

Abstract
In human and experimental models of heart failure, sarcoplasmic reticulum Ca2+ ATPase (SERCA2a) activity is decreased, resulting in abnormal calcium handling. The disturbances in calcium metabolism have been shown to contribute significantly to the contractile dysfunction observed in heart failure. We investigated whether increasing SERCA2a expression can improve ventricular function in an animal model of heart failure obtained by creating ascending aortic constriction in rats. After 19–23 wk of banding during the transition from compensated hypertrophy to heart failure (documented by >25% decrease in fractional shortening), rats were randomized to receive either an adenovirus carrying the SERCA2a gene (Ad.SERCA2a, n = 13) or β-galactosidase (Ad.βgal, n = 14) by using a catheter-based technique. The failing hearts infected with Ad.βgal were characterized by a significant decrease in SERCA2a expression and a decrease in SERCA2a activity compared with nonfailing sham-operated rats (n = 11). In addition, these failing hearts had reduced left-ventricular systolic pressure, maximal rate of left-ventricular pressure rise and decline (+dP/dt, −dP/dt), and rate of isovolumic relaxation (τ). Overexpression of SERCA2a restored both SERCA2a expression and ATPase activity to nonfailing levels. Furthermore, rats infected with Ad.SERCA2a had significant improvement in left-ventricular systolic pressure, +dP/dt, −dP/dt, and rate of isovolumic relaxation (τ) normalizing them back to levels comparable to sham-operated rats. In this study, we show that in an animal model of heart failure where SERCA2a protein levels and activity are decreased and severe contractile dysfunction is present, overexpression of SERCA2a in vivo restores both systolic and diastolic function to normal levels.
Both contraction and relaxation abnormalities at the cardiac myocyte level have been identified in human and animal models of heart failure (1, 2). Trabeculae and isolated cardiac cells from failing hearts have characteristic functional abnormalities, which include an increase in diastolic Ca2+, an increase in the time course of Ca2+ transient, and a decrease in sarcoplasmic reticulum (SR) Ca2+ release (1–3). These abnormalities are especially accentuated at high frequencies of stimulation leading to the characteristic negative force-frequency relationship in failing myocardium (4).
Because the SR plays a central role in controlling Ca2+ movements in myocardial cells during excitation–contraction coupling, a large number of studies have been performed examining the expression and function of the SR Ca2+ ATPase (SERCA2a)(5–9). SERCA2a activity and SR Ca2+ uptake are reduced in failing hearts (9). This reduction in activity is generally, but not invariably, associated with reductions in SERCA2a mRNA and protein. Taken together, these results support the hypothesis that abnormal Ca2+ handling in failing hearts is caused in part by a decrease in SERCA2a activity.
To examine the importance of SERCA2a in the development of decompensated heart failure, we overexpressed SERCA2a in an animal model of heart failure by using gene transfer. In the rat model of pressure-overload hypertrophy induced by ascending aortic banding, the decrease in SERCA2a mRNA content parallels a decrease in SERCA2a protein concentration and Ca2+-ATPase activity (10). These changes occur during the transition from compensated hypertrophy to decompensated heart failure characterized by premature death, left-ventricular (LV) enlargement, reduction in fractional shortening, myocyte hypertrophy, elevated plasma norepinephrine levels, and depressed LV-developed pressure as a function of perfusate [Ca2+] (11–14). This alteration in SERCA2a gene expression provides an early marker for the development of LV decompensation. We have recently shown that overexpression of SERCA2a in neonatal myocytes by adenoviral gene transfer results in increased contractility and a faster relaxation rate of the calcium transient (15, 16). More recently, we have developed a catheter-based technique to deliver adenoviruses to the heart in a homogeneous fashion (17). In this study, we tested the hypothesis that cardiac overexpression of SERCA2a could mediate significant functional benefits in rats undergoing the transition to heart failure after aortic banding.
Methods
Construction of Recombinant Adenoviruses.
Three first-generation type 5 recombinant adenoviruses were used in these studies: Ad.βgal, Ad.E1-SERCA2a, and Ad.E3-GFP.E1-SERCA2a (GFP, green fluorescent protein). The construction of Ad.E1-SERCA2a has been described earlier (15). We also used an alternative adenoviral backbone system that allows transgene expression from E1 with simultaneous expression of GFP from E3 (18).This backbone was derived from pBHG11 by direct ligation of the GFP expression cassette into a unique PacI site in E3. Because we found no differences in terms of expression or physiological parameters between the groups of animals infected with Ad.E1-SERCA2a or Ad.E3-GFP.E1-SERCA2a, we refer to the adenoviruses carrying SERCA2a (with or without GFP) as Ad.SERCA2a.
Experimental Protocol.
Four-week-old Wistar rats (70–80 g) were obtained from Charles River Breeding Laboratories. After 2–3 days of acclimatization, the rats were anesthetized with i.p. pentobarbital (65 mg/kg) and placed on a ventilator. After thoracotomy, a tanatalum clip with an internal diameter of 0.58 mm (Weck Hemoclip, Research Triangle Park, NC) was placed on the ascending aorta. Animals in the sham group underwent a similar procedure without insertion of a clip. The animals were initially randomized in two groups: one group of 30 animals with aortic banding and a second group of 32 animals that were sham operated. All animals survived the initial operation. These groups of animals were studied 2–3 days after gene transfer.
