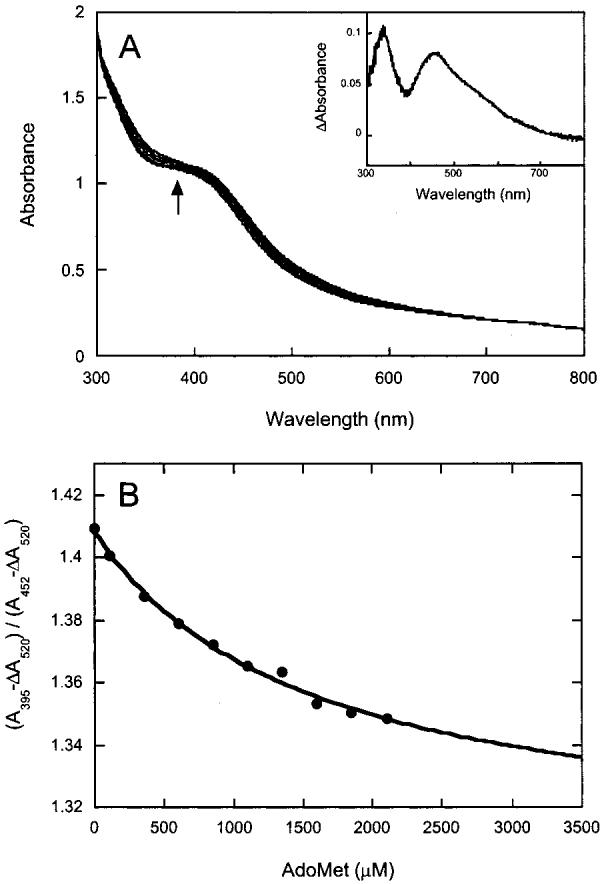
UV-visible spectral changes observed following addition of AdoMet to biotin synthase reconstituted with one [4Fe-4S]2+ cluster per monomer. (A) Biotin synthase (∼60 μM) was converted to a form that contains approximately one [4Fe-4S]2+ cluster (see Materials and Methods), and AdoMet (0-2 mM) was added in small aliquots. After each addition, the protein was equilibrated for 5 min and a spectrum recorded. A slight but noticeable increase in turbidity correlated with the observed increase in absorbance at all wavelengths, suggesting that the protein was not completely stable in this reconstitution state and slowly precipitated during the experiment. In the inset, the difference spectrum (2 mM AdoMet - initial protein) shows a decrease in absorbance at 395 nm that is superimposed upon a general increase due to precipitation. (B) The absorbance at 395 nm was corrected for the turbidity by subtracting the change in absorbance at 520 nm, and then divided by the corrected absorbance at 452 nm to yield a curve reflecting the apparent decrease in absorbance at 395 nm due to AdoMet binding. The data are fit to a binding isotherm with a Kd of 1.4 + 0.3 mM.
AdoMet Binds Slowly to Biotin Synthase in the Absence of DTB. The spectral changes that accompany AdoMet binding are sufficient in magnitude to allow kinetic characterization of this binding event using a stopped-flow UV-visible spectrophotometer. If the strong cooperativity of DTB and AdoMet binding observed in equilibrium experiments is due to one substrate forming a portion of the binding site for the second substrate, then binding would likely be kinetically ordered and the rate of AdoMet binding would be affected by the presence of DTB. However, before attempting to interpret the potentially complex effect of DTB on the kinetics of AdoMet binding, we examined how AdoMet alone binds to biotin synthase (in the absence of DTB). Biotin synthase reconstituted to contain [2Fe-2S]2+ and [4Fe-4S]2+ clusters in a 1:1 ratio per monomer (25 μM after mixing) was rapidly mixed with AdoMet (50-4000 μM after mixing) in 100 mM Tris and 100 mM NaCl (pH 8). Spectral changes were monitored at 410 nm for 4 min and are shown in Figure 4A. Both the apparent rate and the magnitude of the observed absorbance changes are increased as the concentration of AdoMet is increased. These traces are fit to four exponential phases, including two major phases that are clearly dependent on the concentration of AdoMet, and two phases that appear to be independent of AdoMet. First, an initial rapid decrease is observed in all traces with a rate constant of ∼6 s-1 and a ΔA410 of ∼0.005; the rate and magnitude of this small spectral change were unchanged with increasing AdoMet concentrations, and a similar spectral change was also observed in a control where reconstituted protein is mixed with anaerobic buffer. We suspect that this small initial decrease in absorbance is due to a slight dissociation of Fe from the protein upon dilution, and we have ignored this kinetic phase in further analyses. Also observed was a very slow kinetic phase (kobs ≈ 0.002 s-1, ΔA410 ∼ 0.005) that is only observed at high AdoMet concentrations (>1 mM); we suspect this phase corresponds to slow destruction of the [4Fe-4S]2+ cluster and chelation of the resulting free iron by excess free AdoMet. However, in addition to these two phases that likely result from cluster instability, all of the kinetic traces show two additional phases whose associated rate constants (Figure 5A) and magnitudes vary with increasing AdoMet concentrations. At low AdoMet concentrations, the traces are dominated by a slow phase with rate constants that vary with increasing AdoMet concentrations from 0.02 to 0.2 s-1 [Figure 5A (■)]. Our kinetic simulations suggest that this phase corresponds to a slow second-order binding of AdoMet as indicated by the linear fit from 500 to 3000 μM, yielding the following observed rate constants: kon = 30 M-1 s-1 and koff = 0.09 s-1 [Figure 5A (- - -)]. Deviation from linearity at low AdoMet concentrations is likely due to partial depletion of the pool of free AdoMet. At high AdoMet concentrations, the traces are dominated by a fast kinetic phase with rate constants that vary from 0.5 to 2.3 s-1 [Figure 5A (•)]. Saturation of the observed rate constant at high AdoMet concentrations suggests that this phase corresponds to a rapid equilibrium binding of AdoMet with an initial Kd of ≈1000 μM followed by a conformational change with the following observed rate constants: kf = 1.53 s-1 and kr = 0.54 s-1 [Figure 5A (s)].
