

Abstract
Background
Nuclear objects that have in common the property of being recognized by monoclonal antibodies specific for phosphoprotein epitopes and cytoplasmic intermediate filaments (in particular, SMI-31 and RT-97) have been reported in glial and neuronal cells, in situ and in vitro. Since neurofilament and glial filaments are generally considered to be restricted to the cytoplasm, we were interested in exploring the identity of the structures labeled in the nucleus as well as the conditions under which they could be found there.
Results
Using confocal microscopy and western analysis techniques, we determined 1) the immunolabeled structures are truly within the nucleus; 2) the phosphoepitope labeled by SMI-31 and RT-97 is not specific to neurofilaments (NFs) and it can be identified on other intermediate filament proteins (IFs) in other cell types; and 3) there is a close relationship between DNA synthesis and the amount of nuclear staining by these antibodies thought to be specific for cytoplasmic proteins. Searches of protein data bases for putative phosphorylation motifs revealed that lamins, NF-H, and GFAP each contain a single tyrosine phosphorylation motif with nearly identical amino acid sequence.
Conclusion
We therefore suggest that this sequence may be the epitope recognized by SMI-31 and RT-97 mABs, and that the nuclear structures previously reported and shown here are likely phosphorylated lamin intermediate filaments, while the cytoplasmic labeling revealed by the same mABs indicates phosphorylated NFs in neurons or GFAP in glia.
Background
Objects in nuclei recognized by antibodies specific for phosphoprotein epitopes, cytoplasmic IFs, or both, have been reported in glial and neuronal cells, in situ and in vitro. The nuclear structures appear spherical or rod-like and may have a positional relationship with nuclear pores [1-4]. Morphologically, these structures appear similar to the nuclear "speckles" that are thought to be storage sites for RNA splicing factors [5-7]. However, while intermediate filament (IF) phosphoproteins could be components of nuclear speckles, they are immunologically distinct. Investigations of intermediate filaments (IF) in the nucleus have focused on lamins (see Goldman for a current review) [8], but many reports of in situ nuclear localization of cytoplasmic IFs also exist, e.g., vimentin in association with nuclear DNA in cultured fibroblasts [9,10], and an estrogen-sensitive cytokeratin association with nuclear DNA in human breast cancer cells [11].
In a recent study, Glass et al. [12], using the SMI-31 monoclonal antibody (mAB) to identify phosphorylated neurofilament proteins, reported discrete SMI-31 labeling within nuclei of SH-SY5Y neuroblastoma cells. SH-SY5Y cells are a subclone of the SK-N-SH human neuroblastoma cell line derived from neoplastic neural crest cells and under certain growth conditions, generate neuritic processes [12,13]. Sternberger and Sternberger [14] describe SMI-31 mAB as specific for phosphorylated epitopes on the heavy neurofilaments peptide (NF-H) and to a lesser extent medium neurofilament peptide (NF-M). The RT-97 mAB [9] has been characterized as recognizing phosphorylated epitopes on the 210 kDa NF-H peptide [15], and used similarly to SMI-31 to identify neurites in vitro and in situ [16-18]. One would predict, therefore, that labeling with RT97 would produce staining patterns, including nuclear, similar to those of SMI-31 in SH-SY5Y cells.
The nuclear localization of RT-97 and SMI-31 mAB could be the result of an association of phosphorylated NFs with nuclear components. Alternatively, it could be that lamins or other nuclear proteins have a phosphorylated epitope also found on NFs. For example, Schilling et al. [1] identified nuclear structures using SMI-31 mAB in rat glial nuclei in vitro and in vivo, and Shea et al. [19] showed both SMI-31 and RT-97 strongly labeled nuclei of NB2a neuroblastoma cells. Herrera [20] demonstrated nuclear localization patterns, similar to those obtained by Glass et al. [12], using rat glioma cells (9L) immunolabeled with the J1-31 mAB, which appears to recognize a phosphorylated form of GFAP [21,22].
These observations prompted us to further investigate nuclear antigens in SH-SY5Y neuroblastoma cells and to attempt to determine the relationship between these nuclear objects and cellular growth dynamics. We asked the following questions: 1) are the immuno-labeled structures within the nucleus or just closely associated; 2) is the phosphoepitope labeled by SMI-31 and RT-97 mABs specific to NFs or can it be identified on other IFs in other cell types; and 3) is there a relationship between the cell cycle as determined by DNA synthesis and the amount of nuclear labeling by SMI-31 and RT-97?
Results
The immunolabeled structures are within the nucleus
As visualized by confocal microscopy, the SMI-31 and RT-97 mABs labeled discrete locations apparently within nuclei and revealed a filamentous network in the cytoplasm. (Figure 1A,1B). The nuclear structures were clearly seen to be located seen within nuclei when visualized by confocal z-projections, and this location was confirmed by demonstrating mAB staining and DNA in single optical planes 300 nm thick. The nuclei were found to be 3 to 5 μm thick, or about 10 to 17 optical section thick. The nuclear structures were also found to co-localize with nuclear DNA using the co-localization function of the Bio-Rad LaserSharp software (data not shown). Therefore, within the limits of resolution of confocal microscopy, SMI-31 mAB and RT-97 mAB label epitopes in SH-SY5Y cells that are inside the nucleus.
