

Abstract
Screening in mixtures is a common approach for increasing the efficiency of high-throughput screening. Here we investigate how the “compound load” of mixtures influences promiscuous aggregate-based inhibition. We screened 764 molecules individually and in mixtures of 10 at 5 μM each, comparing the observed inhibition of the mixtures to that predicted from single-compound results. Synergistic effects on aggregation predominated, although antagonism was also observed. These results suggest that screening mixtures can increase aggregation-based inhibition in a nonadditive manner.
High-throughput screening (HTS) is widely used to interrogate large libraries of small molecules for biological reagents or new leads for drug discovery. A common approach in HTS is the screening of mixtures of small molecules.1-6 This approach is efficient from the standpoint of reagent consumption, increases throughput, and is often required when screening natural products7 or combinatorial libraries. Typically, the screening of mixtures is followed by a deconvolution step where active agents in promising mixtures are identified. Several deconvolution methods are used, including iterative,8,9 positional scanning,10 and self-deconvoluting matrices.2,11
One problem in HTS is the occurrence of false-positive hits; molecules that appear to inhibit the target but on investigation do so for what turn out to be uninteresting mechanisms. These include chemical modification of the target, interference in the assay readout, and promiscuous aggregate-based inhibition. Such phenomena have inspired considerable literature.12-17 Our own interest has focused on the formation of promiscuous aggregates. These are large particles, 100-1000 nm in diameter, formed by the self-association of small molecules at micromolar concentrations in aqueous media.18-21 They nonspecifically interact with enzymes, sequestering them from substrate. Intruigingly, this inhibition is reversed in the presence of low concentrations of nonionic detergents. Recently we exploited this effect to develop a high-throughput assay for detecting aggregates via detergent-sensitive inhibition. When we used this assay to screen a diverse set of drug-like organic molecules, we found that 19% of them formed promiscuous aggregates at 30 μM.22
Anecdotal evidence suggests that screening molecules in mixtures, as opposed to individually, may lead to its own set of artifacts: synergistic or antagonistic effects on inhibition that can obscure the presence of true actives. This issue has been the source of some controversy.6,23,24 Our interest in this debate stems from the effects of “compound load,” which may be considered the total concentration of organic material in a mixture, on the formation of promiscuous aggregates. Whereas most molecules might be well-behaved at low concentrations in isolation, upon mixing they might affect each other non-additively, specifically through aggregation. Here, we investigate how the behavior of mixtures of known promiscuous aggregators and nonaggregators deviates from a simple summation model for combinations of mutually exclusive inhibitors.
A set of 764 soluble, diverse, drug-like molecules were purchased from Chemical Diversity, Inc., and randomly combined into 80 mixtures of, on average, 10 molecules. Each molecule was present at a concentration of 5 μM for a total chemical load of 50 μM. The mixtures were assayed for inhibition against the model enzyme β-lactamase as previously described.18-22 Each of the molecules was also screened individually at 5 μM, both in the presence and absence of nonionic detergent.22 These experiments indicated that the 764 molecules consisted of 128 aggregators, 564 nonaggregators, and 72 molecules with intermediate inhibition. An aggregator was defined as a molecule that showed detergent-reversible inhibition greater than 24%, and a nonaggregator inhibited less than 11%.22 There were no molecules whose inhibition was not reversible by detergent in this set.
These individual screening results were fed into a model of the null hypothesis that inhibitors in a mixture would act exclusively and reversibly and exist in equilibrium with free enzyme (Figure 1). According to this model, the total percent of enzyme inhibited by a mixture of n mutually exclusive inhibitors is given by the following equation, adapted from refs 25-27:
Figure 1.

Equilibrium diagram for a mixture of mutually exclusive inhibitors. Total inhibition can be determined from eq 1.
| (1) |
One consequence of this classical summation model is that a combination of inhibitors very quickly yields diminishing returns. For example, a mixture of three well-behaved competing inhibitors that individually have percent inhibitions of 75%, 25%, and 50% (i.e., ([Ii · E]/[E]) ) 3, 1/3, and 1, respectively) will only have an overall inhibition of 81% as a mixture. To be conservative, we included what would normally be considered insignificant levels of inhibition (<11%)22 when calculating predicted inhibition by eq 1. This resulted in increased levels of predicted inhibition in the mixtures and thus reduction of the apparent synergistic effects.