Figure 4.
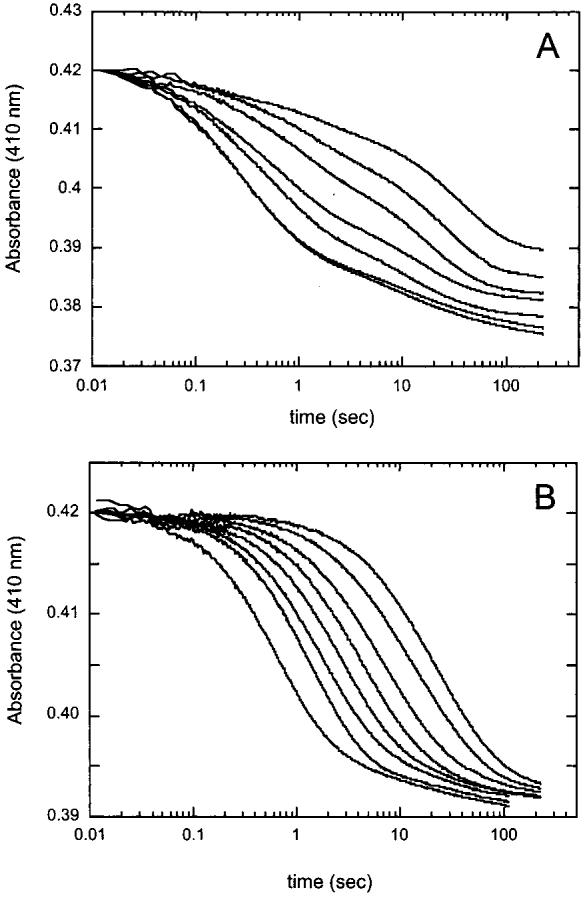
Transient kinetic analysis of absorption changes accompanying AdoMet binding to biotin synthase. (A) Biotin synthase (25 μM monomer after mixing) was reconstituted (32) and rapidly mixed with AdoMet in 100 mM Tris and 100 mM NaCl (pH 8.0). Traces are, from right to left, for 50, 100, 250, 500, 1000, 2000, and 3000 μM AdoMet after mixing. (B) Biotin synthase (25 μM monomer after mixing) was reconstituted (32) and rapidly mixed with AdoMet in 100 mM Tris and 100 mM NaCl (pH 8.0) containing DTB (200 μM). Traces are, from right to left, for 50, 100, 200, 300, 400, 600, 800, and 2000 μM AdoMet after mixing. Minor variations in the initial absorbance were corrected for each trace to give an average initial value for A410 of 0.42.
Figure 5.
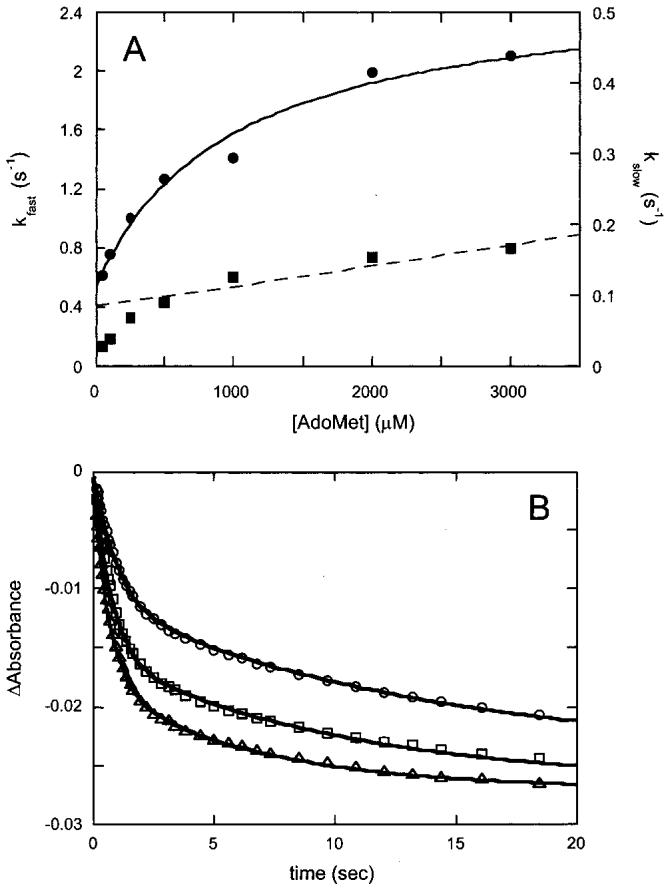
Data modeling for AdoMet binding to biotin synthase. (A) The major spectral changes accompanying AdoMet binding in Figure 4A (0.2-20 s) are fit to two exponential kinetic phases. Rate constants for the fast (•, left axis) and slow (■, right axis) phases are shown as a function of the AdoMet concentration. The fast rate constant is fit assuming rapid equilibrium binding followed by a slower conformational change, while the slow rate constant is fit assuming slower second-order binding behavior coupled with a slow conformational change, yielding the following kinetic parameters: Kd = 1000 μM, kf = 1.53 s-1, and kr = 0.54 s-1 for the fast phase and kon = 30 M-1 s-1 and koff = 0.09 s-1 for the slow phase. (B) Changes in absorbance from 0.2 to 20 s extracted from Figure 4A are modeled using the rate constants from panel A as a starting point for kinetic simulations. Experimental data are shown for 250 (○), 500 (□), and 1000 μM (△), and each modeled curve is generated by adjusting only the AdoMet concentration in the simulation. The ordered binding mechanism as shown in Figure 9 (top) gave reasonable simulations using an extinction coefficient Δε410 of -1400 M-1 cm-1 per monomer and the following refined parameters: Kdl = 900 ± 50 μM, k2 = 3 ± 0.2 s-1, k-2 = 0.3 ± 0.05 s-1, k3 = 10-50 M-1 s-1, k-3 = 0.002 ± 0.001 s-1, k4 = 0.5 ± 0.4 s-1, and k-4 = 0.05 ± 0.04 s-1.