Figure 1.
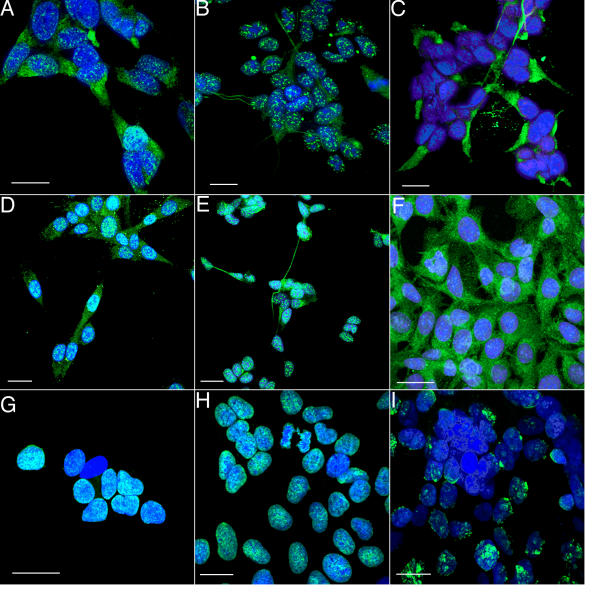
Immunolocalization of mABs SMI-31, SMI-32, anti-BrdU, and pAB anti-GFAP. A. A micrograph showing a single, confocal image plane of SH-SY5Y cells labeled with SMI-31 mAB followed by an Alexa-Fluor-488 conjugated secondary antibody (green). Nuclei were revealed by the TO-PRO-3 DNA probe (blue). The green SMI-31 mAB labeling appears as white dots within the blue nuclei, and as filamentous green structures in the cytoplasm. B. A single image plane of SH-SY5Y cells grown on glass coverslips and labeled with RT97 mAB followed by an Alexa-Fluor-488 conjugated secondary antibody (green). Nuclei were revealed by TO-PRO-3 probe (blue). RT97 mAB labeling appears as white dots in nuclei and as filamentous or clumpy green structures in the cytoplasm C. A confocal projection of SH-SY5Y cells grown on glass coverslips and labeled with SMI-32 mAB followed by an Alexa-Fluor-488 conjugated secondary antibody (green). Nuclei were revealed by TO-PRO-3 probe (blue). SMI-32 mAB did not label nuclei but did reveal filamentous green structures in the cytoplasm. D. A confocal projection of F98 rat glioma cells grown on glass coverslips and labeled with SMI-31 mAB followed by an Alexa-Fluor-488 conjugated secondary antibody (green). Nuclei were revealed by TO-PRO-3 probe (blue). SMI-31 mAB labeling appears as white dots in nuclei and as filamentous green structures in the cytoplasm. Labeling in these cells was so intense that the nuclei appeared completely full of the labeled epitope. E. A confocal projection of F98 rat glioma cells grown on glass coverslips and labeled with RT97 mAB followed by an Alexa-Fluor-488 conjugated secondary antibody (green). Nuclei were revealed by TO-PRO-3 probe (blue). SMI-31 mAB labeling appears as white dots in nuclei. RT97 staining of nuclear and cytoplasmic structures in F98 rat glioma cells was similar but less intense than seen with SMI-31 on F98 cells. F. A confocal projection of F98 cells grown on glass coverslips and labeled with anti-GFAP polyclonal AB followed by an Alexa-Fluor-488 conjugated secondary antibody (green). Nuclei were revealed by TO-PRO-3 probe (blue). The anti-GFAP revealed the expected filamentous green structures in the cytoplasm but did not label structures in nuclei. The white areas that look as if they are in the nucleus are actually green staining in the cytoplasm below (seen through) the blue nucleus. G, H, I. Confocal projections of SH-SY5Y cells showing nuclei labeled with TO-PRO-3 (blue) and anti-BrdU (green, white when co-localized with blue). These panels show nuclei of cells in culture for 1, 3, and 6 days, respectively. Labeling intensity appeared to decline as the culture became confluent, suggesting a corresponding decline in DNA synthesis and cells exiting the cell cycle. See also Figure 4. *the calibration bar in each panel represents 5 μm.
The phosphoepitope labeled by SMI-31 and RT-97 is not specific to NFs, and it can be identified on other IFs in other cell types
SMI-31 mAB labeled F98 rat glioma nuclei and cytoplasm with equal or greater avidity than in SH-SY5Y cells (Figure 1D). The cytoplasmic staining produced by SMI-31 was not distinguishable from that produced by anti-GFAP in the F98 glioma cells (Figure 1F). F98 glioma cells are not known to express neurofilament protein, and SMI-32, a mAB characterized as specific for non-phosphorylated NFs [19], stained the expected cytoplasmic network in SH-SY5Y cells (Figure 1C) but did not label any structures in F98 glioma cells (data not shown). Thus, it appears that SMI-31 recognizes a phosphoepitope that occurs on proteins in addition to NF-H and NF-M, as previously reported (see the Discussion for other reports of SMI-31 staining glial cells in situ). RT-97 also labeled nuclear and cytoplasmic structures in F98 cells (Figure 1E).