A second group of 16 animals that were banded were examined for long-term effects of Ad.SERCA2a vs. Ad.βgal infection. These animals were followed for 7 days after gene transfer.
A third group of 12 control animals was used to examine the expression level of SERCA2a at 2, 7, 14, and 28 days after infection with Ad.SERCA2a.
Serial Echocardiographic Assessment.
At intervals of 6, 12, and 18 wk after banding and then on a weekly basis between 18 and 22 wk, serial echocardiograms were performed. Animals were anesthetized with pentobarbital 40 mg/kg i.p. and the anterior chest shaved. Transthoracic M-mode and two-dimensional echocardiography was performed with a Hewlett–Packard Sonos 5500 imaging system with a 12-MHz broadband transducer. Gene transfer was performed in all animals within 3 days of detection of a drop in fractional shortening of >25% compared with the fractional shortening at 6 wk after banding. In the sham-operated rats, gene delivery was performed at 20 wk.
Adenoviral Delivery Protocol.
The group of 30 animals with aortic banding was subdivided in two groups of fifteen, with each group receiving either Ad.SERCA2a or Ad.βgal. Two animals in the SERCA2a group and one animal in the β-galactosidase group died during the gene transfer surgery. The group of 24 sham-operated animals was subdivided into two groups of twelve, with each group receiving either Ad.SERCA2a or Ad.βgal. One animal in the SERCA2a group and one animal in the β-galactosidase group died during the gene transfer surgery. The adenoviral delivery system has been described previously in detail by our group (17). Briefly, after the rats were anesthetized and a thoracotomy performed, a 22-G catheter containing 200 μl of adenovirus was advanced from the apex of the left ventricle to the aortic root. The aorta and main pulmonary artery were clamped for 10 sec distal to the site of the catheter and the solution injected, then the chest was closed. The rats underwent hemodynamic studies 2–3 or 7 days after the injection of the adenovirus.
LV Pressure and Dimension Measurements.
Rats in the different treatment groups had French high-fidelity pressure transducer (Millar Instruments, Houston, TX) introduced into the left ventricle. LV systolic pressure, end-diastolic pressure, the maximal rates of pressure rise (+dP/dt) and of pressure fall (−dP/dt), and the time constant of isovolumic relaxation were measured or derived in the different groups. For the pacing studies, an epicardial lead was placed at on atrial appendage connected to a stimulator (Grass Instruments, Quincy, MA). In a subset of animals, multiple 0.7-mm piezoelectric crystals (Sonometrics, Canada) were placed over the surface of the left ventricle along the short axis of the ventricle to measure the intercrystal distance. LV pressure–dimension loops were generated under different loading conditions by clamping the inferior vena cava. The end-systolic pressure–dimension relationship was obtained by producing a series of pressure dimension loops over a range of loading conditions to generate the maximal slope.
Histochemistry.
Hearts were examined by immunohistochemistry to evaluate the expression of β-galactosidase. Hearts were fixed with PBS containing 0.5% glutaraldehyde and then permeabilized. They were then incubated overnight in a solution containing 5-bromo-4-chloro3-indolyl β-d-galactopyranoside (X-Gal), and 10-μm sections were then cut and examined under light microscopy.
Isolation of Single Cardiac Myocytes.
The isolation procedure has been previously described in detail (19). The isolated myocytes were then fixed with 0.5% glutaraldehyde for 15 min and stained with a solution containing X-Gal for 2 hr. To quantify the percentage of cells with blue nuclei, we counted 100 cells for each heart preparation.
Preparation of SR membranes from Left Ventricles.
We isolated SR membranes from the left ventricles of hearts as previously described (20). Protein concentration was determined by a modified Bradford assay (21).
Western Blot Analysis.
SDS/PAGE was performed on the isolated membranes from cell cultures under reducing conditions on 7.5% or 12% separation gels with a 4% stacking gel in a Miniprotean II cell (Bio-Rad). For immunoreaction, the blot was incubated with 1:2,500 diluted monoclonal antibodies to either SERCA2a, 1:1,000 diluted anticalsequestrin (Affinity BioReagents), or 1:2,500 diluted anticardiac phospholamban monoclonal IgG (Upstate Biotechnology) for 90 min at room temperature. The densities of the bands were evaluated by using NIH image. Normalization was performed by dividing densitometric units of each membrane preparation by the calsequestrin densities in each of these preparations. Serial dilution of the membrane preparations revealed a linear relationship between amounts of protein and the densities of the immunoreactive bands (data not shown).
SERCA2a Activity.
SERCA2a activity assays were carried out based on pyruvate/NADH-coupled reactions, as previously described (16). Ca2+-ATPase activity was calculated as: Δ absorbency/(6.22 × protein × time) in nmol ATP/(mg protein × min).
Statistics.
All values are presented as mean ± SD. A two-factor ANOVA was performed to compare the different hemodynamic parameters among the different groups. For the echo data, where the variables were examined at various intervals, ANOVA with repeated measures was performed. Statistical significance was accepted at the level of P < 0.05.
Results
Characterization of Animals.
Aortic banding induced an increase in posterior wall thickness and an increase in fractional shortening at 6 wk and 12 wk without a significant change in LV diastolic diameter (LVDD) as shown in Table 1. After 18 wk, LVDD increased, and fractional shortening fell. Once the fractional shortening decreased by 25% from the 6-wk baseline measurement, gene transfer was performed.
Table 1.