We have used the rate constants derived from Figure 5A as an initial set of values to simulate AdoMet binding in the absence of DTB, using the mechanism depicted in Figure 9. In conducting these simulations, we have made several assumptions about the nature of the AdoMet-BS interaction. First, we assume that initial binding of AdoMet involves noncovalent interactions that do not contribute to the spectral change, and that this initial complex undergoes a slow conformational change that leads to covalent interaction of AdoMet with the [4Fe-4S]2+ cluster (25, 38), resulting in the observed decrease in absorbance. Second, we assume that binding of the first AdoMet per dimer results in a change in the protein that alters the kinetic parameters for interaction with the second equivalent of AdoMet. Finally, to simplify the kinetic analysis, we have assumed that the rate constants for binding of the second equivalent of AdoMet are unchanged when the first equivalent of AdoMet participates in the intervening conformational change (both paths around the kinetic box in Figure 9 are equivalent). The experimental and simulated data for kinetic traces at 250, 500, and 1000 μM are shown in Figure 5B. In general, we find that the rapid kinetic phase is simulated well with a single site per BS dimer that binds AdoMet with weak initial affinity (Kdl = 900 μM, Figure 9), but that undergoes a reversible conformational change (E → E*) with a k2 of 3 s-1 and a k-2 of 0.3 s-1, resulting in a decreased apparent dissociation constant [Kdl+2 = Kdl(k-2/k2) = 90 μM]. We have simulated the slower kinetic phase with a second AdoMet binding site per BS dimer; however, in this case, the concentration dependence indicates that formation of the initial AdoMet-BS complex is comparable in rate to the subsequent conformational change. Although we initially attempted to perform the simulation using rates for the conformational change equivalent to those determined above (k2 and k-2), we were not able to obtain satisfactory fits, and have instead allowed for a somewhat slower conformational change (k4 ≈ 0.5 s-1, and k-4 ≈ 0.05 s-1). Using these parameters for the conformational change, the binding step is simulated with a k3 of 10 M-1 s-1 and a k-3 of 0.002 s-1. The kinetic parameters for this slow binding phase are not precisely determined by the data, in part due to the similar magnitudes of the rate constants for steps 3 and 4 and the resulting difficulty in distinguishing the concentration-dependent and -independent components of this phase. Using the modeled rate constants, we calculate the initial dissociation constant for binding to the second site (Kd3) as 200 μM with an effective dissociation constant Kd3+4 [=Kd3(k-4/k4)] of 20 μM. Note that this predicts binding that is much tighter than that observed by either equilibrium dialysis (Figure 1A) or spectrophotometric titrations (Figure 2B inset). At least for the spectrophotometric titrations, the observation of weak binding may be partially a kinetic effect due to the very slow second-order binding constant determined for this second binding site. When we kinetically simulate a complete titration using the rate constants described above, we find that equilibrium is not reached even after 5 min between additions, and on the basis of simulations, we estimate a nonequilibrium “observed binding constant” of ∼500 μM for the experimental conditions cited for the Figure 2B inset.
AdoMet Binds More Rapidly in the Presence of DTB. Having previously shown that the affinity of biotin synthase for AdoMet is increased in the presence of DTB (Figure 1A), we expected the kinetics of AdoMet binding to also be altered by DTB. We repeated the experiments described above, mixing reconstituted biotin synthase (25 μM after mixing) with AdoMet (25-3000 μM after mixing) in a stopped-flow spectrophotometer, but now with DTB (200 μM) added to the AdoMet sample. We observed a decrease in absorbance at 410 nm similar to that observed in the absence of DTB; the resulting kinetic traces (Figure 4B) are again fit to four exponential phases. The fastest (kobs ≈ 6 s-1) and slowest (kobs ≈ 0.002 s-1) phases are again found to be minor contributors to the total absorbance changes, similar in magnitude in all of the samples, and not dependent on the concentration of AdoMet. Two kinetic phases are observed whose rates and magnitudes are affected by the concentration of AdoMet. At low AdoMet concentrations, the kinetics are dominated by a slow phase with rate constants that vary with increasing AdoMet concentrations from 0.020 to 0.21 s-1 [Figure 6A (■)]. This phase likely corresponds to a slow second-order binding of AdoMet as indicated by the linear fit from 200 to 2000 μM, yielding the following rate constants: kon = 680 M-1 s-1 and koff = 0.05 s-1 [Figure 6A (- - -)]. At high AdoMet concentrations, the traces are dominated by a fast phase with a rate constant that increases linearly from 0.068 to 1.5 s-1 as the AdoMet concentration is increased from 50 to 2000 μM [Figure 6A (•)]. Although this phase also would appear to correspond to a second-order binding of AdoMet with a kon of 680 M-1 s-1 and a koff of 0.1 s-1 [Figure 6A (—)], our subsequent data simulation suggests that this is likely due to a peculiar coupling between rapid equilibrium AdoMet binding and slow DTB binding.