Electrophoretic and western analysis of SH-SY5Y and F98 cell extracts corroborated the finding that SMI-31 recognized an epitope on proteins other than NFs. In SH-SY5Y cell extracts, the SMI-31 and SMI-32 lanes show strong labeling of bands corresponding to the MW of NF-H and NF-M. Additional bands, not seen in the SMI-32 lane, are present in the SMI-31 and RT-97 lanes between and above the NF-H and NF-M, and probably represent forms of NF-M and NF-H with varying degrees of phosphorylation. SMI-31 and RT-97 also revealed a band or closely spaced bands at 70 kDa which corresponds approximately to the MW of lamins. Western analysis using anti-lamin B revealed a similar 70 kDa band in extracts of SH-SY5Y cells, but no bands were labeled by anti-lamin A/C. Labeled bands below 65 kDa may represent degradation products produced during cell extraction. Normal serum controls were negative (data not shown).
Western analysis with anti-GFAP pAB, SMI-31 mAB, RT-97 mAB, and anti-lamins mAB was performed on F98 cell extracts subjected to SDS-PAGE. The anti-GFAP lane shows a strong band at the expected MW, and multiple bands above it as well as minor bands below. The higher MW bands most likely represent GFAP multimers, while the lower MW bands are possibly degradation products. Multiple bands also appear in both the SMI-31 and RT-97 lanes, although for the most part the antibodies do not appear to label the same bands. In lanes labeled with SMI-31, RT-97 or anti-lamin B, a weak but well defined band appears at approximately 70 kDa (Figure 2). Controls (normal mouse serum) were negative (data not shown).
Figure 2.
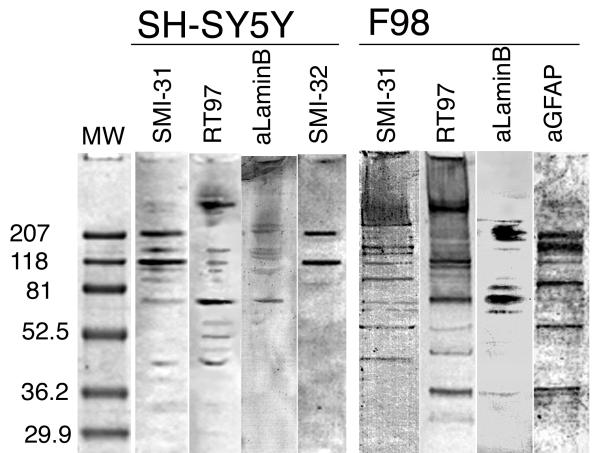
Western analysis with SMI-31, RT97, anti-lamin B, SMI-32 on SH-SY5Y, and SMI-31, RT97, anti-lamin B, anti-GFAP on F98 cell extracts subjected to SDS-PAGE. On SH-SY5Y, the SMI-31 and SMI-32 lanes show strong labeling of bands corresponding to the MW of NF-H, and NF-M. Additional bands, not seen in the SMI-32, are present in the SMI-31 and RT97 lanes between and above the NF-H and NF-M, and probably represent phosphorylated forms of NF-M and NF-H. SMI-31, RT-97 and anti-lamin B also revealed a band or closely spaced bands at approximately 70 kD which corresponds approximately to the MW of lamin B. Western analysis with anti-lamin A/C revealed no bands in either SH-SY5Y or F98 extracts (not shown). On F98 cells, SMI-31 labeled several well defined bands, including those of MW corresponding to GFAP and lamin B. The RT97 mAB also strongly labeled several F98 bands, including bands corresponding to GFAP and lamin B. The anti-lamin B mAB strongly labeled bands at the appropriate MW (~70 kD) and also a cluster of bands in the range of the heavy NF MW (~207 kD). The anti-GFAP pAB labeled a band at the appropriate MW (~52 kD) and also band higher of MW appropriate for multimers of GFAP (~100, 150, 200 kD). Labeled bands lower than 65 kD may represent degradation products produced during cell extraction. Negative controls (control blots stained with appropriate normal serum) showed no labeling (data not shown).
Numerous putative phosphorylation motifs were identified in the amino acid sequences of NF-H, GFAP and nuclear lamins A, B, and C when analyzed by the NetPhos server, including a single tyrosine kinase motif, LNDRFAGYIDKVR, found in the N-terminal region of all five proteins (see Figure 3). In addition, 74 putative serine (= 0.99), 26 threonine (= 0.9) and 7 other tyrosine (= 0.5) phosphorylation sites were identified; however, only the tyrosine phosphorylation site shown was shared among the five proteins analyzed. Therefore it is possible that this sequence constitutes or contains the epitope recognized by the SMI-31 and RT-97 mABs, and may account for the cross-reactivity of these antibodies with respect to NF-H, GFAP, and lamins.