The apparent IC50 of any single, noncooperative and well-behaved inhibitor is given by the concentration of that inhibitor divided by the ratio of its percent inhibition to its percent activity, i.e., ([Ii]/([Ii·E]/[E])). It is appropriate to use this ratio-based apparent IC50 when comparing the predicted and observed behavior of mixtures since, unlike raw percent inhibition, this term is linear with potency (e.g., at a fixed concentration, a molecule exhibiting 80% inhibition is actually sixteen-fold more potent than an inhibitor exhibiting 20% inhibition). We define antagonism and synergy as the measured inhibition of mixtures that is less than and more than predicted by eq 1, respectively.
The 80 mixtures were screened in quadruplicate against β-lactamase (Figure 2a). Predicted potencies, calculated from single-molecule assays, are compared to observed potencies in Figure 2b. In the ideal case that a mixture behaved as a collection of simple, mutually exclusive inhibitors, the ratio between the predicted potency and the observed potency would be one, and the Log of the ratio would equal zero. In reality, large deviations between predicted and experimental results were observed for most mixtures; 45% of the mixtures were at least 2-fold more potent than predicted and 16% of mixtures showed at least a 2-fold diminution. Large deviations were seen for 24% of the mixtures, which were greater than 10-fold more potent than predicted, and 10% of the mixtures, which were at least 10-fold less potent than expected. Overall, the deviation ranged from 200-fold less potent to 447-fold more potent than predicted. In an effort to keep extreme levels of inhibition from skewing these results, maximum inhibition was arbitrarily set to 95%. Had we not done this, the bias toward extreme synergism and antagonism would have been higher still. Modeling mixtures as being comprised of mutually nonexclusive25 or cooperative inhibitors27did not explain these nonadditive effects; synergistic effects are larger than even a nonexclusive model predicts, and neither of these alternate treatments of the data explain the observed antagonism (analyses not shown).
Figure 2.
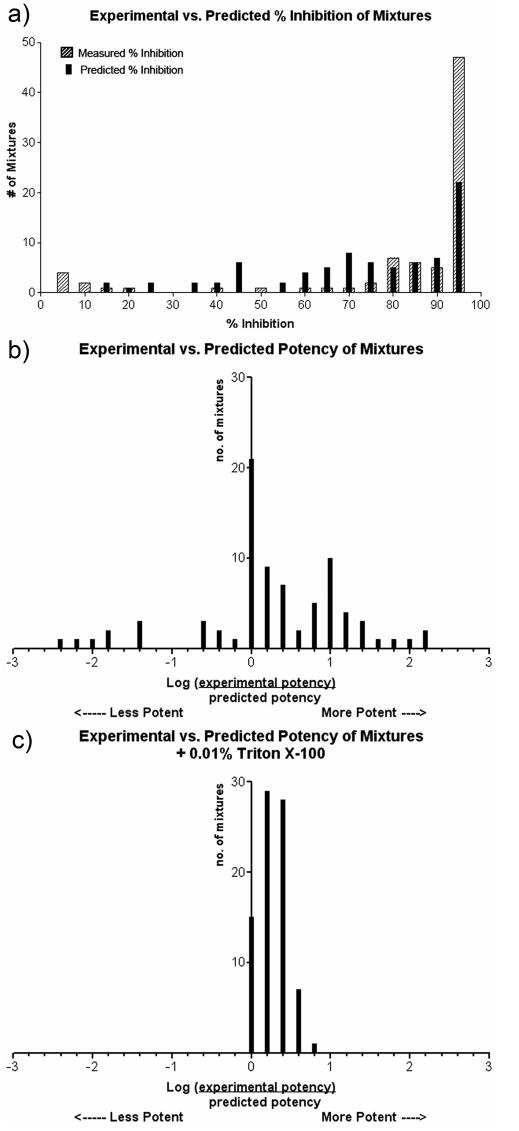
(a) 80 mixtures binned according to their observed and predicted percent inhibition. (b) 80 mixtures of molecules binned according to the deviation between the observed and predicted results. The deviation is a comparison of the predicted and observed potency (see text). (c) Identical analysis as in part b for 80 mixtures of molecules screened in the presence of 0.01% Triton X-100.
Deviations from prediction were largely eliminated when the multicompound assays were repeated in the presence of 0.01% Triton X-100, a nonionic detergent that disrupts aggregates (Figure 2c).19,21,22 As expected, some mixtures retained low levels of inhibition at this concentration of detergent. As before, the minimum predicted inhibition of each mixture was set to 5% to minimize the effect of dividing small numbers. In the presence of Triton X-100, effects ranged from 0 (no effect) to 6-fold more potent than expected (Figure 2c), indicating almost total reversal of inhibition, synergy, and of antagonism for most mixtures.