Figure 6.
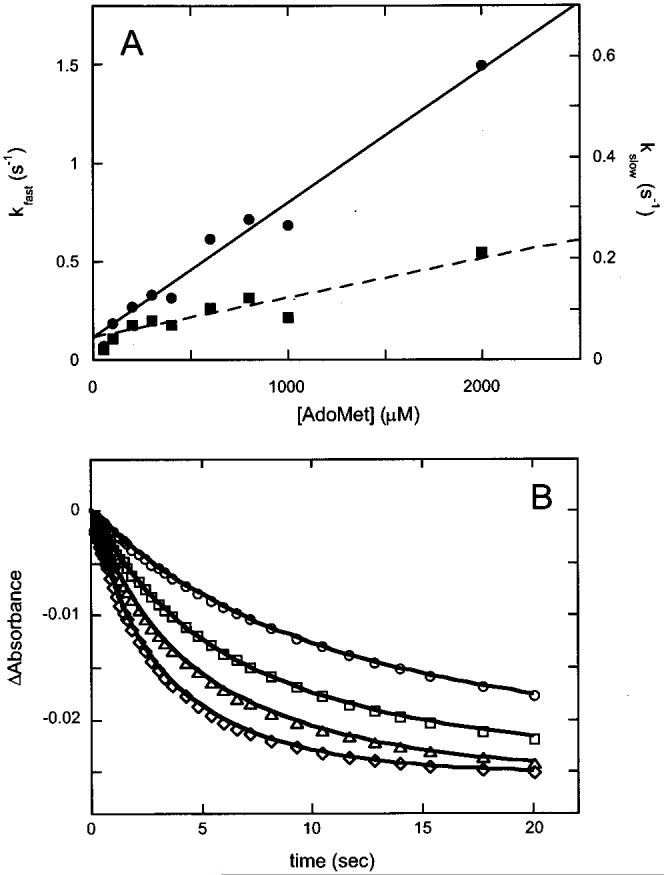
Data modeling for AdoMet binding to biotin synthase in the presence of DTB. (A) The major spectral changes accompanying AdoMet binding in Figure 4B (0.2-20 s) are fit to two exponential kinetic phases. Rate constants for the fast (•, left axis) and slow (■, right axis) phases are shown as a function of the AdoMet concentration. The fast phase is fit to apparent second-order binding coupled with a fast conformational change (s), while the slow phase is fit assuming slower second-order binding behavior coupled with a fast conformational change (- - -), yielding the following kinetic parameters: kon = 680 M-1 s-1 and koff = 0.1 s-1 for the fast phase and kon = 80 M-1 s-1 and koff = 0.05 s-1 for the slow phase. (B) Changes in absorbance from 0.2 to 20 s extracted from Figure 4B are modeled using the rate constants from the previous simulation of AdoMet binding alone as a starting point for kinetic simulations based upon the mechanism in Figure 9. Experimental data are shown for 100 (○), 200 (□), 300 (△), and 400 μM (◇), and each modeled curve is generated by adjusting only the AdoMet concentration in the simulation. The ordered binding mechanism as shown in Figure 9 (bottom) gave reasonable simulations using an extinction coefficient Δε410 of -1400 M-1 cm-1 per monomer and the following refined parameters: Kdl = 900 ± 50 μM, k5 = (1 ± 0.2) × 104 M-1 s-1, k-5 < 1 s-1, k6 > 10 s-1, k-6 < 1 s-1, k7 = 450 ± 100 M-1 s-1, k-7 < 0.3 s-1, k8 > 10 s-1, and k-8 < 1 s-1.
We have performed kinetic simulations using several alternate kinetic models to determine the order of AdoMet and DTB binding and to determine the effect that DTB has upon the kinetics of AdoMet binding. In addition to assumptions cited above regarding AdoMet binding, we have included one additional assumption: since DTB binding was not observed in the absence of AdoMet, we have assumed that DTB must bind at some point after binding of the first AdoMet molecule. We started these simulations using microscopic rate constants derived from simulations of AdoMet binding above, and then made the smallest changes to these rate constants that are consistent with the data. The experimental and simulated data for kinetic traces at 100, 200, 300, and 400 μM are shown in Figure 6B, and the mechanism that gives the best simulation over the entire concentration and time range is depicted in Figure 9 (bottom). In this mechanism, the first molecule of AdoMet binds weakly as before (Kdl = 900 μM), but is then “trapped” by the second-order binding of DTB with the following rate constants: k5 = 104 M-1 s-1 and k-5 < 1 s-1. The AdoMet-DTB-BS complex then undergoes a rapid conformational change (E → E*), resulting in the observed spectral change with a rate constant k6 of > 10 s-1. The second equivalent of AdoMet can then bind with slow second-order rate constants (k7 ≈ 450 M-1 s-1 and k-7 < 0.3 s-1), followed by an associated rapid conformational change (k8 > 10 s-1 and k-8 < 1 s-1) that results in further spectral changes. In this mechanism, the observed rate constants are largely determined by Kdl and k5 (fast phase) and k7 (slow phase), and we can only place limits on values for other rate constants. In particular, the rate constant for the associated conformational change must be at least 10 s-1, but larger values are also consistent. The range of values for several rate constants limits our ability to calculate predicted dissociation constants; however, we have again simulated a titration as depicted in Figure 2B and obtain an average “simulated” dissociation constant of ∼10 μM. In the presence of DTB, we predict that following each addition of AdoMet, the sample would reach equilibrium in ∼2 min, suggesting that the binding experiment shown in Figure 2B reflects a true equilibrium titration.