Echocardiographic measures in rats after sham-surgery or aortic banding
| Treatment | PW, mm | LVDD, mm | LVSD, mm | FS, % |
|---|---|---|---|---|
| Sham | ||||
| 6 wk | 1.83 ± 0.12 | 8.15 ± 0.22 | 4.80 ± 0.22 | 41.4 ± 3.3 |
| 12 wk | 1.89 ± 0.11 | 8.32 ± 0.31 | 4.97 ± 0.28 | 44.2 ± 4.9 |
| 18 wk | 1.93 ± 0.10 | 8.55 ± 0.21 | 5.09 ± 0.25 | 40.1 ± 2.8 |
| 20 wk | 1.91 ± 0.10 | 8.61 ± 0.24 | 5.01 ± 0.24 | 41.4 ± 2.3 |
| LVH | ||||
| 6 wk | 2.42 ± 0.16* | 7.93 ± 0.18 | 4.11 ± 0.21 | 46.8 ± 2.1* |
| 12 wk | 2.66 ± 0.14* | 8.44 ± 0.24 | 4.30 ± 0.16 | 48.2 ± 2.0* |
| 18 wk | 2.60 ± 0.18* | 8.87 ± 0.22* | 5.55 ± 0.13 | 36.4 ± 2.6*† |
| 20–23 wk | 2.68 ± 0.20* | 9.44 ± 0.28* | 6.36 ± 0.10* | 32.6 ± 1.8*† |
PW, posterior wall thickness during diastole; LVDD, left ventricular diameter during diastole; LVSD, left ventricular systolic diameter during systole; FS, fractional shortening; *, P < 0.05 compared to sham at similar time period; †, P < 0.05 compared to values at 6 wk.
Cardiac Gene Transfer.
As we have previously reported, adenoviral gene transfer with the catheter-based technique induced an expression pattern that is grossly homogeneous throughout the ventricles in failing and nonfailing hearts by tissue immunostaining. We also isolated single cardiomyocytes from failing and control rat hearts after in vivo infection with Ad.βgal, as shown in Fig. 1. After in vivo transduction with Ad.βgal, there was a robust transduction of cardiomyocytes in both the sham control-operated rats and in the failing hearts. We also isolated single cardiomyocytes from failing and control rat hearts after in vivo infection with Ad. SERCA2a and the majority of the cells exhibit bright green fluorescence, with little background fluorescence in noninfected cardiomyocytes.
Figure 1.
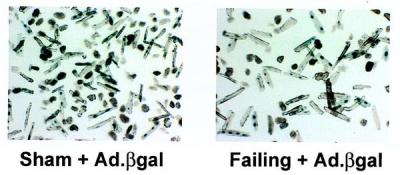
Cardiomyocytes isolated from hearts from Sham + Ad.βgal, and Failing + Ad.βgal, which were also stained with X-Gal.
Quantification of Calcium Regulatory Proteins.
We first examined the expression of SERCA2a expression serially 2 days, 7 days, and 14 days after adenoviral gene transfer in sham rats. As shown in Fig. 2A, there was a clear increase of SERCA2a at 2 and 7 days after infection, but at 14 days the expression was back to baseline (Fig. 2A). Because phospholamban exerts a modulatory effect on SERCA2a, and calsequestrin is an important SR buffer of calcium, we also measured their expression, which did not change at these time points. The protein expression of SERCA2a was significantly decreased in failing rat left ventricles when compared with SERCA2a levels of nonfailing left ventricles from age-matched sham-operated rats, as shown in Fig. 2 B and C. Adenoviral gene transfer of SERCA2a in failing hearts increased SERCA2a protein expression 2-fold, restoring it to levels observed in the nonfailing hearts. Adenoviral gene transfer of SERCA2a (normalized to calsequestrin) in sham-operated rats also increased SERCA2a by 50 ± 5%.
Figure 2.

(A) Immunoblots of SERCA2a and phospholamban at days 0, 2, 7, 14, and 28 after infection with Ad.SERCA2a in control rats. (B) Immunoblots of SERCA2a, phospholamban, and calsequestrin from crude membranes of left ventricles from control, sham rats infected with Ad.βgal, sham rat hearts infected with Ad.SERCA2a, failing rat hearts infected with Ad.βgal, and failing hearts infected with Ad.SERCA2a at day 2–3. (C) Protein levels of SERCA2a in preparations from sham rats infected with Ad.βgal (n = 7), sham rat hearts infected with Ad.SERCA2a (n = 8), preparations from failing rat hearts infected with Ad.βgal (n = 8), and preparations of failing hearts infected with Ad.SERCA2a at day 2–3 (n = 9). *, P < 0.05 compared with Sham + Ad.βgal; †, P < 0.05 compared with Failing + Ad.βgal; ‡, P < 0.05 compared with Sham + Ad.SERCA2a.
SERCA2a Activity.
Failing hearts had a significant decrease in maximal Ca2+ ATPase activity when compared with age-matched nonfailing hearts (Fig. 3). In addition, the affinity of SR Ca2+-ATPase for Ca2+ was significantly depressed in failing hearts. Adenoviral gene transfer Ad.SERCA2a restored the maximal Ca2+ ATPase activity.
Figure 3.

ATPase activity measured vs. [Ca2+] in membrane preparations from control uninfected sham rats (n = 6, ♦), sham rats infected with Ad.βgal (n = 7, ●), sham rat hearts infected with Ad.SERCA2a (n = 8, ○), preparations from failing rat hearts infected with Ad.βgal (n = 8, ▴), and preparations of failing hearts infected with Ad.SERCA2a at day 2–3 (n = 9, ▵).