Figure 3.

A portion of the amino acid sequences for NF-H, GFAP, and lamin A, B, and C. The Match-Box server prints aligned sequences in lower case, an unaligned as uppercase. The numbers printed below each column of aligned amino acids indicates the reliability of the alignment, where 1 = 10%, 3 = 20%, and 5 = 50% chance of equal occurrence in related or unrelated sequences (low scores are best, Depiereux et al, 1997). While we found many phosphorylation motifs in the sequences examined, the indicated tyrosine kinase motif was the only one shared with a high degree of identity among all 5 sequences. As it occurs only once, the remainder of the sequences are not shown. The darker gray shading indicates the predicted motif for NF-H (the reference sequence), while the light gray shows regions of GFAP and lamins that include the tyrosine (bold-face "Y") in a motif with very high sequence identity.
DNA synthesis and the volume and intensity of nuclear labeling are related
Analyses of nuclear labeling intensity and comparison to proliferation index as a function of days in culture revealed a strong correlation. Comparison of nuclear labeling of SH-SY5Y cells immuno-stained with SMI-31 and RT-97 mABs to the culture proliferation index shows that nuclear labeling reached its greatest extent when DNA synthesis was also maximal. Both values fell as cells exited the cell cycle as indicated by lack of BrdU uptake, and by day 5, when the cultures appear confluent, essentially all cell replication had stopped (see Figure 1G,1H,1I and Figure 4).
Figure 4.
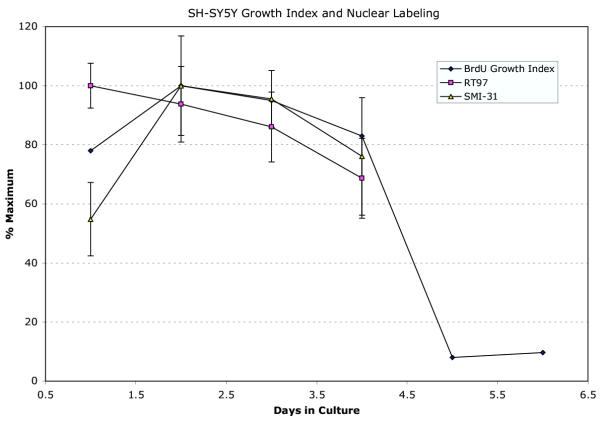
Graph showing a strong correlation between the culture proliferation index (percent nuclei showing BrdU incorporation) and the extent of nuclear labeling as a function of days in culture for SH-SY5Y cells immuno-stained for SMI-31, RT97. Values for SMI-31 and RT-97 labeling represent averages (± SEM) of the percentage of nuclear area labeled as determined by confocal projection. Proliferation index data is shown through day 5 and 6 to illustrate that the culture enters a confluent, stationary phase. See also Figure 1G,1H and 1I.
Discussion
SH-SY5Y cells immunolabeled with SMI-31 mAB or RT-97 mAB demonstrated discrete nuclear localizations in nuclei with pronounced staining in neuronal processes and diffuse staining within cell bodies. Confocal microscopy demonstrated clearly that the antibodies label structures within the nucleus. Comparison among antibodies that are specific for phosphorylated IFs (SMI-31, RT-97) and those not (SMI-32, anti-GFAP) suggests that the nuclear structures are a phosphorylated IF. Cross reactivity of both SMI-31 and RT-97 on glial cytoplasm and nuclei indicates the epitope is not cell type specific. Examination of western blots indicate SMI-31 and RT-97 recognize, among others, a protein in the 66 – 74 kDa range in extracts of both neuroblastoma and glioma cells. Western analysis with anti-lamin B labeled a similar band in the same extracts. The calculated molecular weights of lamins A, B and C are approximately 74, 66, and 65 kDa, respectively; therefore, it seems most likely that lamins, NF-H, and GFAP all share a common motif that, when phosphorylated, is recognized by SMI-31 and RT-97 mABs. In support of this, a search for phosphorylation motifs among NF-H, GFAP and lamins A, B, and C revealed a single, high probability tyrosine site that occurs once on each of these proteins (Figure 3), further suggesting the possibility that this site constitutes or is contained within the epitope recognized by these two antibodies.
Based on the finding of a shared, tyrosine phosphorylation site, it seems likely that the nuclear structures reported in this and previous studies [1,12] that employed SMI-31 are phosphorylated lamins. Furthermore, nuclear labeling and DNA synthesis appeared tightly associated in SH-SY5Y cultures, consistent with the suspected role of lamins in DNA replication [8]. Others have reported SMI-31 labeling in glial as well as neuronal nuclei. For example, Schilling et al. [1] identified developmentally regulated nuclear antigens recognized by SMI-31 mAB in C6 rat glioma cells in vitro as well as glial and neuronal nuclei in vivo, suggesting the presence non-neurofilament epitopes. In addition, Shea et al. [19], in a study of neurite outgrowth from NB2a cells, showed that both the SMI-31 and RT-97 mABs strongly labeled nuclei of undifferentiated cells, and the nuclear labeling declined as differentiation progressed. These results are similar to the results reported here. In a recent paper by Glass et al. [12], SMI-31 was described as labeling nuclear epitopes in cells in culture and in neurons of mature rat brain as visualized by cryo-sectioning and confocal microscopy. However, in light of the findings reported here, the nuclear objects reported by Glass et al. in rat brain sections may have been in mitotically active glial cells as well as in neurons.