To investigate the reliability of these results, six mixtures were rescreened alongside their individual components in the high-throughput, plate-based assay as well as in a low-throughput version of the assay (each in duplicate).22 This format allowed for the most precise individual compound data to be used in predicting the inhibition of the mixtures. The low-throughput assay was conducted in 1 mL cuvettes, on a HP 8453 spectrophotometer. Of the six mixtures, three were chosen from among the most synergistic, two were chosen from among the most antagonistic and one mixture was chosen that lacked nonadditive effects. These mixtures behaved identically when rescreened with the high-throughput assay: the three mixtures displaying synergistic effects continued to inhibit significantly more than predicted, based on single compound assays that were conducted at the same time, on the same plate. Correspondingly, the antagonistic mixtures continued to inhibit less than expected (Supporting Information Table 1). The low-throughput results were qualitatively the same, though the actual magnitude of inhibition both in the one-at-a-time and mixture assays was somewhat diminished, reflecting a general tendency of low-volume, plate-based assays to be more sensitive to promiscuous aggregation than high-volume cuvette-based assays. Nevertheless, the low-throughput assay essentially reproduced the high-throughput results.
Taken together, these results suggest a role for “compound load” in aggregation-based inhibition, typically potentiating, though occasionally antagonizing it. Unexpectedly, the magnitude of these effects were uncorrelated with the number of “active” (known aggregator or ambiguous) molecules in the well (data not shown). Extreme examples of synergy included Mixtures 1 and 2, which were composed entirely of molecules inactive at 5 μM individually that when combined potently inhibited β-lactamase (98% and 97% inhibition, respectively). The simplest interpretation of these effects is that the amount of overall concentration of organic materials-the compound loads-has passed a point where aggregation occurs. Thus, whereas none of the components in Mixtures 1 and 2 were promiscuous aggregators at 5 μM, four molecules from Mixture 1 and three molecules from Mixture 2 did form aggregates when measured individually at 30 μM, as determined in our previous study of these molecules.22 This supports the role of chemical load in these nonadditive effects, though it does not suggest an obvious distinction between synergistic or antagonistic mixtures.
To confirm that aggregates were present in the mixtures, we used dynamic light scattering (DLS) to measure particle formation in two of them as well as in solutions of the respective individual molecules. Consistent with their lack of inhibition, no particles were observed in the individual solutions (Figure 3). However, mixtures of these molecules exhibited intense light scattering, and particles were observed with radii in the 100 nm size range. Admittedly, a caveat to these observations is that light scattering was also observed in a mixture that did not significantly inhibit β-lactamase (data not shown). This result is, however, consistent with earlier observations that light scattering is a necessary-but-not-sufficient condition for the presence of promiscuous aggregates.22 The inhibition displayed by the mixtures was largely eliminated by 0.01% Triton X-100, also consistent with an aggregation-based mechanism.
Figure 3.
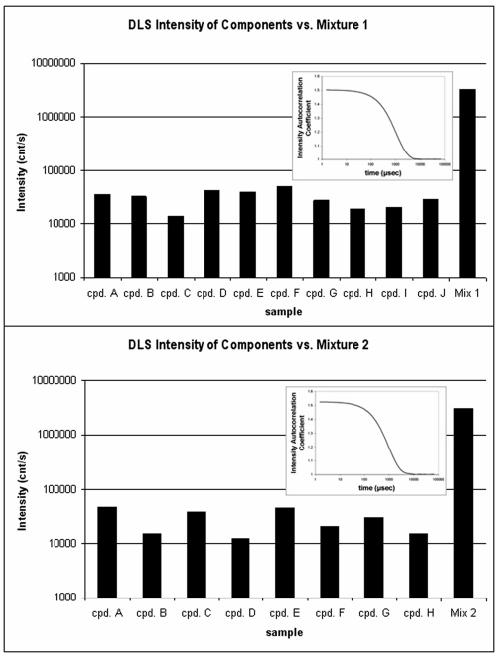
Light scattering study for two mixtures comprised of inactive molecules. Both mixtures scattered > 10-fold more intensely than any individual components. Inset: Autocorrelation functions for each mixture, consistent with the presence of large particles.