Hemodynamic Effects of SERCA2a Overexpression.
As shown in Table 2, LV systolic function was adversely affected in the failing group compared with the sham group, as evidenced by a decrease in LV systolic pressure, a depression of the maximal rate of LV pressure rise, and an increase in heart rate. Overexpression of SERCA 2a significantly increased the rate of rise of pressure and peak LV systolic pressure in rats with heart failure. Diastolic parameters were also significantly altered in the failing rat hearts compared with sham-operated rats, as evidenced by a decrease in the maximal rate of decline of LV systolic pressure, an increase in diastolic pressure, and a significantly prolonged time course of pressure decline (τ). Overexpression of SERCA2a normalized both the maximal rate of decline of LV systolic pressure and the time course of pressure decline (τ); however it did not restore diastolic pressure to normal levels. Overexpression of SERCA2a in sham-operated nonfailing hearts significantly increased the maximal rate of LV pressure rise and significantly decreased τ. Furthermore, it resulted in a trend toward an increase in maximal rate of LV pressure decline (P = 0.07).
Table 2.
Hemodynamic effects of SERCA2a overexpression after 2–3 days
| Treatment | HR, min−1 | LVSP, mm Hg | +dP/dt, mm Hg/sec | −dP/dt, mm Hg/sec | LVEDP, mm Hg | τ, msec |
|---|---|---|---|---|---|---|
| Control Sham | 348 ± 76 | 95 ± 13 | 6,929 ± 1,353 | −5,967 ± 1,594 | 4 ± 3 | 20 ± 3 |
| Sham + Ad.βgal | 363 ± 41 | 91 ± 11 | 6,909 ± 2,020 | −5,572 ± 1,227 | 4 ± 3 | 19 ± 4 |
| Failing + Ad.βgal | 453 ± 35† | 129 ± 20† | 4,830 ± 1,497† | −3,813 ± 1,567† | 9 ± 5† | 23 ± 7† |
| Sham + SERCA2a | 357 ± 36 | 97 ± 10 | 8,438 ± 1,770† | −6,452 ± 1,553† | 6 ± 3 | 13 ± 4 |
| Failing + SERCA2a | 420 ± 42*† | 165 ± 27*† | 7,332 ± 2,584* | −6,110 ± 1,711* | 11 ± 9 | 14 ± 3*† |
HR, heart rate; LVSP, left-ventricular systolic pressure; +dP/dt, maximal rate of rise of left-ventricular pressure; −dP/dt, maximal rate of decline of left-ventricular pressure; LVEDP, left-ventricular end-diastolic pressure; τ, time course of isovolumic relaxation; *, P < 0.05 compared to Failing + Ad.βgal; †, P < 0.05 compared to Sham + Ad.βgal.
In the groups of animals that were followed long term, at 7 days after infection there were four deaths of eight in the failing group receiving Ad.βgal, whereas in the failing group receiving Ad.SERCA2a, there were two deaths, also of eight. We performed hemodynamic measurements on these animals and, as shown in Table 3, there was a similar profile of improved +dP/dt, −dP/dt, and τ 7 days after infection with Ad.SERCA2a in the failing group.
Table 3.
Hemodynamic effects of SERCA2a overexpression after 7 days
| Treatment | HR, min−1 | LVSP, mm Hg | +dP/dt, mm Hg/sec | −dP/dt, mm Hg/sec | LVEDP, mm Hg | τ, msec |
|---|---|---|---|---|---|---|
| Failing + Ad.βgal | 442 ± 27 | 112 ± 24 | 5,387 ± 818 | −3,748 ± 458 | 12 ± 6 | 23 ± 2 |
| Failing + SERCA2a | 440 ± 21 | 147 ± 22* | 7,924 ± 1,857* | −6,782 ± 971* | 9 ± 5 | 14 ± 2* |
HR, heart rate; LVSP, left-ventricular systolic pressure; +dP/dt, maximal rate of rise of left-ventricular pressure; −dP/dt, maximal rate of decline of left-ventricular pressure; LVEDP, left-ventricular end-diastolic pressure; τ, time course of isovolumic relaxation; *, P < 0.05 compared to Failing + Ad.βgal.
Pressure–Dimension Relationship.
To define further ventricular function, pressure–dimension analysis was performed in a subset of animals. The maximal slope of the end-systolic pressure dimension relationship was lower in failing hearts infected with Ad.βgal compared with control, indicating a diminished state of intrinsic myocardial contractility: 56 ± 17 mmHg/mm vs. 113 ± 30 mmHg/mm (n = 6, P < 0.01). Overexpression of SERCA2a restored the slope of the end-systolic pressure–dimension relationship to control levels [101 ± 29 mmHg/mm (n = 7), P < 0.01 compared with failing + Ad.βgal; P > 0.1 compared with control].
Contractile Reserve and Effect of Isoproterenol.