Although these nuclear epitopes appear similar to nuclear speckles, the latter have not been reported as IF-associated structures, but rather storage sites of RNA processing proteins (see Sanford 2002, for a current, brief review [5]). However, as it is know that lamins are involved in nucleo-skeletal organization [23] and organization of DNA replication sites [4], it would not be unexpected to find lamins associated with nuclear speckles, perhaps as structural organizers.
Conclusions
Both RT-97 and SMI-31 may recognize the phosphorylated tyrosine motif, LNDRFAGYIDKVR, which occurs in NF-H and thus provides the basis for the claim of these antibodies being specific for phosphorylated NFs [14]. However, this same motif occurs in GFAP and lamins A, B, and C and possibly other proteins, and is not restricted to neurons as shown by the glial staining reported here and in other studies [1]. It would be interesting to look at nuclei from a variety of tissue types using these antibodies as probes.
In this study and in previous studies performed in other laboratories [1,19], a close relationship between the nuclear phosphoepitopes and growth phase has been reported. Because of the association of phosphorylated lamins with DNA replication and the cell cycle [8], and the sharing of phosphoepitopes among lamins, NFs, and GFAP, it seems most probable that the nuclear epitopes revealed in neuroblastoma and glioma cells by SMI-31 and RT-97 are contained on lamin B.
Methods
Preparation and Maintenance of Cell Cultures
All culture reagents were obtained from Sigma, St. Louis, MO, unless otherwise noted. SH-SY5Y cells were maintained in culture as previously described [12]. Initial cell density was determined using a hemacytometer (Fisher Scientific, Pittsburgh, PA) and diluted appropriately to maintain a seeding density of 4.0 × 104 cells/cm2 of surface area. Cells were grown for approximately 72 hours and fixed in cold methanol unless otherwise indicated. F98 rat glioma cells (ATCC, http://www.atcc.org/, Manassas, VA) were also used for comparative purposes in this study, and were cultured as described for SH-SY5Y cells.
Fixation and Immunofluorescence Staining for Confocal Microscopy
Cells grown on coverslips were washed briefly in a phosphate-buffered saline (PBS) solution (pH 7.4, 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4) and then either fixed in 100% methanol at -20°C for 1 minute or at room temperature in a 4% formaldehyde (PFA) solution freshly made from paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, PA) and buffered with PBS (or PBS with 0.2% Tween-20, PBST) for 10 minutes followed by 3 washes in PBS/PBST for 10 minutes each. As a previous study [13] had suggested nuclear staining by antibodies targeted to cytoplasmic proteins might be fixation dependent [4] or an artifact of fixation, we did a preliminary comparison of SMI-31 labeling of methanol vs. paraformaldehyde fixed cells. We found minor differences in the staining pattern between methanol and paraformaldehyde fixed cells, but immuno-labeled nuclear structures were visible regardless of fixation. No differences in the localization of mAB staining in the cells was noted when comparing between the two methods of fixation.
After fixation the cell-bearing coverslips were incubated in 20% non-fat powdered milk in PBS to block non-specific protein-protein interactions for 2 hours at room temperature. The coverslips were then washed 3 times, 10 minutes each, and incubated with the desired primary antibody (Table 1) for 16–24 hours at 4°C. The cells were again washed three times in PBS (10 minutes each) and incubated with the appropriate secondary antibody (Table 2) for 2 hours at room temperature. During the final 30 minutes of secondary antibody incubation 40 μl of TO-PRO-3 (T-3605, Molecular Probes, Eugene, OR), a DNA specific probe was added for identification of nuclei (Table 1). Negative controls for all immuno-preparations were prepared by use of the appropriate normal serum in place of the primary antibody. The coverslips were then mounted on glass slides in a medium consisting of 90% glycerol in water containing 1 mg/ml p-phenylenediamine, and stored at -20°C in the dark.
Table 1.
Primary antibodies.
| Antibody | Species/Isotype | Dilution for Fluorescence | Dilution for Western Analysis | Supplier |
| Anti-BrdU mAB | Mouse IgG | 1:400 in PBS | Molecular Probes, Eugene, OR (#A-21300) | |
| Anti-GFAP pAB | Rabbit IgG | 1:86 in PBS | 1:400 in PBS | Sigma Chem. Co., St. Louis, MO (#G-9269) |
| Anti-lamin A/C mAB | Mouse IgG | 1:500 | Chemicon International, Temecula CA (MAB3538) | |
| Anti-lamin B mAB | Mouse IgG | 1:500 | Chemicon International, Temecula CA (MAB3536) | |
| Anti-neurofilament, RT-97 | Mouse IgG | Undiluted culture supernatant | Undiluted culture supernatant | Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, IA |
| Anti-nonphospho-neurofilament, SMI-32 mAB | Mouse IgG1 | 1:1000 in PBS | 1:1000 in PBS | Sternberger Monoclonals, Inc. Lutherville, MD |
| Anti-phospho-neurofilament, SMI-31 mAB | Mouse IgG1 | 1:1000 in PBS | 1:2000 in PBS | Sternberger Monoclonals, Inc., Lutherville, MD |
Table 2.