These results raise the question of what effect aggregators may have on classical, well-behaved enzyme inhibitors in a mixture. We turned to the transition-state analogue benzo[b]-thiophene-2-boronic acid (BZB), a pure competitive inhibitor of β-lactamase that has been extensively characterized.28 The potency of two aggregate-containing mixtures increased when they were doped with BZB; when these same mixtures were assayed in the presence of Triton X-100, inhibition fell to the level expected BZB alone (data not shown). These results suggest that synergistic or antagonistic effects, caused by aggregates, may obscure the presence of well-behaved inhibitors in mixtures. Artifactual synergy or antagonism is largely eliminated by addition of nonionic detergent, leaving the effects of the classical inhibitor intact. It should also be noted, however, that sensitivity to detergent varies from aggregate to aggregate, and residual inhibition detected in the presence of detergent (e.g. Figure 2c) may be caused by a stubborn aggregator rather than a bona fide inhibitor.
Multicompound mixtures are widely used in HTS, considerably improving throughput and conserving sometimes scarce assay resources. Three key observations emerge from this study with implications for this common strategy. First, “compound load” is a critical consideration in mixtures. Even at low concentrations of the individual components, here 5 μM, mixtures can display nonideal, nonadditive behaviors resulting from formation of large aggregates that inhibit enzymes. Second, and less expected, sometimes mixtures of compounds can act antagonistically, leading to less-than-expected inhibition. We do not fully understand this phenomenon, but believe it to be real-addition of detergent eliminates both synergistic and antagonistic effects of mixtures. We note that the majority of effects that we observed were synergistic. Finally, since the addition of detergent largely eliminates aggregate-based inhibition while leaving classical inhibition intact, it can be used to identify a classical competitive inhibitor from among a mixture of aggregates. However, since the reversal of inhibition is dependent on detergent concentration, and varies by compound, the persistence of reduced inhibition must be interpreted carefully. Overall, these results suggest heightened caution when interpreting the results of mixture-based high-throughput screening. This is merited by the widespread occurrence of aggregators among drug-like molecules and their nonclassical, nonadditive behavior in aqueous media.
Supplementary Material
Acknowledgments
Acknowledgment. This work was supported by GM71630. We thank Kristin Coan, Sarah Boyce, and Michael Keiser for reading this manuscript, and Michael Snider for interesting discussions.
Footnotes
Supporting Information Available: High-throughput and low-throughput data is available for the follow-up experiments. This material is available free of charge via the Internet at http://pubs.acs.org
References
- (1).Houghten R, Pinilla C, Appel J, Blondelle S, Dooley C, Eichler J, Nefzi A, Ostresh J. Mixture-Based Synthetic Combinatorial Libraries. J. Med. Chem. 1999;42:3743–3777. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- (2).Devlin J, Lian A, Trinh L, Polokoff M, Senator D, Zheng W, Kondracki J, Kretschmer P, Morser J, Lipson S, Spann R, Loughlin J, Dunn K, Morrissey M. High Capacity Screening of Pooled Compounds: Identification of the Active Compound Without Re-Assay of Pool Members. Drug Dev. Res. 1996;37:80–85. [Google Scholar]
- (3).Pinilla C, Appel J, Borras E, Houghten R. Advances in the Use of Synthetic Combinatorial Chemistry: Mixture-based Libraries. Nature Med. 2003:9. doi: 10.1038/nm0103-118. [DOI] [PubMed] [Google Scholar]
- (4).Taylor E, Gibbons J, Braeckman R. Intestinal Absorption Screening of Mixtures from Combinatorial Libraries ni the Caco-2 Model. Pharm. Res. 1997;14:572–577. doi: 10.1023/a:1012140709158. [DOI] [PubMed] [Google Scholar]
- (5).Wu X, Glickman J, Bowen B, Sills M. Comparison of Assay Technologies for a Nuclear Receptor Assay Screen Reveals Differences in the Sets of Identified Functional Antagonists. J. Biomol. Screening. 2003;8:381–392. doi: 10.1177/1087057103256466. [DOI] [PubMed] [Google Scholar]
- (6).Snider M. Screening of Compound Libraries...Consomme or Gumbo. J. Biomol. Screening. 1998;3:169–170. [Google Scholar]