To test whether SERCA2a overexpression augmented inotropic reserve, we studied the effect of incremental atrial pacing on hemodynamic parameters in failing hearts and sham-operated hearts. As shown in Fig. 4a, +dP/dt increased with a rise in heart rate, but this rate-dependent response was significantly blunted and became negative in the failing hearts. Overexpression of SERCA2a in the failing hearts restored the frequency response to values near sham-operated animals. Failing hearts are characterized by having a decreased responsiveness to β-adrenergic agents mainly because of a down-regulation of β1-receptors and uncoupling of these receptors to adenylyl cyclase. To examine to what extent SERCA2a overexpression can restore the responsiveness, we infused 0.1 μg/kg/min of isoproterenol in sham rats, failing rats + Ad.βgal, and failing rats + Ad.SERCA2a. As shown in Fig. 4b, the response to isoproterenol was substantially reduced in failing rats when compared with sham rats. SERCA2a overexpression improved the response to β-adrenergic stimulation compared with failing hearts but did not restore it to normal levels.
Figure 4.

(A) Dependence of LV +dP/dt on heart rate in the different groups of animals: control uninfected sham rats (n = 6, ■), sham rats infected with Ad.βgal (n = 5, ●), sham rat hearts infected with Ad.SERCA2a (n = 4, ○), preparations from failing rat hearts infected with Ad.βgal (n = 5, ▴), and preparations of failing hearts infected with Ad.SERCA2a (n = 5, ▵ ). *, P < 0.001 compared with sham + Ad.βgal; †, P < 0.005 compared with Failing + Ad.βgal; ‡, P < 0.05 compared with Sham + Ad.SERCA2a. (B) LV pressure +dP/dt measurements at baseline and during infusion of 0.1 μg/kg/min of isoproterenol in the different groups of animals: control rats (n = 4), sham rats infected with Ad.βgal (n = 5), sham rat hearts infected with Ad.SERCA2a (n = 4), failing rat hearts infected with Ad.βgal (n = 4), and failing hearts infected with Ad.SERCA2a (n = 4). *, P < 0.001 compared with Sham + Ad.βgal; †, P < 0.005 compared with Failing + Ad.βgal.
Effect on Morphological Parameters.
As shown in Table 4, the failing hearts had a significant increase in LV mass when normalized to either tibial length or body mass. Tibial length that was used as an index of growth independent of body weight was uniformly constant across the different groups. Body mass was also not significantly different across the different groups. Overexpression of SERCA2a in the failing heart did not have a significant effect on LV mass, whether normalized to tibial length or body mass.
Table 4.
Morphometric analyses
| Treatment | Body weight, mm | LV weight, g | LV/BW × 103 | Lung wet/dry weight ratio | LV/TL × 103, g/mm |
|---|---|---|---|---|---|
| Sham + Ad.βgal | 578 ± 34 | 1.09 ± 0.14 | 1.9 ± 0.1 | 3.7 ± 0.5 | 26.4 ± 5 |
| Failing + Ad.βgal | 596 ± 41 | 1.82 ± 0.22* | 3.1 ± 0.1* | 5.6 ± 0.7* | 44.3 ± 6* |
| Sham + SERCA2a | 559 ± 44 | 1.14 ± 0.15 | 2.1 ± 0.1 | 3.6 ± 0.4 | 28.5 ± 4 |
| Failing + SERCA2a | 611 ± 51 | 1.85 ± 0.17*† | 3.0 ± 0.2*† | 4.7 ± 0.5*† | 45.1 ± 5*† |
LV, left ventricle; *, P < 0.05 compared to Sham + Ad.βgal; †, P < 0.05 compared to Sham + Ad.SERCA2a.
Discussion
In this study, we show that in an animal model of heart failure, where SERCA2a protein levels and activity are decreased and severe contractile dysfunction is present, restoration of SERCA2a expression in vivo improves both systolic and diastolic function to normal levels.
Cardiac Gene Transfer.
We have previously reported that the catheter-based adenoviral technique induces global gene transfer in adult rat hearts with an expression pattern that is grossly homogeneous throughout the ventricles (17). Because these observations were made in healthy rats, we sought to quantify the efficiency of gene transfer in failing rat hearts. To assess the degree of gene expression using the delivery method described, we used three approaches. First, we visualized the whole heart after immunostaining with X-Gal after injection with Ad.βgal. Second, we isolated single cardiomyocytes from sham and failing rat hearts after in vivo transduction with Ad.βgal. And finally, we isolated single cardiomyocytes from sham and failing rat hearts after in vivo transduction with Ad.SERCA2a. With the tissue immunostaining technique, we found that certain areas had diffuse staining, whereas in others a more patchy distribution of expression was observed. This staining may underestimate the extent of transgene expression both because the nuclear-localized β-gal construct used exhibits cytoplasmic activity only in highly expressing cells and because histochemical staining of β-gal is relatively insensitive. To evaluate further the transduction efficiency of our technique, we also isolated single cardiomyocytes from infected failing hearts. A high percentage of cardiomyocytes demonstrated specific nuclear β-gal activity reflecting transgene expression in both sham and failing rat hearts, 2–3 days after in vivo transduction with Ad.βgal. In addition, a majority of isolated myocytes exhibited green fluorescence after gene transfer of SERCA2a. Even though each method for evaluating gene transfer is different and may yield different absolute results, the efficiency of our gene transfer method was sufficient to induce physiological changes. Together our data using reporter constructs demonstrates highly effective gene transfer to adult rat hearts in vivo.
