

Abstract
Gene expression profiling of tumors using DNA microarrays is a promising method for predicting prognosis and treatment response in cancer patients. It was recently reported that expression profiles of sporadic breast cancers could be used to predict disease recurrence better than currently available clinical and histopathological prognostic factors. Having observed an overlap in those data between the genes that predict outcome and those that predict estrogen receptor-α status, we examined their predictive power in an independent data set. We conclude that it may be important to define prognostic expression profiles separately for estrogen receptor-α-positive and estrogen receptor-α-negative tumors.
Keywords: breast cancer, estrogen receptor, gene expression, microarray, prognosis
Introduction
Recent studies have shown that microarray analysis of tumors generate molecular phenotypes that can be used to classify tumors into subgroups that are not evident by traditional histopathological methods, to improve diagnosis, and to predict outcomes of disease [1-6]. In a recent study, van 't Veer et al. [7] reported, among other important conclusions, that expression profiles of sporadic primary breast tumors could be used to predict disease recurrence better than currently available clinical and histopathological prognostic factors (i.e. tumor grade, tumor size, angioinvasion, age, and estrogen receptor [ER]-α). Those investigators identified a set of 231 genes, from which they extracted 70 that were used to predict recurrences within 5 years with an accuracy of 89% for a test set of 20 tumors. However, our experience suggests that, in breast cancer patient populations, predicting clinical variables from gene expression data is complicated by a common correlation between ER-α status and the clinical parameters studied. ER-α status has been shown to leave a remarkable imprint on the expression profile of a tumor, and may have confounding effects on the clinical factor studied [7-9]. Therefore, we believe that it is necessary to be cautious when interpreting the results from the study of van 't Veer et al. and similar studies.
In the set of tumors analyzed by Van't Veer et al. [7] there is an evident correlation between clinical outcome for the patients and ER-α status of the tumors. For that reason, we investigated whether their results can be applied to a data set from a different patient cohort in which there is no correlation between ER-α status and prognosis.
Estrogen receptor-α and gene expression in breast cancer
ER-α status is by far the most obvious and noticeable subdivision of breast tumors based on gene expression profiles. This has become evident in several studies [4,7-9]. In a previous study [8] we used the gene expression profiles of 47 tumors and artificial neural networks to generate a prediction model based on the 100 highest ranked discriminator genes that could accurately predict the ER-α status of 11 breast tumors from an independent test set. Furthermore, we found that sets of genes much further down our ranked list still carried substantial information about ER-α status. Using a similar approach, West et al. [9] also succeeded in predicting the ER-α status of a set of breast tumors. In their study, van 't Veer et al. [7] also found that the main subdivision of tumors by unsupervised clustering was into two groups based on ER-α status. Indeed, there is considerable overlap between the genes that discriminate ER-α status in their data and those that were reported by us and by West et al. In addition, van't Veer et al. also found that as many as 2460 genes (out of 24,479 included in their arrays) carried information on ER-α status, confirming our finding that a large number of genes expressed in breast tumors are associated with ER-α status.
Prediction of prognosis from gene expression profiles
In the study by van't Veer et al. [7], disease recurrence was better predicted using the expression signature of 70 identified genes as compared with currently available clinical and histopathological prognostic factors (tumor grade, tumor size, angioinvasion, age, and ER-α status). We investigated the expression of approximately 7000 genes in a patient cohort of 44 primary breast tumors (size 20–50 mm, from lymph node-negative patients all treated with adjuvant tamoxifen). In contrast to the findings of van't Veer et al., we could not predict relapse with statistical confidence. The tumors we studied were selected to comprise four nearly equal sized groups: ER-α-positive or ER-α-negative, each subdivided into good (no distant metastases within a follow-up period of 5–11 years, median 8.0 years) and poor (distant metastases within 6 years, median 2.6 years) prognosis groups (Fig. 1). Using either artificial neural networks [5,8] or the signal-to-noise statistic [1] and random permutation tests, we were unable to develop a statistically reliable outcome classifier in these data. This may suggest that, because ER-α-positive and ER-α-negative tumors have such distinct expression profiles, the molecular pathways that are characteristic of the aggressive tumors within these two groups may not overlap to a substantial degree. Hence, it would probably be beneficial for those two groups to be considered separately when predicting prognosis on the basis of gene expression profiles. Unfortunately, such an analysis could not be performed satisfactorily in this data set because the number of tumors in the ER-α subgroups was too low. Additionally, it should be noted that the effect of ER-α status on outcome might be even more pronounced in this data set because the patients received endocrine treatment after primary surgical resection.