DTB Binding Increases the Affinity for AdoMet and the Rate of the AdoMet-Induced Conformational Change. The equilibrium binding data depicted in Figures 1 and 2 demonstrate that the presence of DTB increases the affinity of BS for AdoMet, and comparison of the kinetic curves in panels A and B of Figure 4 suggests that DTB also increases the rate of AdoMet binding. To further probe this phenomenon, we examined the effect of varying the DTB concentration (from 0 to 100 μM after mixing) on the rate of the observed spectrophotometric change, while holding fixed both the BS (25 μM monomer after mixing) and AdoMet (500 μM after mixing) concentrations. We find that DTB appears to increase the rate of the overall spectrophotometric change (Figure 7). The concentration dependence of this effect is most easily observed by plotting the change in absorbance (0.1-2 s) associated with the fast kinetic phase (Figure 7 inset), which can be fit to one DTB binding site per dimer with a Kd of ∼1 μM. We simulated the kinetic traces using the mechanism depicted in Figure 9 to better understand the apparent kinetic effect of DTB (data not shown). From these simulations, we conclude that the primary effect of DTB is to increase the rate of the AdoMet-associated conformational change such that, whereas in the absence of DTB this conformational change is partially rate-limiting (k2 = 3 s-1 and k4 = 0.5 s-1), in the presence of DTB the conformational change is no longer rate-limiting and DTB binding and AdoMet binding (k5 and k7, respectively) become rate-limiting. A secondary effect of DTB binding, which may be related, is that AdoMet and DTB binding appear to be kinetically ordered such that bound DTB blocks dissociation of the first equivalent of AdoMet.
Figure 7.
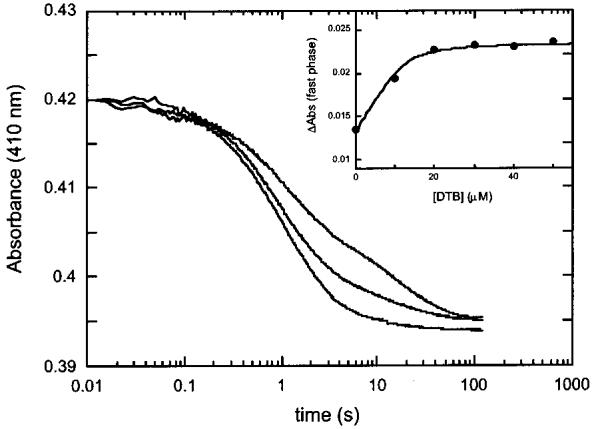
Effect of varying DTB concentration on the rate of absorption changes accompanying AdoMet binding to biotin synthase. Reconstituted biotin synthase (25 μM) was mixed with AdoMet (500 μM) and DTB (0-50 μM) in a stopped-flow spectrophotometer. Traces are for 0, 10, and 30 μM DTB. The inset shows the effect of DTB on the change in absorbance associated with the fast AdoMet binding phase. Traces were fit to four exponential decays as described in the legend of Figure 4, and the magnitude of absorbance changes associated with the primary fast kinetic phase (0.2-2 s) is replotted as a function of the DTB concentration. Data are fit to a quadratic binding isotherm holding the concentration of DTB binding sites at 12.5 μM (one per dimer) and the Kd at 1 μM.
Bound Substrates Slow the Oxidation of the [4Fe-4S]2+ Cluster. The [4Fe-4S]2+ cluster in biotin synthase is sensitive to oxygen degradation; this is likely why aerobic purification yields a protein that contains only [2Fe-2S]2+ clusters. Since substrate binding induces a decrease in the extinction coefficient of the [4Fe-4S]2+ cluster, it is likely that one or both of the substrates bind in the proximity of or coordinates directly to this cluster. In particular, covalent coordination of AdoMet to the [4Fe-4S]2+ cluster, such as that observed for PFL activase (38), may block access of oxygen to the cluster and slow the resulting oxidative degradation. To test this, we exposed reconstituted biotin synthase to oxygen by vigorously stirring an anaerobic solution of the protein with an equal volume of aerobic 100 mM Tris and 100 mM NaCl (pH 8.0) at 20 °C and monitored oxidation by recording UV-visible spectra over time. In the absence of substrates, the [4Fe-4S]2+ cluster is completely lost in 5 min and the resulting spectral changes are well fit to a single rate constant of 0.01 s-1 [Figure 7 (•)]. The addition of 1 equiv of DTB and 2 equiv of AdoMet per dimer is sufficient to significantly protect most of the [4Fe-4S]2+ cluster [Figure 7 (□)]. In this case, we observe a small fraction of the protein that is oxidized with a comparable rate constant (∼0.007 s-1), which we suspect is protein that does not contain bound substrates. However, the majority of the protein oxidizes at a much slower rate that we estimate to be ∼1 × 10-5 s-1 (t1/2 ∼ 19 h), assuming a comparable total spectral change after complete oxidation. Protection of the [4Fe-4S]2+ cluster from oxidation also protects the enzyme against inactivation. When a sample was mixed with aerobic buffer in the presence of substrates, and then assayed anaerobically in the absence of DTT, FeCl3, and Na2S (32), we obtain ∼0.7 equiv of biotin per monomer, compared with ∼0.8 equiv of biotin obtained from a sample that was maintained under constant anaerobic conditions. In contrast, a sample that was exposed to aerobic buffer in the absence of substrates produced no biotin. This substrate-induced protection has proven to be useful in our laboratory in allowing manipulation of reconstituted biotin synthase for short periods of time with full exposure to an aerobic atmosphere.