In the nonfailing rats using adenoviral gene transfer, we were able to increase SERCA2a protein levels by 1.5-fold, whereas in the failing rats, the increase was closer to 2-fold. In transgenic animals overexpressing SERCA2a, the increase in protein levels has been variable. He et al. reported an increase of 1.2-fold in their transgenic mice by using the cytomegalovirus enhancer and the chicken β-actin reporter (22); however, even with this small increase in SERCA2a expression, they documented beneficial effects on contractility. More recently, Baker et al. generated mice overexpressing SERCA2a with 1.55-fold increase in protein by using the α myosin heavy-chain promoter (23). These values are in the range of the level of overexpression we found in nonfailing rats after gene transfer of SERCA2a but lower than that achieved in the failing hearts. Rat cardiomyocytes rely heavily on the SR for calcium removal (≈92%), and their SR is saturated with ATPase pumps (24). In heart-failure state, the number of functional ATPase pumps decreases, and even though the amount of pumps overexpressed is constant, the result is a larger relative increase in SERCA2a. Alternatively, in failing hearts there may be more space for ATPase pumps to be inserted (25), allowing the level of overexpression that can be achieved to be higher.
SERCA2a Expression and ATPase Activity in the Failing Heart.
In this model of heart failure in the rat, SERCA2a protein levels decreased significantly in the failing heart when compared with sham-operated animals. These results are consistent with previous studies that demonstrated a decrease in the mRNA steady-state level for SERCA2a in transition from LV hypertrophy to failure after aortic banding (10). In addition, we found that the decrease in SERCA2a protein level is accompanied by a reduction in ATPase activity at varying levels of calcium concentrations. A reduction in the ATPase activity and Ca2+ uptake has been documented in all models of heart failure, including human failing ventricles (5, 25–28). Furthermore, SERCA2a activity has been shown to be directly related to both intracellular Ca2+ release from the SR and systolic tension (increasing ATPase results in increase Ca2+ uptake and release), while being inversely related to diastolic Ca2+ and diastolic tension (decreasing ATPase results in a decrease in Ca2+ uptake and an increase in diastolic Ca2+) (20, 25, 26).
Effect of SERCA2a on Systolic and Diastolic Function.
In nonfailing hearts, overexpression of SERCA2a resulted in an enhancement in contractility, as evidenced by increases in +dP/dt and −dP/dt as well as faster relaxation, without affecting heart rate. These results correlate well with those from transgenic animals (23), where the rates of contraction and relaxation are faster in mice overexpressing SERCA2a. In the present study, overexpression of SERCA2a resulted in an increase in systolic pressure, a decrease in the time constant of isovolumic relaxation, and an increase in both the maximal rate of rise and the maximal rate of fall of LV systolic pressure in the failing hearts. Isovolumic relaxation, which is an index of active relaxation and reflects the removal of Ca2+ from the myofilaments into the SR, was significantly prolonged in the failing hearts and was restored to normal levels by overexpression of SERCA2a. In the failing hearts, diastolic pressure was not decreased by overexpression of SERCA2a. This may be explained by increased passive stiffness of the senescent hearts, which results from the presence of fibrosis in this model of heart-failure myocardium (11). Overexpression of SERCA2a would not be expected to affect these extramyocardial parameters.
Frequency Potentiation.
The potentiation of cardiac contractility by increases in heart rate is an important regulatory mechanism in the working heart (4, 29, 30). This is because of an enhanced calcium release from the SR and increased SERCA2a activity. The force-frequency response is blunted in human and experimental heart failure and contributes to impaired cardiac reserve (31, 32). A number of investigators have shown that abnormal calcium handling underlies the blunting of the frequency potentiation of force by demonstrating correlations between optimum frequency of stimulation and SERCA2a activity and expression (4, 20). In our studies, the frequency response was clearly abnormal in the failing heart. Overexpression of SERCA2a in these hearts restored the frequency potentiation.
β-Adrenergic Responsiveness.
Failing hearts have decreased β-adrenergic responsiveness mainly because of a decrease in β1 receptors and uncoupling of the β1 receptors to adenylyl cyclase caused by an increase in inhibitory G proteins and an up-regulation of β-adrenergic kinase (33). The downstream mechanisms of β-adrenergic signaling pathways involve an increase in cAMP and subsequent phosphorylation of phospholamban, which results in an increase in SERCA2a activity and enhancement in SR Ca2+ uptake (34). Therefore, abnormalities in SERCA2a would further decrease the responsiveness to β-adrenergic agents. We found that isoproterenol induced a much smaller increase of +dP/dt in failing hearts when compared with sham hearts, as has been shown before. Overexpression of SERCA2a enhanced this response in the failing hearts but did not restore it to normal. These results would indicate that down-regulation of SERCA2a contributes to the decrease in responsiveness of β-adrenergic agents.
Potential Therapeutic Implications.
In this model of heart failure, SERCA2a overexpression improved parameters of inotropy and lusitropy and normalized contractile reserve. These effects translate into an inotropic intervention. However, other inotropic interventions have been shown clinically to increase mortality in chronic heart failure in numerous trials. Unlike agents that increase cAMP, thereby increasing intracellular Ca2+, overexpression of SERCA2a decreases diastolic intracellular Ca2+ by increasing uptake into the SR and enhancing Ca2+ release. Beyond the contractile benefits of lowering diastolic Ca2+, it has been shown that sustained elevations of resting Ca2+ lead to activation of serine–threonine phosphatases including calcineurin-inducing hypertrophy and cell death (35). Therefore, a decrease in diastolic Ca2+ may in effect decrease the stimulation of phosphatases and reduce the proapoptotic and prohypertrophy signaling. Although overexpression of SERCA2a would be anticipated to increase ATP hydrolysis, elevated resting Ca2+ also incurs energy costs, because other transporters are recruited to remove Ca2+. In balance, it is not clear how SERCA2a overexpression will affect the energy cost of contractility. More detailed analyses such as NMR spectroscopy would be needed to directly address this question.