Secondary antibodies and probes used for immunofluorescence and western blot analysis.
| Antibody/Probe (Catalog #) | Conjugate | Dilution | Supplier |
| Goat anti-mouse IgG (#A-11001) | Alexa-fluor 488 | 1:200 in PBS | Molecular Probes, Eugene, OR |
| Goat anti-rabbit IgG (#T-6778) | Alexa-fluor 488 | 1:200 in PBS | Molecular Probes, Eugene, OR |
| Goat anti-mouse polyvalent (#A-0162) | Alkaline Phosphatase | 1:4000 in PBS | Sigma Chem. Co, St. Louis, MO |
| Goat anti-rabbit IgG (#A-3812) | Alkaline Phosphatase | 1:30,000 in PBS | Sigma Chem. Co., St. Louis, MO |
| Nucleic Acid binding probe; TO-PRO-3 iodine (#T-3605) | --- | 1:1000 in PBS | Molecular Probes, Eugene, OR |
Microscopy and Quantitative Analysis of Immunolabeling
An Olympus IX-70 fitted with a Bio-Rad MRC 1024 confocal scanhead via the Keller port was used for all image acquisition. Initial image processing was done using Bio-Rad LaserSharp software running on a Compaq PC; final processing and printing of images was done using Adobe Photoshop 7.0 software.
Culture dynamics were determined for SH-SY5Y neuroblastoma cells at an initial density of 4.0 × 104 cells/cm2. Cells were introduced into five 25 cm2 flasks, then harvested from one flask each day using 5 ml Puck's saline solution (pH 7.3, 137 mM NaCl, 5.4 mM KCl, 0.7 mM Na2HPO4, 9.5 mM HEPES, 5.6 mM dextrose, and 0.8 mM EDTA). Detached cells were pelleted at 2500 rpm for 5 minutes in a clinical centrifuge and resuspended in 2 ml of PBS. Cell density was determined each day by counting the number of cells present in representative samples using a two-chambered hemacytometer. A proliferation index was calculated as the percentage of nuclei present engaged in DNA synthesis, using BrdU uptake as an indicator (see part D below for BrdU labeling method; also see Figure 4).
Quantitation of fluorescent labeling was performed on confocal images collected using calibrated microscope channel settings for cells grown 1–5 days. All images were collected from cells stained at the same time with the same batch of antibody. Thus any differences in staining intensity and area stained were expected to be a function of time in culture and/or culture density. This entire process was performed in duplicate.
Confocal stacks were projected and then imported into NIH Image 1.6 software (freeware at http://rsb.info.nih.gov/nih-image/). Density slice filtering was used to remove background autofluorescence and then record the area stained in each nucleus in the flattened image. Intensity values above background were also determined, but not used as they were found insensitive to growth conditions and more sensitive to nuclear thickness (the number of optical sections in the projected stack) than to experimental parameters. The area per nucleus stained is not affected by the number of sections in the projected stack, and was significantly related to culture conditions. These data were used to relate nuclear area stained to time in culture. Statistical analysis was done using linear regression and single factor analysis of variance (ANOVA).
Identification of Actively Growing Cells using 5-Bromo-2'-Deoxyuridine
Cells were seeded onto glass coverslips at an initial density of 4.0 × 104 cells/cm2 and allowed to grow for 1–5 days. Twenty-four hours prior to fixation, the cells were incubated with 10 μM 5-bromo-2'-deoxyuridine in MEM (see Table 1). At the appropriate time, the cells were fixed in methanol at -20°C for 1–2 minutes, allowed to air dry, then stored at -20°C until all coverslips were ready for processing. The cells were rehydrated in PBS for 5 minutes followed by immersion in 2N HCl for 1 hour at room temperature. The cells were incubated in 0.1 M borate buffer (pH 8.5, 0.1 M boric acid, 25 mM Na2B4O7, and 75 mM NaCl) twice for 5 minutes each (10 minutes total), followed by three 10 minute washes in PBS. Next, cells were immunolabeled using anti-BrdU monoclonal mouse IgG primary antibody followed by goat anti-mouse IgG conjugated to Alexa-Fluor 488 (see Tables 1 and 2). All antibody incubations were 1 hour at room temperature in reduced light. During the final 30 minutes of secondary antibody incubation TO-PRO-3 nucleic acid stain (Molecular Probes) was added at a 1:1000 dilution. The coverslips were then washed and mounted for microscopic analysis. The intensity and area/nucleus of BrdU labeling was quantitated as described above for immunolabeling.