Figure 1.
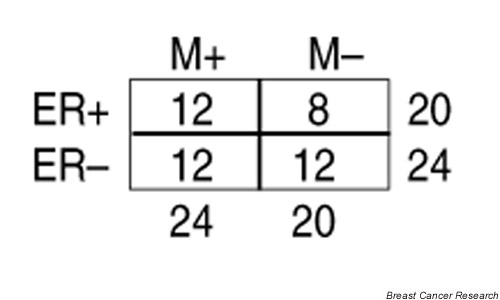
The distribution of clinical characteristics in our 44 sporadic breast tumors. Estrogen receptor-α status is denoted as ER+ and ER-. Clinical outcome for the patients is represented by M+ (distant recurrences within 6 years) and M- (no recurrences within a follow-up period of at least 5 years). Microarray data were generated as described by Gruvberger et al. [8].
Furthermore, we investigated whether the outcome predictor gene set generated by van't Veer et al. [7] could be applied to our patient cohort. We found that none of the outcome predictor genes identified by those investigators, which were also represented in our array data (58 out of 231), was a significant outcome discriminator (α < 0.01; as performed in Allander et al.[10]) in our material. In order to verify that the 58 genes still had significant predictive power in the dataset reported by van't Veer et al., we used their 'leave one out' supervised classification method in their data and found that these 58 genes still discriminated the two prognostic classes (odds ratio [OR] = 5.7, 95% confidence interval [CI] 2.1–15; P = 0.0007 [Fisher's exact test]), whereas no discrimination was seen in our data (OR = 1.9, 95% CI 0.5–6; P = 0.4). We used multidimensional scaling analysis to illustrate these results (Fig. 2).
Figure 2.
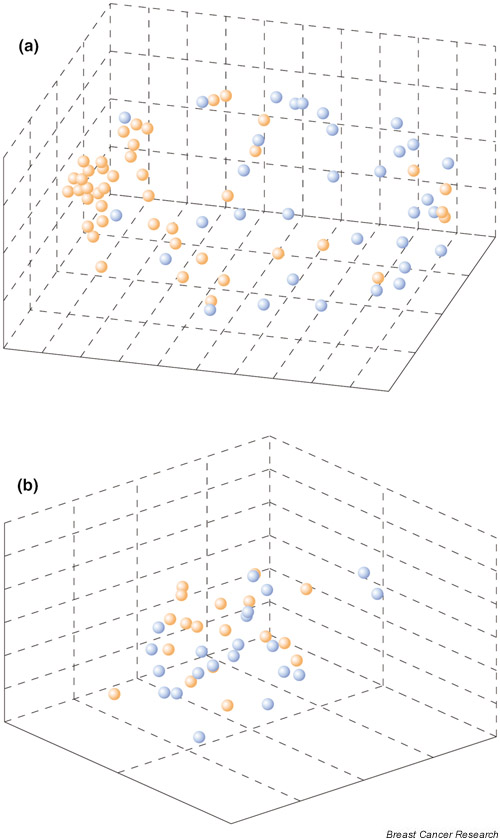
Multidimensional scaling (MDS) clustering of gene expression data from breast tumors using 58 out of 231 genes from the outcome predictor gene set identified by van 't Veer et al. [7] that were also included in our array analysis. These genes retain their predictive value in those data but not in our independent patient sample. (a) Fifty-eight primary breast tumors (training set) from the study by van't Veer et al. and (b) 44 from our array study are plotted. Tumors with a poor prognosis (distant recurrences within 6 years) are colored blue and tumors with a good prognosis (no recurrences within a follow-up period of 5–14 years) are orange. MDS displays the position of each tumor sample in a three-dimensional euclidean space, with the distance between the samples reflecting their approximate degree of correlation [11].
Intriguingly, for the training set of 58 sporadic tumor samples that van't Veer et al. [7] analyzed, their supervised ER-α predictor gene set also had predictive power for outcome (OR = 3.7, 95% CI 1.3–11; P = 0.02), perhaps owing to the predominance (80%) of ER-α-positive tumors in the good prognosis group (Fig. 3). Likewise, in their independent test set, all of the tumors with a good prognosis were ER-α-positive except one (86%; Fig. 4). In addition, 71% of the 231 prognostic genes identified by van't Veer et al. were also listed by them as ER-α status reporter genes, confirming an overlap between the predictors of prognosis and ER-α status. It is important to note, however, that they were able to achieve better prognostic predictions using the 231 selected genes (OR = 15, 95% CI 4–56; P = 0.000004) than with the ER-α predictor gene set. Nevertheless, we believe that the correlation between prognosis and ER-α status in samples reported by van't Veer et al. (Figs 3 and 4) might have led to the selection of a prognostic gene set that may not be broadly applicable to other breast tumor cohorts in which no correlation between ER-α status and prognosis exists.