- (7).Hamers T, Molin K, Koeman J, Murk A. A Small-Volume Bioassay for Quantification of the Esterase Inhibiting Potency of Mixtures of Organophosphate and Carbamate Insecticides in Rain-water: Development and Optimization. Toxicol. Sci. 2000;58:60–67. doi: 10.1093/toxsci/58.1.60. [DOI] [PubMed] [Google Scholar]
- (8).Geysen H, Rodda S, Mason T. A Priori Delineation of a Peptide which Mimics a Discontinuous Antigenic Determinant. Mol. Immunol. 1986;23:709–715. doi: 10.1016/0161-5890(86)90081-7. [DOI] [PubMed] [Google Scholar]
- (9).Houghten R, Pinilla C, Blondelle S, Appel J, Dooley C, Cuervo J. Generation and Use of Synthetic Peptide Combinatorial Libraries for Basic Research and Drug Discovery. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- (10).Pinilla C, Appel J, Blanc P, Houghten R. Rapid Identification of High Affinity Peptide Ligands Using Positional Scanning Synthetic Peptide Combinatorial Libraries. Biotechniques. 1992;13:901–905. [PubMed] [Google Scholar]
- (11).Ferrand S, Schmid A, Engeloch C, Glickman J. Statistical Evaluation of a Self-Deconvoluting Matrix Strategy for High-Throughput Screening of the CXCR3 Receptor. Assay Drug Dev. Technol. 2005;3:413. doi: 10.1089/adt.2005.3.413. [DOI] [PubMed] [Google Scholar]
- (12).Rishton Reactive compounds and in vitro false positives in HTS. DDT. 1997;2:382–384. [Google Scholar]
- (13).Roche O, Schneider P, Zuegge J, Guba W, Kansy M, Alanine A, Bleicher K, Danel F, Gutknecht EM, Rogers-evans M, Neidhart W, Stalder H, Dillon M, Sjogren E, Fotouhi N, Gillespie P, Goodnow R, Harris W, Jones P, Taniguchi M, Tsujii S, Vvon Der Saal W, Zimmermann G, Schneider G. Development of a Virtual Screening Method for Identification of “Frequent Hitters” in Compound Libraries. J. Med. Chem. 2002;45:137–142. doi: 10.1021/jm010934d. [DOI] [PubMed] [Google Scholar]
- (14).Walters WP, Ajay, Murcko MA. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 1999;3:384–387. doi: 10.1016/s1367-5931(99)80058-1. [DOI] [PubMed] [Google Scholar]
- (15).Rishton Nonleadlikeness and leadlikeness in biochemical screening. DDT. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- (16).Walters WP, Namchuk M. Designing screens: how to make your hits a hit. Nat. Rev. Drug Discovery. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- (17).Huth J, Mendoza R, Olejniczak E, Johnson R, Cothron D, Liu Y, Lerner C, Chen J, Hajduk P. ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 2005;127:217–224. doi: 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- (18).McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- (19).McGovern SL, Helfand BT, Feng BY, Shoichet BK. A Specific Mechanism of Nonspecific Inhibition. J. Med. Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- (20).McGovern SL, Shoichet BK. Kinase Inhibitors: Not Just for Kinases Anymore. J. Med. Chem. 2003;46:1478–1483. doi: 10.1021/jm020427b. [DOI] [PubMed] [Google Scholar]
- (21).Seidler J, McGovern SL, Doman T, Shoichet BK. Identification and Prediction of Promiscuous Aggregating Inhibitors among Known Drugs. J. Med. Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- (22).Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nature Chem. Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- (23).Chung T. Screen Compounds Singly: Why Muck It Up. J. Biomol. Screening. 1998;3:171. [Google Scholar]
- (24).Wilson-Lingardo L, Davis PW, Ecker DJ, Hebert N, Acevedo O, Sprankle K, Brennan T, Schwarcz L, Freier SM, Wyatt JR. Deconvolution of Combinatorial Libraries for Drug Discovery: Experimental Comparison of Pooling Strategies. J. Med. Chem. 1996;39:2720–2726. doi: 10.1021/jm960169g. [DOI] [PubMed] [Google Scholar]
- (25).Chou T, Talalay P. A Simple Generalized Equation for the Analysis of Multiple Inhibitions of Michaelis-Menten Kinetic Systems. J. Biol. Chem. 1977;252:6438–6442. [PubMed] [Google Scholar]
- (26).Chou T, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. In: Webber G, editor. Advances in Enzyme Regulation. Vol. 22. Pergamon; New York: 1984. pp. 27–55. [DOI] [PubMed] [Google Scholar]
- (27).Chou T, Talalay P. Generalized Equations for the Analysis of Inhibitions of Michaelis-Menten and Higher-Order Kinetic Systems with Two or More Mutually Exclusive and Nonexclusive Inhibitors. Eur. J. Biochem. 1987;115:207–216. doi: 10.1111/j.1432-1033.1981.tb06218.x. [DOI] [PubMed] [Google Scholar]
- (28).Weston GS, Blazquez J, Baquero F, Shoichet BK. Structure-based enhancement of boronic acid-based inhibitors of AmpC beta-lactamase. J. Med. Chem. 1998;41:4577–4586. doi: 10.1021/jm980343w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.