Preparation of Total Cell Lysates, Electrophoresis and Western Blot Analysis
Cells were seeded at an initial density of 4.0 × 104 cells/cm2 and grown in 150 cm2 flasks for 2 days. MEM was removed and cells were detached from the substrate using 10 ml Puck's saline solution and incubated at 37°C for 5 minutes. The detached cells were transferred to a centrifuge tube and spun at 2500 rpm for 5 minutes. The pellet was resuspended in 5 volumes of 2 × sample buffer (60 mM Tris-HCl, 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, 0.1% bromphenol blue, and 1 mM EGTA) then diluted 1:1 in PBS. Samples were incubated in boiling water for 5 minutes then passed through a 20-gauge and 26-gauge needle repeatedly to shear chromosomal DNA. The lysates were centrifuged at 4000 × g for 15 minutes, then aliquoted and stored at -20°C prior to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western analysis as previously described [12]. Protein concentrations of the cell lysates were determined using the Bio-Rad Protein Assay (Hercules, CA), and sample volumes adjusted so that 5–10 μg of total protein were loaded into wells of a discontinuous (12% separating, 4% stacking) polyacrylamide gel. Gels were prepared in duplicate and one of each pair stained in Coomassie blue and the other used for electrophoretic transfer onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The identities of the proteins bound to the nitrocellulose membrane were revealed using the appropriate primary and secondary antibodies (Tables 1 and 2). Molecular weight values were determined by calculating Rf ratios as the distance from the bottom of the well to the protein band of interest divided by the distance from the bottom of the well to the dye front. Standard curves were developed using log MW of protein standards versus Rf ratio of those standards. Apparent molecular weights of proteins identified by western blot analysis were determined by extrapolating MW from the standard curve using measured Rf ratios.
Identification of Consensus Phosphorylation Motifs in IF proteins
Amino acid sequences for human NF-H (Accession QFHUH), lamin A/C (Accession P02545), and B (Accession P20700), and human GFAP (Accession XP_050159) were obtained from the National Center for Biotechnology Information Protein Data Base http://www.ncbi.nlm.nih.gov:80/entrez/query.fcgi?db=Protein. These were searched for potential phosphorylation sites using PhosphoBase v2.0 http://www.cbs.dtu.dk/databases/PhosphoBase/, a Web based analysis tool provided by the Center for Biological Sequence Analysis, BioCentrum-DTU, Technical University of Denmark [24]. The five amino acid sequences were aligned to NF-H using the Match-Box server located on the Internet at http://www.fundp.ac.be/sciences/biologie/bms/matchbox_submit.html. Amino acid sequences predicted by PhosphoBase as serine, threonine, or tyrosine phosphorylation motifs for NF-H were then compared to the aligned proteins in regions of high identity, as determined by reliability scores generated by the Match-Box server [25].
Authors' contributions
SW designed and performed the experimental aspects of this work as her master's thesis. DG provided guidance in conceptual interpretation of the results and performed the quantitative measurements of fluorescence and statistical analyses. TR provided expertise with respect to neuroblastoma culture and antibodies. NC was responsible for overseeing the immunological part of this work. JK was responsible for the coordination of the overall project, supervised the design, and provided material support for this project carried out in his laboratory. The original manuscript draft was prepared by SW, edited by all the authors, and prepared for publication by JK. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
The authors gratefully acknowledge the contributions of summer students Grace Gonzalez, Ben Yock and Beth Borkosky. This work has been supported in part by grants from the National Science Foundation (NSF DUE # 96-50654, ESI 9731321), National Institute of Health Bridges to the Baccalaureate (NIH GM58375-01A1), Texas Higher Education Coordinating Board (ATP003658-0193-1999 and ARP003658-0496-2001), and the Faculty Research Enhancement Program and Department of Biology of Southwest Texas State University.
Contributor Information
Shannon E Weigum, Email: sweigum@mail.utexas.edu.
Dana M García, Email: dana_garcia@swt.edu.
Timothy D Raabe, Email: traabe@stmarytx.edu.
Nicholas Christodoulides, Email: DrNick@mail.utexas.edu.
Joseph R Koke, Email: jrkoke@swt.edu.