Figure 3.
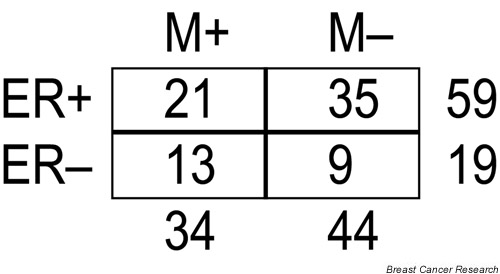
The distribution of clinical characteristics of the 78 sporadic breast tumors used in the training/validation set in the study by van 't Veer et al. [7]. Estrogen receptor-α status is denoted as ER+ and ER-. Clinical outcome for the patients is represented by M+ (distant recurrences within 5 years) and M- (no recurrences within a follow-up period of at least 5 years).
Figure 4.
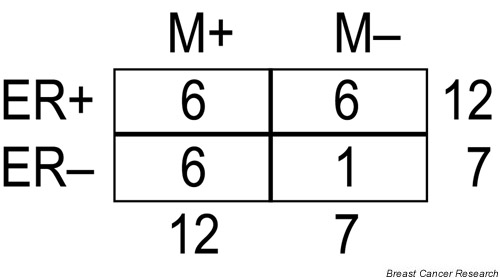
The distribution of clinical characteristics of the 19 sporadic breast tumors used as an independent test set in the study by van't Veer et al. [7]. Estrogen receptor-α status is denoted as ER+ and ER-. Clinical outcome for the patients is represented by M+ (distant recurrences within 5 years) and M- (no recurrences within a follow-up period of at least 5 years).
Conclusion
Most breast cancer gene expression studies to date have examined limited sample numbers and tend not to have sufficient power to allow analysis within standard clinicopathologically defined subsets. Moreover, the majority of breast cancer patients today receive some form of postoperative treatment (radiation, endocrine and/or chemotherapy) that influences the clinical course and significance of prognostic factors. In general, there are risks inherent in the development of a prognostic classifier from a sample set comprised of patients who are heterogeneous for clinicopathological variables of known prognostic significance. Although the resulting predictive classifier may accurately describe the sample set used for its development, that result may merely reflect the influence of known prognostic variables on gene expression, and it may fail when applied to an independent test set containing patients who are indistinguishable on the basis of all standard clinical and pathological markers, but who still differ in terms of outcome. Although it is still of great importance to gain an understanding of which patterns of gene expression are linked to known variables such as ER-α status, studies should be designed to reveal rather than obscure these links, and to uncover any potential gene expression patterns that predict outcome within uniform groups. If microarray based prognostic tools are to become widely used in the clinical setting, then additional studies must be conducted that seek and independently validate prognostic information in larger sample sets, which are carefully selected and matched with respect to known prognostic variables, such as ER-α status and therapy.
Abbreviations
CI = confidence interval; ER = estrogen receptor; OR = odds ratio.
The original version of this commentary contained an error in figure 1. This has been corrected. See reply, http://breast-cancer-research.com/content/5/1/57
References
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Staudt LM, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Gruvberger S, Ringner M, Chen Y, Panavally S, Saal LH, Borg A, Ferno M, Peterson C, Meltzer PS. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- West M, Blanchette C, Dressman H, Huang E, Ishida S, Spang R, Zuzan H, Olson JA, Jr, Marks JR, Nevins JR. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci USA. 2001;98:11462–11467. doi: 10.1073/pnas.201162998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander SV, Nupponen NN, Ringner M, Hostetter G, Maher GW, Goldberger N, Chen Y, Carpten J, Elkahloun AG, Meltzer PS. Gastrointestinal stromal tumors with KIT mutations exhibit a remarkably homogeneous gene expression profile. Cancer Res. 2001;61:8624–8628. [PubMed] [Google Scholar]
- Khan J, Simon R, Bittner M, Chen Y, Leighton SB, Pohida T, Smith PD, Jiang Y, Gooden GC, Trent JM, Meltzer PS. Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res. 1998;58:5009–5013. [PubMed] [Google Scholar]