DISCUSSION
The reaction catalyzed by biotin synthase requires a complex sequence of events that involve two substrates, AdoMet and DTB, and two iron-sulfur clusters. The reaction is initiated by electron transfer from flavodoxin into the [4Fe-4S]2+ cluster, generating a [4Fe-4S]+ cluster that catalyzes the reductive cleavage of AdoMet to methionine and a high-energy 5′-deoxyadenosyl radical. This high-energy radical can be productively quenched by abstracting hydrogen atoms from the substrate, DTB, generating a substrate radical on the path to product, but in the absence of DTB could also be unproductively quenched by abstracting hydrogen atoms from nearby protein residues or could dissociate from the enzyme and abstract hydrogen atoms from small molecules or even DNA or RNA. To avoid these potentially damaging side reactions, AdoMet-dependent radical enzymes likely are evolved to exercise kinetic control of radical generation, accelerating reductive cleavage of AdoMet in the presence of a bound substrate. In biotin synthase, this control could take several potential forms: DTB binding could alter the redox potential of the [4Fe-4S]2+ cluster, could induce close association of AdoMet with the [4Fe-4S]2+ cluster, or could be tightly coupled to AdoMet binding. Minimally, we find that biotin synthase employs the last mechanism to reduce and or slow binding of AdoMet in the absence of DTB. At physiologic AdoMet concentrations [∼80 μM in E. coli (39)], biotin synthase binds weakly to 1 equiv of AdoMet [Kd(exp) = 100 μM (Figure 1A), Kdl+2(calcd) = 90 μM] and probably binds with comparable affinity but much slower kinetics to a second equivalent of AdoMet per dimer [kon = 10 M-1 s-1 (Figure 9)]. However, in the presence of DTB, biotin synthase binds 2 equiv of AdoMet with high affinity [apparent Kd = 1-5 μM (Figures 1A and 2B)]. DTB binding is not observed in the absence of AdoMet, suggesting an ordered binding mechanism in which at least one molecule of AdoMet binds prior to DTB. Thus, AdoMet binding and DTB binding appear to be kinetically and thermodynamically coupled, with a >20-fold increase in the affinity for AdoMet in the presence of DTB and a >10000-fold increase in the affinity for DTB in the presence of AdoMet.
In addition to the tight coupling of DTB and AdoMet binding, the high-affinity binding of these substrates appears to require the presence of both [2Fe-2S]2+ and [4Fe-4S]2+ clusters (36). Mechanisms have been proposed for biotin formation which require only the [4Fe-4S]2+ cluster for activity (28, 40), while the initial [2Fe-2S]2+ cluster is considered a degradation product resulting from aerobic purification. However, in our hands two alternate methods for reconstituting protein containing only [4Fe-4S]2+ clusters (22, 28) result in protein samples that bind substrates weakly or not at all. When protein reconstituted with one [4Fe-4S]2+ cluster was titrated with AdoMet under anaerobic conditions, a decrease in absorbance at 395 nm was detected that was partially obscured by minor precipitation of the protein; assuming this spectral change also corresponds to AdoMet binding, we estimate a dissociation constant for AdoMet binding to BS containing one [4Fe-4S]2+ cluster of ∼1.4 mM. Dethiobiotin did not increase the affinity of this preparation for AdoMet, consistent with our general failure to observe DTB binding in the absence of the [2Fe-2S]2+ cluster (Table 1). We conclude that AdoMet binding requires the presence of the [4Fe-4S]2+ cluster and is greatly improved by the presence of DTB, while DTB binding requires the presence of the [2Fe-2S]2+ cluster and AdoMet (and by extension the [4Fe-4S]2+ cluster). Thus, although this study does not address the issue of catalytic activity, the high affinity of biotin synthase containing both [2Fe-2S]2+ and [4Fe-4S]2+ clusters (36) for AdoMet and DTB is further evidence that this is the cluster state in the active enzyme.
AdoMet binding results in a small decrease in the extinction coefficient at ∼400 nm. This spectral change is probably due to subtle changes in the coordination environment of the [4Fe-4S]2+ cluster. Transient kinetic analysis indicates that these changes occur in a step following formation of the initial AdoMet-biotin synthase complex. Conservatively, we suggest that AdoMet binding induces a conformational change in the protein in the vicinity of the [4Fe-4S]2+ cluster or that alters the geometry of the cluster itself. However, it is tempting to speculate that the observed spectral change may be due to slow coordination of a portion of AdoMet with the fourth ligation position to the [4Fe-4S]2+ cluster. This cluster, coordinated by a conserved CxxxCxxC motif in all AdoMet-dependent radical enzymes, is in close contact with AdoMet in pyruvate formate-lyase activase (25) and with the product methionine in lysine 2,3-aminomutase (24). Addition of AdoMet to pyruvate formate-lyase activase substituted with a single 57Fe at this open ligation position results in an increase in the isomer shift, suggesting a potential ligand substitution by AdoMet (23). Finally, ENDOR spectra of AdoMet labeled with 17O and 15N in the methionyl group bound to PFL activase containing a [4Fe-4S]+ cluster indicate direct coordination of these labeled atoms to at least one Fe in the cluster (42). The minor changes we observe in the UV-visible spectrum of biotin synthase following AdoMet binding may be a macroscopic effect of similar molecular interactions.