Limitations of the Study.
In this study, we used first-generation adenoviral vectors that induce robust immunological responses and may cause myocardial necrosis. The infected rat hearts demonstrated an inflammatory response; however, there was no evidence of disruption of normal myocardial architecture or collagen deposition (data not shown). The results obtained in this rat model of heart failure may not be applicable to human failing cardiac cells. Human myocardium relies less on the SERCA2a pump for Ca2+ removal than rat myocardium (90% vs. 70%) (25). However, it has been clearly shown that a decrease in SERCA2a in human failing hearts contributes directly to contractile failure (20). Therefore, an increase in SERCA2a activity would be expected to improve contractile function in human cardiomyocytes. Another limitation of this study is that even though a majority of the cells were transduced by our method of gene transfer, we did not achieve 100% infection, which could lead to differential expression and abnormal contraction of the ventricle and even arrhythmias. However, the pressure contours were normal, and no unexpected deaths occurred in the hearts overexpressing SERCA2a. Finally, the intervention we performed was acute because SERCA2a expression was transient. Therefore, we could not address the effects of SERCA2a overexpression chronically. However, it was interesting to find that fewer deaths occurred in the SERCA2a group of rats that were banded and failing in the short term. Clearly, we cannot draw any conclusion regarding effect on mortality with this small group. The use of newer vectors (i.e., E1–E4-deleted adenoviruses or adeno-associated virus) that have prolonged expression will allow us in the future to study the effects on mortality (18, 36, 37).
Conclusions
Our results clearly demonstrate that restoring SERCA2a can improve systolic and diastolic performance in failing hearts. Because disturbed calcium handling plays a significant role in the pathophysiology of ventricular dysfunction, our data provide strong evidence that overexpression of SERCA2a to rescue disturbed calcium cycling and myocardial function of the failing heart is indeed possible. At the cellular level, there are many molecular abnormalities in the failing heart. Targeting one pathway or one transporter may provide a temporary beneficial change in cardiac phenotype, but for sustained improvement a number of pathways will probably need to be targeted. Nevertheless, this study validates the feasibility of cardiac gene transfer in failing hearts as a therapeutic modality.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health [HL 50361 and HL 57623 (R.J.H.); HL 54202, HL 59521, and HL 61557 (A.R.)]; by Small Business Innovation Research (SBIR) grants R43-HL-60323, R44-60323, and RO1 HL-49574 (J.K.G.); and by a Doris Duke Charitable Foundation Clinician Scientist Award and American Federation of Aging Research Grant (R.J.H.). A.R. is an Established Investigator of the American Heart Association.
Abbreviations
- SR
sarcoplasmic reticulum
- SERCA2a
SR Ca2+ ATPase
- GFP
green fluorescent protein
- Ad.SERCA2a
adenovirus carrying the SERCA2a gene
- Ad.βgal
adenovirus carrying β-galactosidase
- X-Gal
5-bromo-4-chloro3-indolyl β-d-galactopyranoside
- LV
left-ventricular
- +dP/dt
−dP/dt, maximal rate of LV pressure rise and decline
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gwathmey J K, Morgan J P. Circ Res. 1985;57:836–843. doi: 10.1161/01.res.57.6.836. [DOI] [PubMed] [Google Scholar]
- 2.Gwathmey J K, Copelas L, MacKinnon R, Schoen F J, Feldman M D, Grossman W, Morgan J P. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 3.Davies C H, Davia K, Bennett J G, Pepper J R, Poole-Wilson P A, Harding S E. Circulation. 1995;92:2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- 4.Gwathmey J K, Slawsky M T, Hajjar R J, Briggs G M, Morgan J P. J Clin Invest. 1990;85:1599–1613. doi: 10.1172/JCI114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arai M, Matsui H, Periasamy M. Circ Res. 1994;74:555–564. doi: 10.1161/01.res.74.4.555. [DOI] [PubMed] [Google Scholar]
- 6.Brillantes A M, Bezprozvannaya S, Marks A R. Circ Res. 1994;75:503–510. doi: 10.1161/01.res.75.3.503. [DOI] [PubMed] [Google Scholar]
- 7.Cheng H, Lederer M R, Xiao R P, Gomez A M, Zhou Y Y, Ziman B, Spurgeon H, Lakatta E G, Lederer W J. Cell Calcium. 1996;20:129–140. doi: 10.1016/s0143-4160(96)90102-5. [DOI] [PubMed] [Google Scholar]