References
- Schilling K, Duvernoy C, Keck S, Pilgrim C. Detection and partial characterization of a developmentally regulated nuclear antigen in neural cells in vitro and in vivo. J Histochem Cytochem. 1989;37:241–247. doi: 10.1177/37.2.2492046. [DOI] [PubMed] [Google Scholar]
- Singh MV, Price KJ, Bhatnagar R, Johnson R, Malhotra SK. J1-31 antigen of astrocytes: cytoplasmic and nuclear localization. Dendron. 1992;1:91–108. [Google Scholar]
- Singh MV, Price KJ, Bhatnagar R, Malhotra SK. Novel rod-shaped structures identified in glioma cell nuclei by immunolabeling and confocal laser fluorescence microscopy. Biomedical Letters. 1994;50:163–172. [Google Scholar]
- Malhotra SK, Bhatnagar R, Shnitka TK, Herrera JJ, Koke JR, Singh MV. Rat glioma cell line as a model for astrogliosis. Cytobios. 1995;82:39–51. [PubMed] [Google Scholar]
- Sanford Jeremy. Nuclear Compartments: Nuclear Splicing Speckles. In: Wendy Bickmore, editor. Nuclear Protein Database (NDP) UK, Medical Research Council Human Genetics Unit; 2002. http://npd.hgu.mrc.ac.uk/compartments/speckles.html [Google Scholar]
- Kruhlak MJ, Lever MA, Fischle W, Verdin E, Bazett-Jones DP, Hendzel MJ. Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J Cell Biol. 2000;150:41–51. doi: 10.1083/jcb.150.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz PJ, Spector DL. Compartmentalization of RNA processing factors within nuclear speckles. J Struct Biol. 2000;129:241–251. doi: 10.1006/jsbi.2000.4213. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes and Development. 2002; 16:533–547. doi: 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- Traub P, Nelson WJ, Kuhn S, Vorgias CE. The interaction in vitro of the intermediate filament protein vimentin with naturally occurring RNAs and DNAs. J Biol Chem. 1983;258:1456–1466. [PubMed] [Google Scholar]
- Hartig R, Shoeman RL, Janetzko A, Tolstonog G, Traub P. DNA-mediated transport of the intermediate filament protein vimentin into the nucleus of cultured cells. J Cell Sci. 1998;111:3573–3584. doi: 10.1242/jcs.111.24.3573. [DOI] [PubMed] [Google Scholar]
- Spencer VA, Coutts AS, Samuel SK, Murphy LC, Davie JR. Estrogen regulates the association of intermediate filament proteins with nuclear DNA in human breast cancer cells. J Biol Chem. 1998;273:29093–29097. doi: 10.1074/jbc.273.44.29093. [DOI] [PubMed] [Google Scholar]
- Glass TL, Raabe TD, Garcia DM, Koke JR. Phosphorylated neurofilaments and SNAP-25 in cultured SH-SY5Y neuroblastoma cells. Brain Res. 2002;934:43–48. doi: 10.1016/s0006-8993(02)02317-x. [DOI] [PubMed] [Google Scholar]
- Hahn M, Glass TL, Koke JR. Extracellular matrix effects on a neuroblastoma cell line. Cytobios. 2000;102:7–19. [PubMed] [Google Scholar]
- Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci U S A. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JN, Anderton BH. Monoclonal antibodies to mammalian neurofilaments. Biosci Rep. 1981;1:263–268. doi: 10.1007/BF01114913. [DOI] [PubMed] [Google Scholar]
- Emerling DE, Lander AD. Laminar specific attachment and neurite outgrowth of thalamic neurons on cultured slices of developing cerebral neocortex. Development. 1994;120:2811–2811. doi: 10.1242/dev.120.10.2811. [DOI] [PubMed] [Google Scholar]
- Greenwood AL, Turner EE, Anderson DJ. Identification of dividing, determined sensory neuron precursors in the mammalian neural crest. Development. 1999;126:3545–3559. doi: 10.1242/dev.126.16.3545. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Ribera B, Goutan E, Pulg B, Rey MJ, Cardozo A, Viñal F, Ribalta T. Phosphorylated Map kinase (ERK1, ERK2) expression is associated with early Tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathology. 2001;11:144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB, Beermann ML, Nixon RA. Appearance and localization of phosphorylated variants of the high molecular weight neurofilament protein in NB2a/d1 cytoskeletons during differentiation. Brain Res Dev Brain Res. 1989;50:142–146. doi: 10.1016/0165-3806(89)90134-x. [DOI] [PubMed] [Google Scholar]
- Herrera JJ, Taylor TA, Malhotra SK, Koke JR. In vitro astrogliosis: chronology of J1-31 antigen expression in reactive 9L astrocytes. Molecular Biology of the Cell. 1995;6:748a. [Google Scholar]
- Stevenson P. Biochemical Characterization of a Possible Precursor to Glial Fibrillary Acidic Protein in Reactive Astrocytes. Thesis, Southwest Texas State University. 1996.
- García DM, Weigum SE, Koke JR. GFAP and nuclear lamins share an epitope recognized by monoclonal antibody J1-31. Brain Research. 2003;In press. doi: 10.1016/s0006-8993(03)02597-6. [DOI] [PubMed] [Google Scholar]
- Singh S, Koke JR, Gupta PD, Malhotra SK. Multiple roles of intermediate filaments. Cytobios. 1994;77:41–57. [PubMed] [Google Scholar]
- Kreegipuu A, Blom N, Brunak S. PhosphoBase, a database of phosphorylation sites: release 2.0. Nucleic Acids Res. 1999;27:237–239. doi: 10.1093/nar/27.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depiereux E, Baudoux G, Briffeuil P, De Bolle X, Vinals C, Feytmans E. Match-Box server: a multiple sequence alignment tool placing emphasis on reliability. CABIOS. 1997;13:249–256. doi: 10.1093/bioinformatics/13.3.249. [DOI] [PubMed] [Google Scholar]