The stoichiometry of substrate binding to biotin synthase containing [2Fe-2S]2+ and [4Fe-4S]2+ clusters in a 1:1 ratio is reproducibly one DTB and two AdoMets per dimer. This stoichiometry is consistent with AdoMet being a stoichiometric oxidant of DTB, as proposed separately by Marquet (9) and Shaw (10). Several studies report the formation of ca. 3 equiv of 5′-deoxyadenosine per biotin (3, 9, 10); the excess 5′-deoxyadenosine is attributed to some uncoupled AdoMet cleavage, and it is proposed that 2 equiv of 5′-deoxyadenosine is formed in the irreversible abstraction of a hydrogen atom from the C6 and C9 positions of DTB. The binding data presented in this paper indicate that a single molecule of DTB is surrounded by two molecules of AdoMet which, following reductive cleavage, generate two 5′-deoxyadenosyl radicals that can independently abstract hydrogen atoms at the C6 and C9 positions of DTB. This arrangement of substrates, and the tight cooperativity of AdoMet and DTB binding, may also help to promote synchronous formation of both C-S bonds in biotin and to limit the production of 6- or 9-mercaptodethiobiotin.
SUPPORTING INFORMATION AVAILABLE
Determination of the stability of AdoMet under dialysis conditions and the titration of [2Fe-2S]2+ BioB with AdoMet. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
Figure 8.
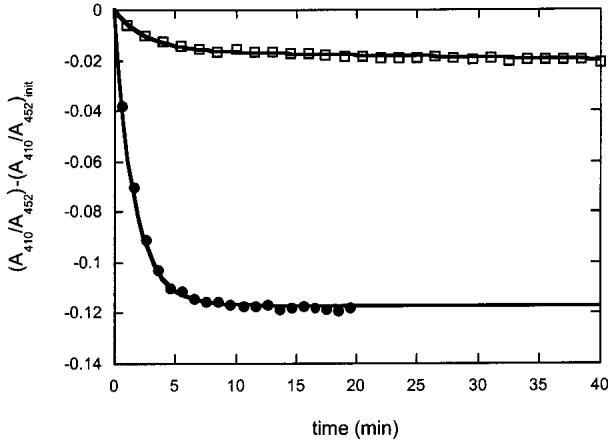
Air oxidation of the [4Fe-4S]2+ cluster in reconstituted biotin synthase in the absence (•) and presence (□) of AdoMet and DTB. Biotin synthase (50 μM) was reconstituted and repurified by gel filtration chromatography in an anaerobic chamber. This protein was divided, and a portion was mixed with an equal volume of air-saturated 100 mM Tris and 100 mM NaCl (pH 8.0) and the absorbance monitored at 410 and 452 nm. A second portion was equilibrated with AdoMet (50 μM) and DTB (25 μM) and then mixed with an equal volume of air-saturated buffer. In the absence of substrates, the data are fit to a single-exponential decay with a rate constant of 0.01 s-1. In the presence of substrates, a small decay is observed at the same rate followed by a very slow decay with an estimated rate constant of 1.5 × 10-5 s-1.
ACKNOWLEDGMENT
We thank Dr. Bruce Palfey (The University of Michigan, Ann Arbor, MI) with assistance in designing and equipping our anaerobic stopped-flow system.
Footnotes
This research has been supported by NIH Research Grant GM59175 (J.T.J.).
- AdoMet
- S-adenosyl-l-methionine
- BS
- biotin synthase (bioB gene product)
- DTB
- dethiobiotin
- DTT
- dithiothreitol
- PFL
- pyruvate formatelyase
- Tris
- tris(hydroxymethyl)aminomethane hydrochloride.
NOTE ADDED IN PROOF
After submission of this paper, Johnson and co-workers (43) presented spectroscopic evidence for AdoMet carboxylate coordination to the [4Fe-4S] cluster in BS.
REFERENCES
- 1.Marquet A. Curr. Opin. Chem. Biol. 2000;5:541–549. doi: 10.1016/s1367-5931(00)00249-0. [DOI] [PubMed] [Google Scholar]
- 2.Marquet A, Tse Sum Bui B, Florentin D. Vitam. Horm. 2001;61:51–101. doi: 10.1016/s0083-6729(01)61002-1. [DOI] [PubMed] [Google Scholar]
- 3.Escalettes F, Florentin D, Tse Sum Bui B, Lesage D, Marquet A. J. Am. Chem. Soc. 1999;121:3571–3578. [Google Scholar]
- 4.Sanyal I, Cohen G, Flint DH. Biochemistry. 1994;33:3625–3631. doi: 10.1021/bi00178a020. [DOI] [PubMed] [Google Scholar]
- 5.Ifuku O, Kishimoto J, Haze S, Yanagi M, Fukushima S. Biosci., Biotechnol., Biochem. 1992;56:1780–1785. doi: 10.1271/bbb.56.1780. [DOI] [PubMed] [Google Scholar]
- 6.Ifuku O, Koga N, Haze S, Kishimoto J, Wachi Y. Eur. J. Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 7.Birch OM, Fuhrmann M, Shaw NM. J. Biol. Chem. 1995;270:19158–19165. doi: 10.1074/jbc.270.32.19158. [DOI] [PubMed] [Google Scholar]
- 8.Ohshiro T, Yamamoto M, Tse Sum Bui B, Florentin D, Marquet A, Izumi Y. Biosci., Biotechnol., Biochem. 1995;59:943–944. doi: 10.1271/bbb.59.943. [DOI] [PubMed] [Google Scholar]
- 9.Guianvarc'h D, Florentin D, Tse Sum Bui B, Nunzi F, Marquet A. Biochem. Biophys. Res. Commun. 1997;236:402–406. doi: 10.1006/bbrc.1997.6952. [DOI] [PubMed] [Google Scholar]
- 10.Shaw NM, Birch OM, Tinschert A, Venetz V, Dietrich R, Savoy LA. Biochem. J. 1998;330(Part 3):1079–1085. doi: 10.1042/bj3301079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frey PA, Booker SJ. Adv. Protein Chem. 2001;58:1–45. doi: 10.1016/s0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- 13.Cheek J, Broderick JB. J. Biol. Inorg. Chem. 2001;6:209–226. doi: 10.1007/s007750100210. [DOI] [PubMed] [Google Scholar]