- 8.Yue D T. Science. 1997;276:755–756. doi: 10.1126/science.276.5313.755. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz K, Mercadier J J. Curr Opin Cardiol. 1996;11:227–236. doi: 10.1097/00001573-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Feldman A M, Weinberg E O, Ray P E, Lorell B H. Circ Res. 1993;73:184–192. doi: 10.1161/01.res.73.1.184. [DOI] [PubMed] [Google Scholar]
- 11.Weinberg E O, Schoen F J, George D, Kagaya Y, Douglas P S, Litwin S E, Schunkert H, Benedict C R, Lorell B H. Circulation. 1994;90:1410–1422. doi: 10.1161/01.cir.90.3.1410. [DOI] [PubMed] [Google Scholar]
- 12.Kagaya Y, Hajjar R J, Gwathmey J K, Barry W H, Lorell B H. Circulation. 1996;94:2915–2922. doi: 10.1161/01.cir.94.11.2915. [DOI] [PubMed] [Google Scholar]
- 13.Lorell B H. Circulation. 1997;96:3824–3827. [PubMed] [Google Scholar]
- 14.Weinberg E O, Lee M A, Weigner M, Lindpaintner K, Bishop S P, Benedict C R, Ho K K, Douglas P S, Chafizadeh E, Lorell B H. Circulation. 1997;95:1592–1600. doi: 10.1161/01.cir.95.6.1592. [DOI] [PubMed] [Google Scholar]
- 15.Hajjar R J, Kang J X, Gwathmey J K, Rosenzweig A. Circulation. 1997;95:423–429. doi: 10.1161/01.cir.95.2.423. [DOI] [PubMed] [Google Scholar]
- 16.Hajjar R J, Schmidt U, Kang J X, Matsui T, Rosenzweig A. Circ Res. 1997;81:145–153. doi: 10.1161/01.res.81.2.145. [DOI] [PubMed] [Google Scholar]
- 17.Hajjar R J, Schmidt U, Matsui T, Guerrero J L, Lee K H, Gwathmey J K, Dec G W, Semigran M J, Rosenzweig A. Proc Natl Acad Sci USA. 1998;95:5251–5256. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bett A J, Haddara W, Prevec L, Graham F L. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davia K, Hajjar R J, Terracciano C M N, Kent N S, Ranu H K, O'Gara P, Rosenzweig A, Harding S E. Physiol Genom. 1999;1:41–50. doi: 10.1152/physiolgenomics.1999.1.2.41. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt U, Hajjar R J, Helm P A, Kim C S, Doye A A, Gwathmey J K. J Mol Cell Cardiol. 1998;30:1929–1937. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- 21.Bradford M. Anal Biochem. 1976;72:248–260. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.He H, Giordano F J, Hilal-Dandan R, Choi D J, Rockman H A, McDonough P M, Bluhm W F, Meyer M, Sayen M R, Swanson E, et al. J Clin Invest. 1997;100:380–389. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker D L, Hashimoto K, Grupp I L, Ji Y, Reed T, Loukianov E, Grupp G, Bhagwhat A, Hoit B, Walsh R, et al. Circ Res. 1998;83:1205–1214. doi: 10.1161/01.res.83.12.1205. [DOI] [PubMed] [Google Scholar]
- 24.Bassani J W, Bassani R A, Bers D M. J Physiol. 1994;476:279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasenfuss G. Circ Res. 1998;83:966–968. doi: 10.1161/01.res.83.9.966. [DOI] [PubMed] [Google Scholar]
- 26.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 27.Hasenfuss G, Reinecke H, Studer R, Pieske B, Meyer M, Drexler H, Just H. Basic Res Cardiol. 1996;91 Suppl 2:17–22. doi: 10.1007/BF00795357. [DOI] [PubMed] [Google Scholar]
- 28.Hasenfuss G, Mulieri L A, Allen P D, Just H, Alpert N R. Circulation. 1996;94:3155–3160. doi: 10.1161/01.cir.94.12.3155. [DOI] [PubMed] [Google Scholar]
- 29.Hasenfuss G, Holubarsch C, Hermann H P, Astheimer K, Pieske B, Just H. Eur Heart J. 1994;15:164–170. doi: 10.1093/oxfordjournals.eurheartj.a060471. [DOI] [PubMed] [Google Scholar]
- 30.Mulieri L A, Hasenfuss G, Leavitt B, Allen P D, Alpert N R. Circulation. 1992;85:1743–1750. doi: 10.1161/01.cir.85.5.1743. [DOI] [PubMed] [Google Scholar]
- 31.Nascimben L, Ingwall J S, Pauletto P, Friedrich J, Gwathmey J K, Saks V, Pessina A C, Allen P D. Circulation. 1996;94:1894–1901. doi: 10.1161/01.cir.94.8.1894. [DOI] [PubMed] [Google Scholar]
- 32.Liao R, Nascimben L, Friedrich J, Gwathmey J K, Ingwall J S. Circ Res. 1996;78:893–902. doi: 10.1161/01.res.78.5.893. [DOI] [PubMed] [Google Scholar]
- 33.Bohm M, Flesch M, Schnabel P. J Mol Med. 1997;75:842–848. doi: 10.1007/s001090050175. [DOI] [PubMed] [Google Scholar]
- 34.Koss K L, Kranias E G. Circ Res. 1996;79:1059–1063. doi: 10.1161/01.res.79.6.1059. [DOI] [PubMed] [Google Scholar]
- 35.Lim H W, Molkentin J D. Nat Med. 1999;5:246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- 36.Svensson E C, Marshall D J, Woodard K, Lin H, Jiang F, Chu L, Leiden J M. Circulation. 1999;99:201–205. doi: 10.1161/01.cir.99.2.201. [DOI] [PubMed] [Google Scholar]
- 37.Gorziglia M I, Lapcevich C, Roy S, Kang Q, Kadan M, Wu V, Pechan P, Kaleko M. J Virol. 1999;73:6048–6055. doi: 10.1128/jvi.73.7.6048-6055.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]