- 14.Frey M, Rothe M, Wagner AFV, Knappe J. J. Biol. Chem. 1994;269:12432–12437. [PubMed] [Google Scholar]
- 15.Harder J, Eliasson R, Pontis E, Ballinger MD, Reichard P. J. Biol. Chem. 1992;267:25548–25552. [PubMed] [Google Scholar]
- 16.Moss M, Frey PA. J. Biol. Chem. 1987;262:14859–14862. [PubMed] [Google Scholar]
- 17.Leuthner B, Leutwein C, Schulz H, Horth P, Haehnel W, Schiltz E, Schagger H, Heider J. Mol. Microbiol. 1998;28:615–628. doi: 10.1046/j.1365-2958.1998.00826.x. [DOI] [PubMed] [Google Scholar]
- 18.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Jr., Marietta MA. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 19.Rebeil R, Sun Y, Chooback L, Pedraza-Reyes M, Kinsland C, Begley TP, Nicholson WL. J. Bacteriol. 1998;180:4879–4885. doi: 10.1128/jb.180.18.4879-4885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broderick JB, Henshaw TF, Cheek J, Wojtuszewski K, Smith SR, Trojan MR, McGhan RM, Kopf A, Kibbey M, Broderick WE. Biochem. Biophys. Res. Commun. 2000;269:451–456. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 21.Lieder KW, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. Biochemistry. 1998;37:2578–2585. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- 22.Duin EC, Lafferty ME, Crouse BR, Allen RM, Sanyal I, Flint DH, Johnson MK. Biochemistry. 1997;36:11811–11820. doi: 10.1021/bi9706430. [DOI] [PubMed] [Google Scholar]
- 23.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. J. Am. Chem. Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 24.Cosper NJ, Booker SJ, Ruzicka F, Frey PA, Scott RA. Biochemistry. 2000;39:15668–15673. doi: 10.1021/bi0022184. [DOI] [PubMed] [Google Scholar]
- 25.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. J. Am. Chem. Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 26.Baraniak J, Moss ML, Frey PA. J. Biol. Chem. 1989;264:1357–1360. [PubMed] [Google Scholar]
- 27.Sun X, Ollagnier S, Schmidt PP, Atta M, Mulliez E, Lepape L, Eliasson R, Graslund A, Fontecave M, Reichard P, Sjoberg BM. J. Biol. Chem. 1996;271:6827–6831. [PubMed] [Google Scholar]
- 28.Ollagnier-De Choudens S, Sanakis Y, Hewitson K, Roach P, Munck E, Fontecave M. J. Biol. Chem. 2002;277:13449–13454. doi: 10.1074/jbc.M111324200. [DOI] [PubMed] [Google Scholar]
- 29.Ugulava NB, Gibney BR, Jarrett JT. Biochemistry. 2000;39:5206–5214. doi: 10.1021/bi9926227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ugulava NB, Gibney BR, Jarrett JT. Biochemistry. 2001;40:8343–8351. doi: 10.1021/bi0104625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jarrett JT, Choi CY, Matthews RG. Biochemistry. 1997;36:15739–15748. doi: 10.1021/bi971987t. [DOI] [PubMed] [Google Scholar]
- 32.Ugulava NB, Sacanell CJ, Jarrett JT. Biochemistry. 2001;40:8352–8358. doi: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patil PV, Ballou DP. Anal. Biochem. 2000;286:187–192. doi: 10.1006/abio.2000.4802. [DOI] [PubMed] [Google Scholar]
- 34.Williams CH, Jr., Arscott LD, Matthews RG, Thorpe C, Wilkinson KD. Methods Enzymol. 1979;62:185–198. doi: 10.1016/0076-6879(79)62217-6. [DOI] [PubMed] [Google Scholar]
- 35.Tse Sum Bui B, Florentin D, Fournier F, Ploux O, Mejean A, Marquet A. FEBS Lett. 1998;440:226–230. doi: 10.1016/s0014-5793(98)01464-1. [DOI] [PubMed] [Google Scholar]
- 36.Ugulava NB, Surerus KK, Jarrett JT. J. Am. Chem. Soc. 2002;124:9050–9051. doi: 10.1021/ja027004j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu SE, Huskey WP, Borchardt RT, Schowen RL. Biochemistry. 1983;22:2828–2832. doi: 10.1021/bi00281a009. [DOI] [PubMed] [Google Scholar]
- 38.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J. Am. Chem. Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 39.Saint-Girons I, Parsot C, Zakin MM, Barzu O, Cohen GN. CRC Crit. Rev. Biochem. 1988;23(Suppl 1):S1–S42. doi: 10.3109/10409238809083374. [DOI] [PubMed] [Google Scholar]
- 40.Hewitson KS, Ollagnier-de Choudens S, Sanakis Y, Shaw NM, Baldwin JE, Munck E, Roach PL, Fontecave M. J. Biol. Inorg. Chem. 2002;7:83–93. doi: 10.1007/s007750100268. [DOI] [PubMed] [Google Scholar]
- 41.Tse Sum Bui B, Florentin D, Marquet A, Benda R, Trautwein AX. FEBS Lett. 1999;459:411–414. doi: 10.1016/s0014-5793(99)01300-9. [DOI] [PubMed] [Google Scholar]
- 42.Cheek J, Broderick JB. J. Am. Chem. Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 43.Cosper MM, Jameson GN, Davydov R, Eidsness MK, Hoffman BM, Huynh BH, Johnson MK. J. Am. Chem. Soc. 2002;124:14006–14007. doi: 10.1021/ja0283044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.